
Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Reef Fish Family Holocentridae (Holocentriformes)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, Library Construction, and Sequencing
2.2. Sequence Assembly, Annotation, and Analyses
2.3. Phylogenetic Analyses
2.4. Positive Selection
3. Results
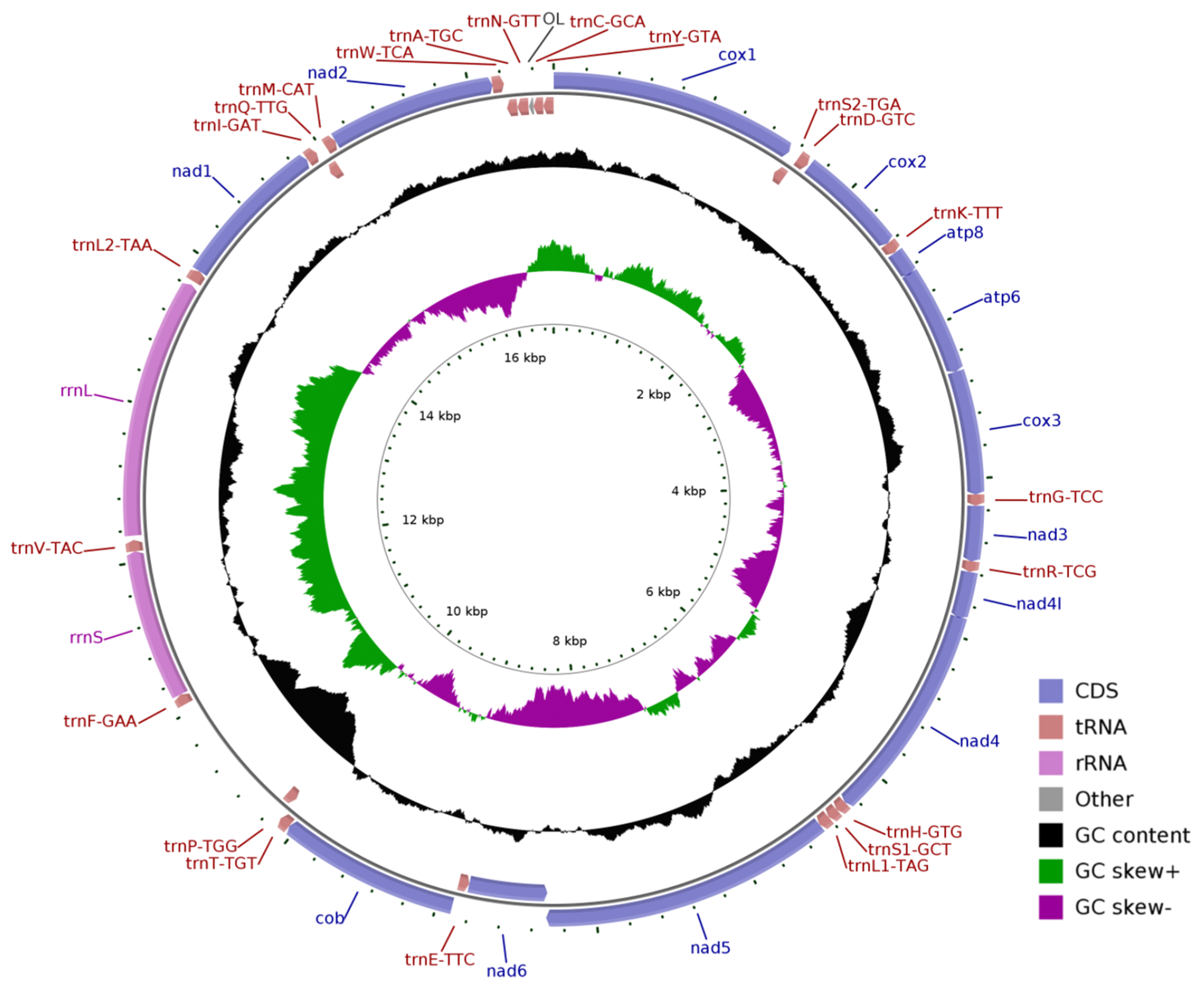
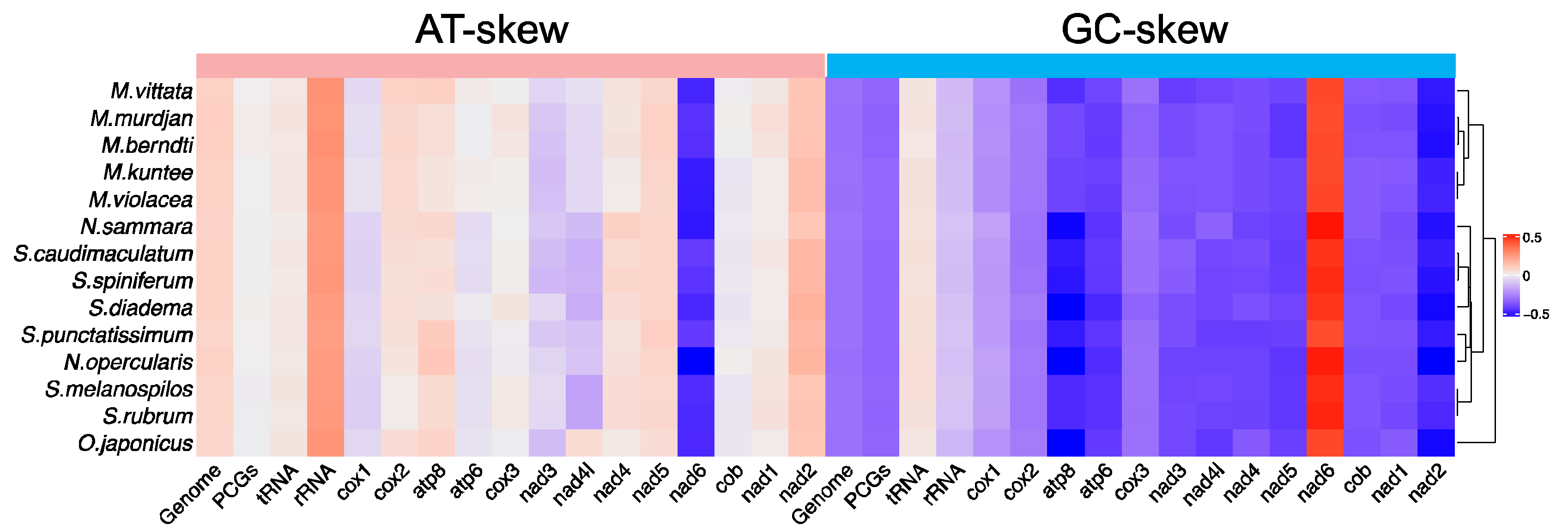
3.1. General Features of Mitochondrial Genomes
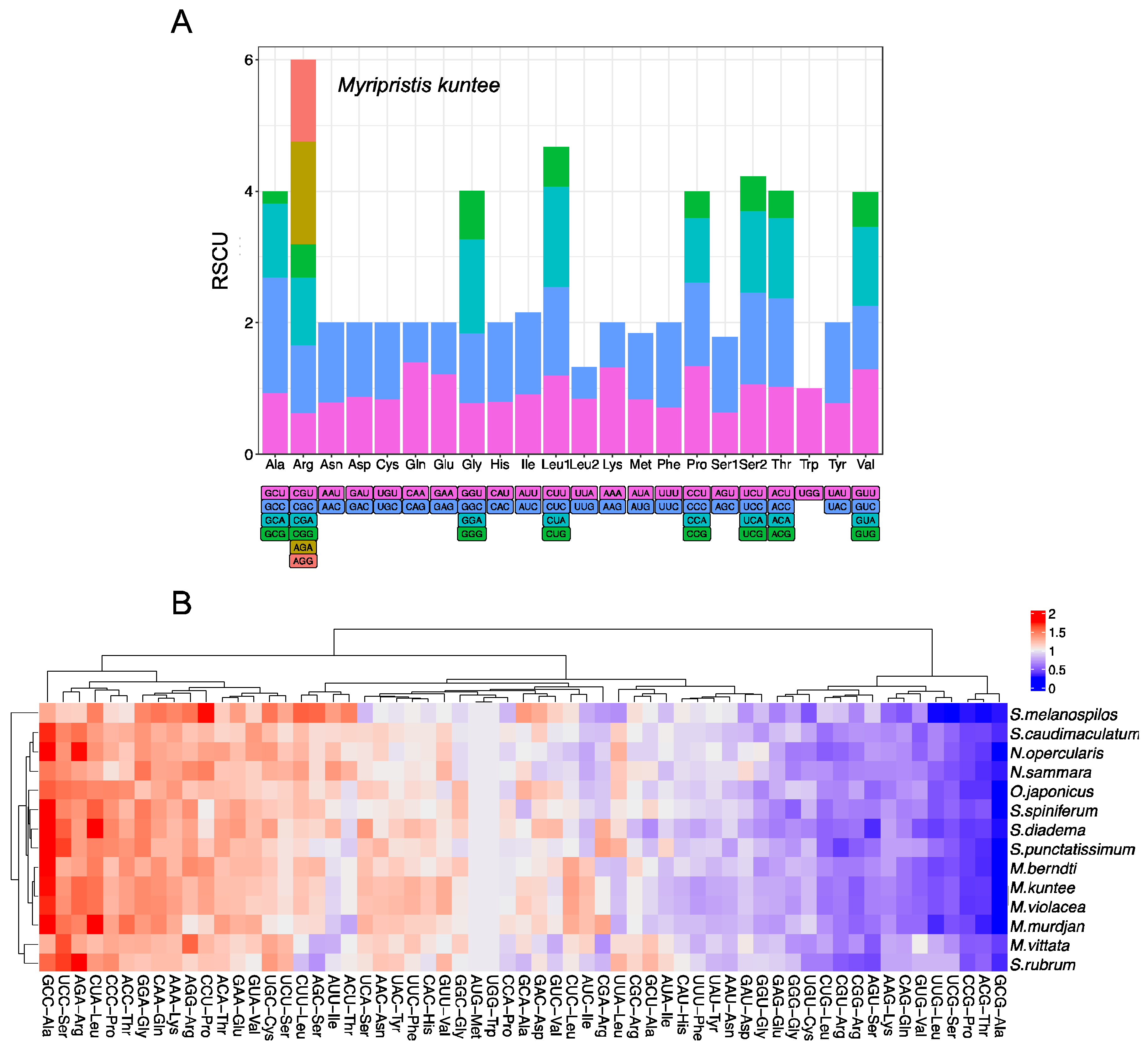
3.2. Nucleotide Composition of Protein-Coding Genes of the Holocentrid Mitogenomes and the Codon Usage
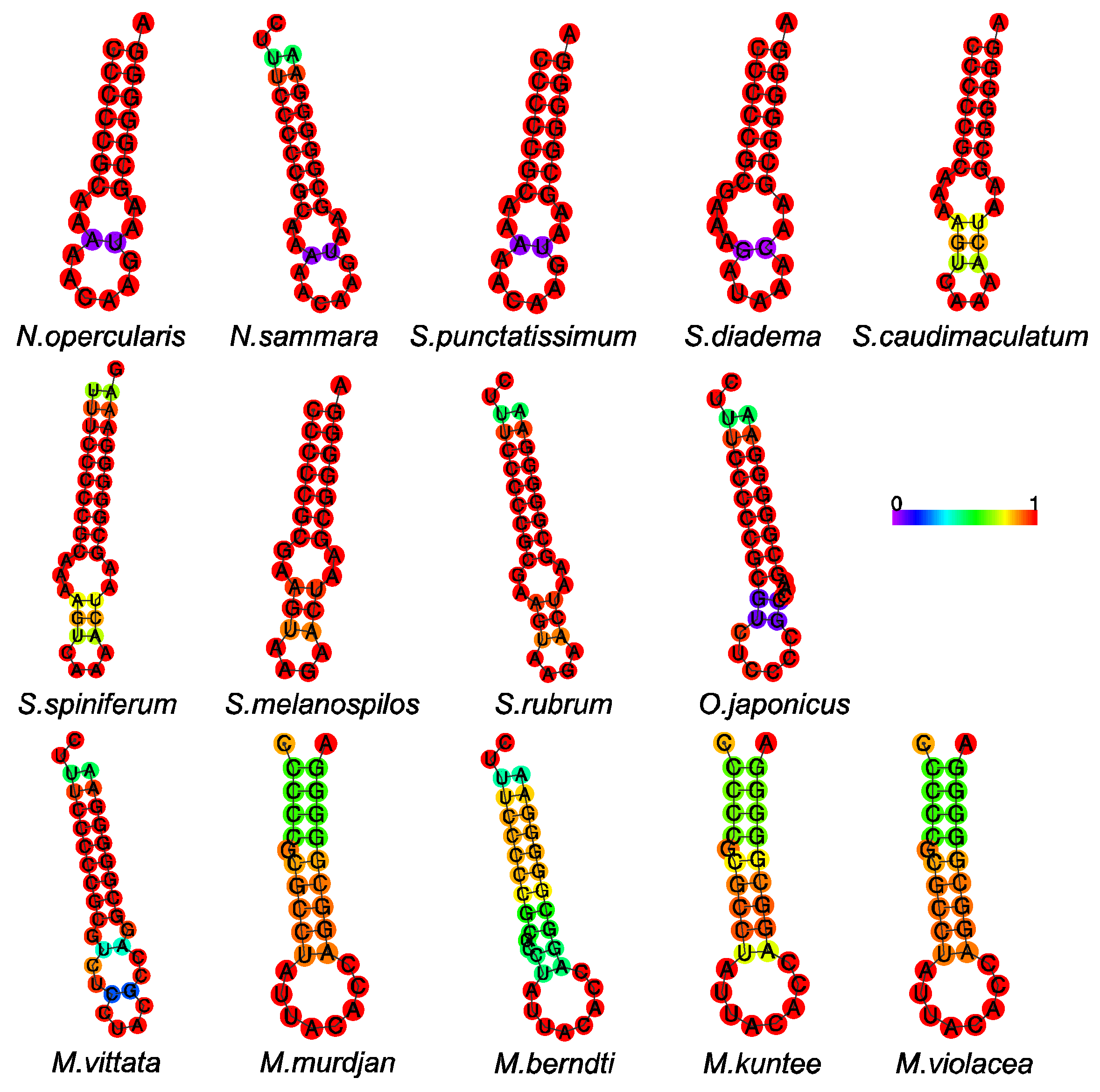
3.3. Transfer RNA and Ribosomal RNA
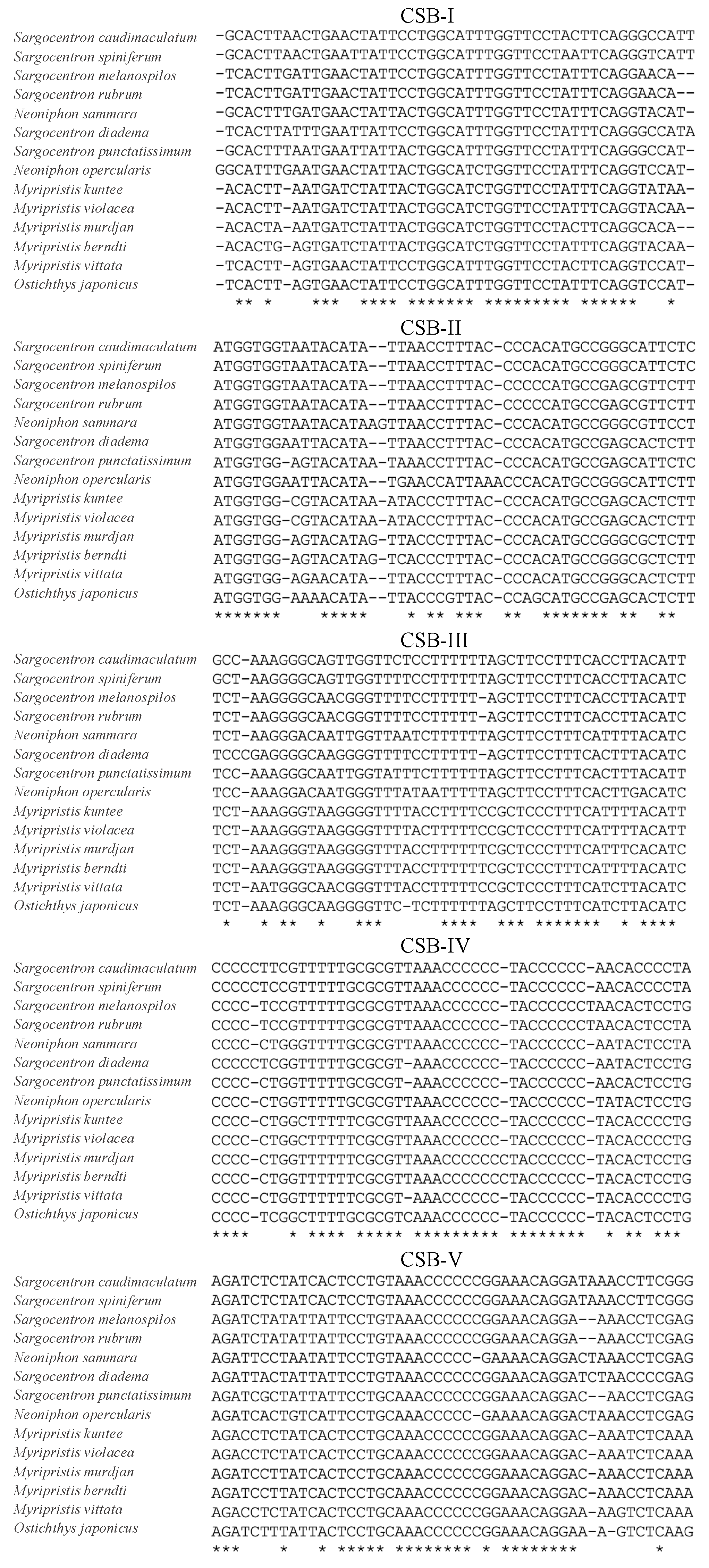
3.4. Overlaps and Control Regions
3.5. Phylogenetic Analysis
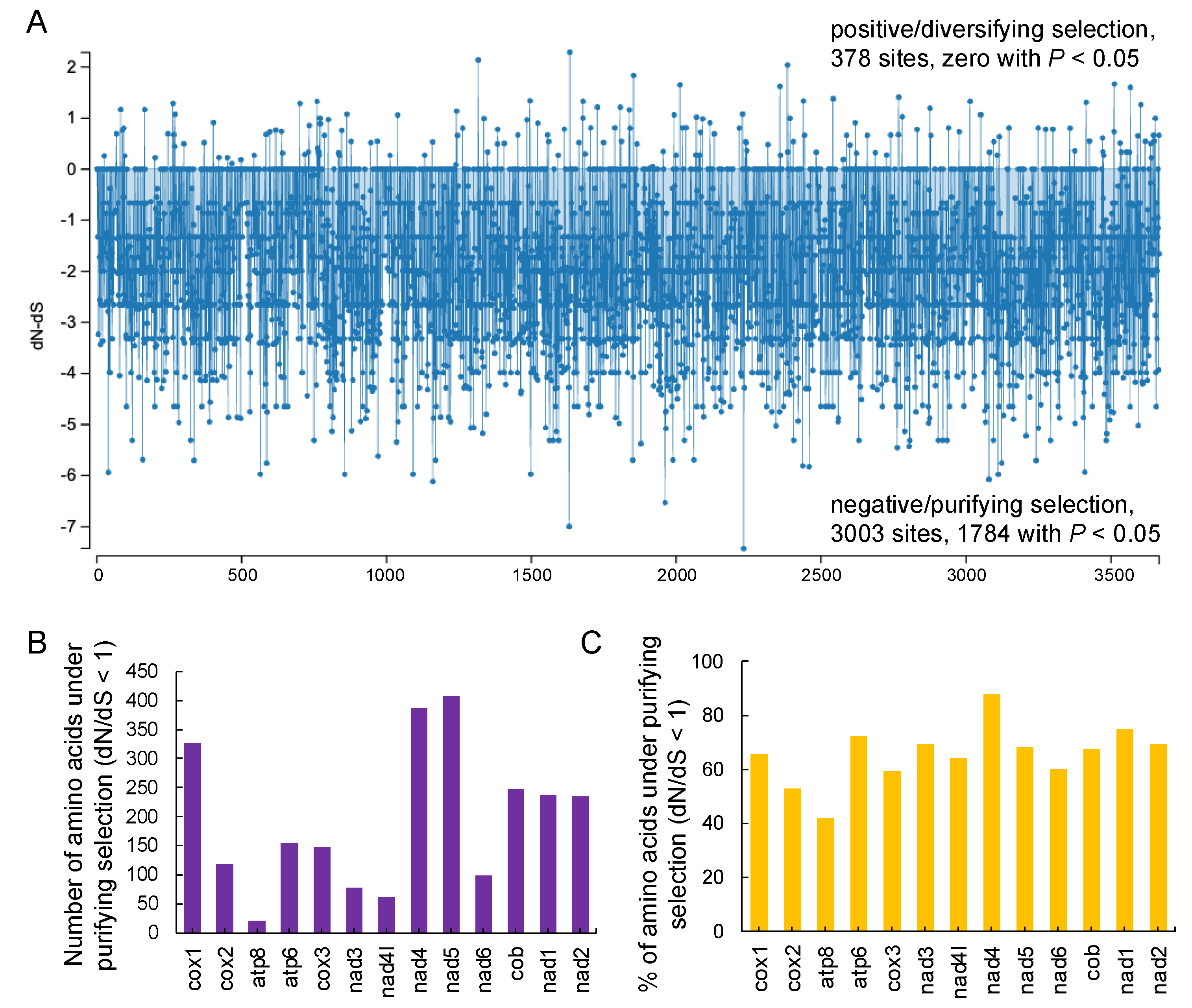
3.6. Non-Synonymous, Synonymous Substitutions, and Positive Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parmentier, E.; Vandewalle, P.; Brie, C.; Dinraths, L.; Lecchini, D. Comparative study on sound production in different Holocentridae species. Front. Zool. 2011, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Fogg, L.G.; Cortesi, F.; Lecchini, D.; Gache, C.; Marshall, N.J.; de Busserolles, F. Development of dim-light vision in the nocturnal reef fish family Holocentridae. I: Retinal gene expression. J. Exp. Biol. 2022, 225, jeb244513. [Google Scholar] [CrossRef] [PubMed]
- de Busserolles, F.; Cortesi, F.; Fogg, L.; Stieb, S.M.; Luehrmann, M.; Marshall, N.J. The visual ecology of Holocentridae, a nocturnal coral reef fish family with a deep-sea-like multibank retina. J. Exp. Biol. 2021, 224, jeb233098. [Google Scholar] [CrossRef]
- Andrews, J.V.; Schein, J.P.; Friedman, M. An earliest Paleocene squirrelfish (Teleostei: Beryciformes: Holocentroidea) and its bearing on the timescale of holocentroid evolution. J. Syst. Palaeontol. 2023, 21, 2168571. [Google Scholar] [CrossRef]
- Schmitz, L.; Wainwright, P.C. Nocturnality constrains morphological and functional diversity in the eyes of reef fishes. BMC Evol. Biol. 2011, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Quimpo, T.J.R.; Cabaitan, P.C.; Olavides, R.D.D.; Dumalagan, E.E., Jr.; Munar, J.; Siringan, F.P. Spatial variability in reef-fish assemblages in shallow and upper mesophotic coral ecosystems in the Philippines. J. Fish. Biol. 2019, 94, 17–28. [Google Scholar] [CrossRef]
- Randall, J.E. Reef and Shore Fishes of the South Pacific; University of Hawaii Press: Honolulu, HI, USA, 2003. [Google Scholar]
- Page, H.M.; Brooks, A.J.; Kulbicki, M.; Galzin, R.; Miller, R.J.; Reed, D.C.; Schmitt, R.J.; Holbrook, S.J.; Koenigs, C. Stable isotopes reveal trophic relationships and diet of consumers in temperate kelp forest and coral reef ecosystems. Oceanography 2013, 26, 181–189. [Google Scholar] [CrossRef]
- Paxton, J.R.; Eschmeyer, W.N. Encyclopedia of Fishes, 2nd ed.; (Natural World); Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Embley, T.M.; Martin, W. Eukaryotic evolution, changes and challenges. Nature 2006, 440, 623–630. [Google Scholar] [CrossRef]
- Martijn, J.; Vosseberg, J.; Guy, L.; Offre, P.; Ettema, T.J.G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 2018, 557, 101–105. [Google Scholar] [CrossRef]
- Yang, M.; Gong, L.; Sui, J.; Li, X. The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS ONE 2019, 14, e0217952. [Google Scholar] [CrossRef]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 719. [Google Scholar] [CrossRef]
- Gao, J.; Li, C.; Yu, D.; Wang, T.; Lin, L.; Xiao, Y.; Wu, P.; Liu, Y. Comparative mitogenome analyses uncover mitogenome features and phylogenetic implications of the Parrotfishes (Perciformes: Scaridae). Biology 2023, 12, 410. [Google Scholar] [CrossRef]
- Ramos, N.I.; DeLeo, D.M.; Horowitz, J.; McFadden, C.S.; Quattrini, A.M. Selection in coral mitogenomes, with insights into adaptations in the deep sea. Sci. Rep. 2023, 13, 6016. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, X.; Liu, G.; Wu, X.; Wei, Q.; Sun, G.; Mei, X.; Dong, Y.; Sha, W.; Zhang, H. Adaptability and evolution of Gobiidae: A genetic exploration. Animals 2022, 12, 1741. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Snelgrove, P.V.; Tyler, P. Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 2014, 29, 465–475. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.M.; Mueller, I.A. The unique mitochondrial form and function of Antarctic channichthyid icefishes. Integr. Comp. Biol. 2010, 50, 993–1008. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Kuck, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, R.; Xu, T. The mitochondrial genome of the spinyhead croaker Collichthys lucida: Genome organization and phylogenetic consideration. Mar. Genom. 2011, 4, 17–23. [Google Scholar] [CrossRef]
- Jin, X.; Wang, R.; Xu, T.; Shi, G. Complete mitochondrial genome of Oxuderces dentatus (Perciformes, Gobioidei). Mitochondrial DNA 2012, 23, 142–144. [Google Scholar] [CrossRef]
- Chen, L.; Lin, Y.; Xiao, Q.; Lin, Y.; Du, Y.; Lin, C.; Ward-Fear, G.; Hu, C.; Qu, Y.; Li, H. Characterization of the complete mitochondrial genome of the many-lined sun skink (Eutropis multifasciata) and comparison with other Scincomorpha species. Genomics 2021, 113, 2526–2536. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, K.; Xiao, L. The complete mitochondrial genome of Pimephales promelas (Cypriniformes: Cyprinidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 3711–3712. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, B.; Li, X.; Lv, Y.; Jiang, X.; Ren, C.; Hu, C.; Luo, P. The complete mitochondrial genome of Stichopus naso (Aspidochirotida: Stichopodidae: Stichopus) and Its Phylogenetic Position. Genes 2022, 13, 825. [Google Scholar] [CrossRef]
- Liu, H.; Sun, C.; Zhu, Y.; Li, Y.; Wei, Y.; Ruan, H. Mitochondrial genomes of four American characins and phylogenetic relationships within the family Characidae (Teleostei: Characiformes). Gene 2020, 762, 145041. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhu, Z.; Zhu, L.; Wang, D.; Wang, J.; Lin, Q. The complete mitogenome of Lysmata vittata (Crustacea: Decapoda: Hippolytidae) with implication of phylogenomics and population genetics. PLoS ONE 2021, 16, e0255547. [Google Scholar] [CrossRef] [PubMed]
- Gun, L.; Yumiao, R.; Haixian, P.; Liang, Z. Comprehensive analysis and comparison on the codon usage pattern of whole Mycobacterium tuberculosis coding genome from different area. Biomed. Res. Int. 2018, 2018, 3574976. [Google Scholar] [CrossRef]
- Sampey, A.; McKinnon, A.D.; Meekan, M.G.; McCormick, M.I. Glimpse into guts: Overview of the feeding of larvae of tropical shorefishes. Mar. Ecol. Prog. Ser. 2007, 339, 243–257. [Google Scholar] [CrossRef]
- Greenfield, D.W.; Randall, J.E.; Psomadakis, P.N. A review of the soldierfish genus Ostichthys (Beryciformes: Holocentridae), with descriptions of two new species from Myanmar. J. Ocean. Sci. Found. 2017, 26, 1–33. [Google Scholar]
- Dornburg, A.; Moore, J.A.; Webster, R.; Warren, D.L.; Brandley, M.C.; Iglesias, T.L.; Wainwright, P.C.; Near, T.J. Molecular phylogenetics of squirrelfishes and soldierfishes (Teleostei: Beryciformes: Holocentridae): Reconciling more than 100 years of taxonomic confusion. Mol. Phylogenet Evol. 2012, 65, 727–738. [Google Scholar] [CrossRef]
- Phung, T.N.; Huber, C.D.; Lohmueller, K.E. Determining the effect of natural selection on linked neutral divergence across species. PLoS Genet. 2016, 12, e1006199. [Google Scholar] [CrossRef]
- Elyashiv, E.; Bullaughey, K.; Sattath, S.; Rinott, Y.; Przeworski, M.; Sella, G. Shifts in the intensity of purifying selection: An analysis of genome-wide polymorphism data from two closely related yeast species. Genome Res. 2010, 20, 1558–1573. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Start | End | Strand | Size (bp) | Start Codon | Stop Codon | Anticodons |
|---|---|---|---|---|---|---|---|
| cox1 | 1 | 1557 | + | 1557 | GTG | AGA | − |
| trnS2 | 1553 | 1623 | − | 71 | − | − | TGA |
| trnD | 1627 | 1698 | + | 72 | − | − | GTC |
| cox2 | 1712 | 2402 | + | 691 | ATG | T | − |
| trnK | 2403 | 2475 | + | 73 | − | − | TTT |
| atp8 | 2477 | 2644 | + | 168 | ATG | TAA | − |
| atp6 | 2635 | 3318 | + | 684 | ATG | TAA | − |
| cox3 | 3318 | 4103 | + | 786 | ATG | TAA | − |
| trnG | 4103 | 4173 | + | 71 | − | − | TCC |
| nad3 | 4174 | 4524 | + | 351 | ATG | TAG | − |
| trnR | 4523 | 4591 | + | 69 | − | − | TCG |
| nad4l | 4592 | 4888 | + | 297 | ATG | TAA | − |
| nad4 | 4882 | 6262 | + | 1381 | ATG | T | − |
| trnH | 6263 | 6331 | + | 69 | − | − | GTG |
| trnS1 | 6332 | 6399 | + | 68 | − | − | GCT |
| trnL1 | 6401 | 6473 | + | 73 | − | − | TAG |
| nad5 | 6474 | 8312 | + | 1839 | ATG | TAA | − |
| nad6 | 8308 | 8829 | − | 522 | ATG | AGG | − |
| trnE | 8830 | 8898 | − | 69 | − | − | TTC |
| cob | 8905 | 10,045 | + | 1141 | ATG | T | − |
| trnT | 10,046 | 10,117 | + | 72 | − | − | TGT |
| trnP | 10,122 | 10,191 | − | 70 | − | − | TGG |
| trnF | 11,054 | 11,121 | + | 68 | − | − | GAA |
| rrnS | 11,122 | 12,070 | + | 949 | − | − | − |
| trnV | 12,071 | 12,142 | + | 72 | − | − | TAC |
| rrnL | 12,170 | 13,814 | + | 1645 | − | − | − |
| trnL2 | 13,839 | 13,912 | + | 74 | − | − | TAA |
| nad1 | 13,913 | 14,887 | + | 975 | ATG | TAA | − |
| trnI | 14,892 | 14,961 | + | 70 | − | − | GAT |
| trnQ | 14,961 | 15,031 | − | 71 | − | − | TTG |
| trnM | 15,031 | 15,100 | + | 70 | − | − | CAT |
| nad2 | 15,101 | 16,147 | + | 1047 | ATG | TAA | − |
| trnW | 16,147 | 16,219 | + | 73 | − | − | TCA |
| trnA | 16,221 | 16,289 | − | 69 | − | − | TGC |
| trnN | 16,291 | 16,363 | − | 73 | − | − | GTT |
| trnC | 16,397 | 16,461 | − | 65 | − | − | GCA |
| trnY | 16,462 | 16,529 | − | 68 | − | − | GTA |
| Base Composition (%) | CSB-I | CSB-II | CSB-III | CSB-IV | CSB-V |
|---|---|---|---|---|---|
| A | 27.66 | 25.53 | 18.37 | 12.77 | 36.73 |
| T | 38.30 | 25.53 | 42.86 | 23.40 | 14.29 |
| G | 14.89 | 17.02 | 16.33 | 10.64 | 12.24 |
| C | 19.15 | 31.91 | 22.45 | 53.19 | 36.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Q.; Liu, Y.; Li, C.-H.; Zhao, J.-F.; Wang, T. Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Reef Fish Family Holocentridae (Holocentriformes). Biology 2023, 12, 1273. https://doi.org/10.3390/biology12101273
Tang Q, Liu Y, Li C-H, Zhao J-F, Wang T. Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Reef Fish Family Holocentridae (Holocentriformes). Biology. 2023; 12(10):1273. https://doi.org/10.3390/biology12101273
Chicago/Turabian StyleTang, Qin, Yong Liu, Chun-Hou Li, Jin-Fa Zhao, and Teng Wang. 2023. "Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Reef Fish Family Holocentridae (Holocentriformes)" Biology 12, no. 10: 1273. https://doi.org/10.3390/biology12101273
APA StyleTang, Q., Liu, Y., Li, C.-H., Zhao, J.-F., & Wang, T. (2023). Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Reef Fish Family Holocentridae (Holocentriformes). Biology, 12(10), 1273. https://doi.org/10.3390/biology12101273