
Deciphering the CircRNA-Regulated Response of Western Honey Bee (Apis mellifera) Workers to Microsporidian Invasion


Abstract
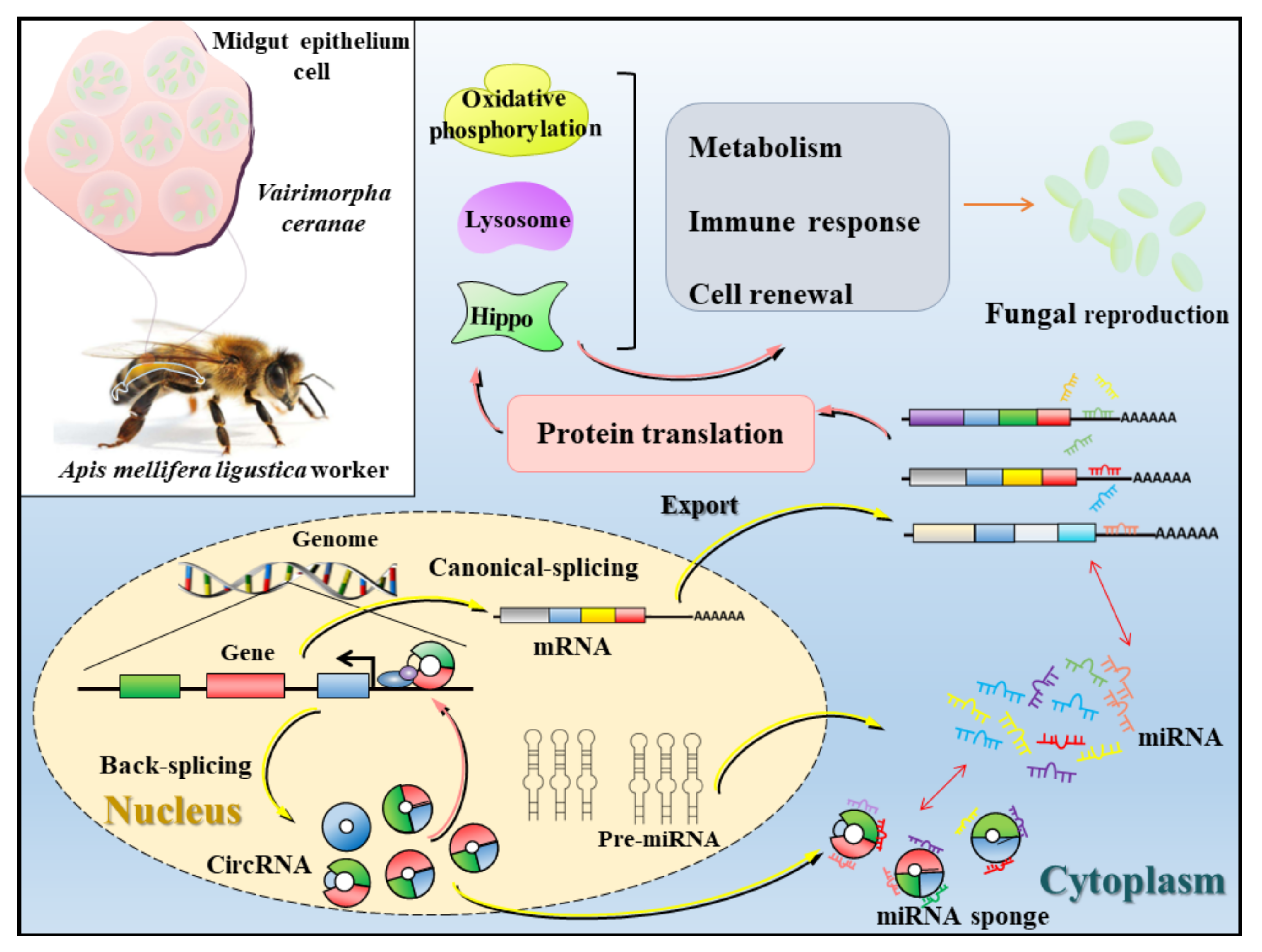
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Samples
2.2. Source of Whole Transcriptome Data
2.3. Identification and Conservative Analysis of CircRNAs
2.4. Investigation of DEcircRNAs and Their Source Genes
2.5. Target Prediction and Regulatory Network Analysis of DEcircRNAs
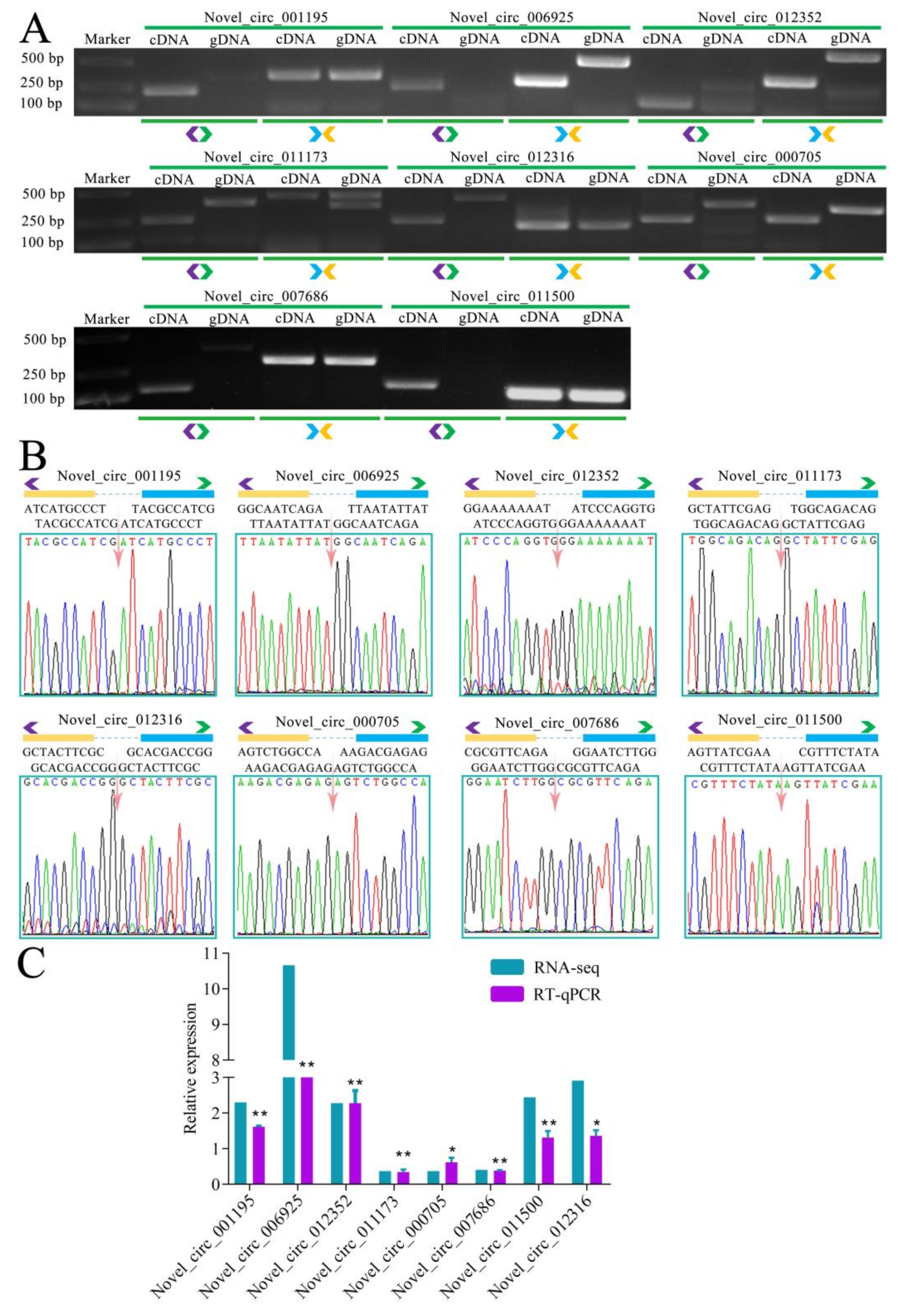
2.6. PCR and Sanger Sequencing Confirmation of Novel circRNAs
2.7. Stem-Loop RT-PCR Verification of miRNAs
2.8. RT-qPCR Validation of DEcircRNAs
3. Results
3.1. Overview of the Transcriptome Data Generated from V. ceranae-Inoculated Midguts of A. m. ligustica Workers
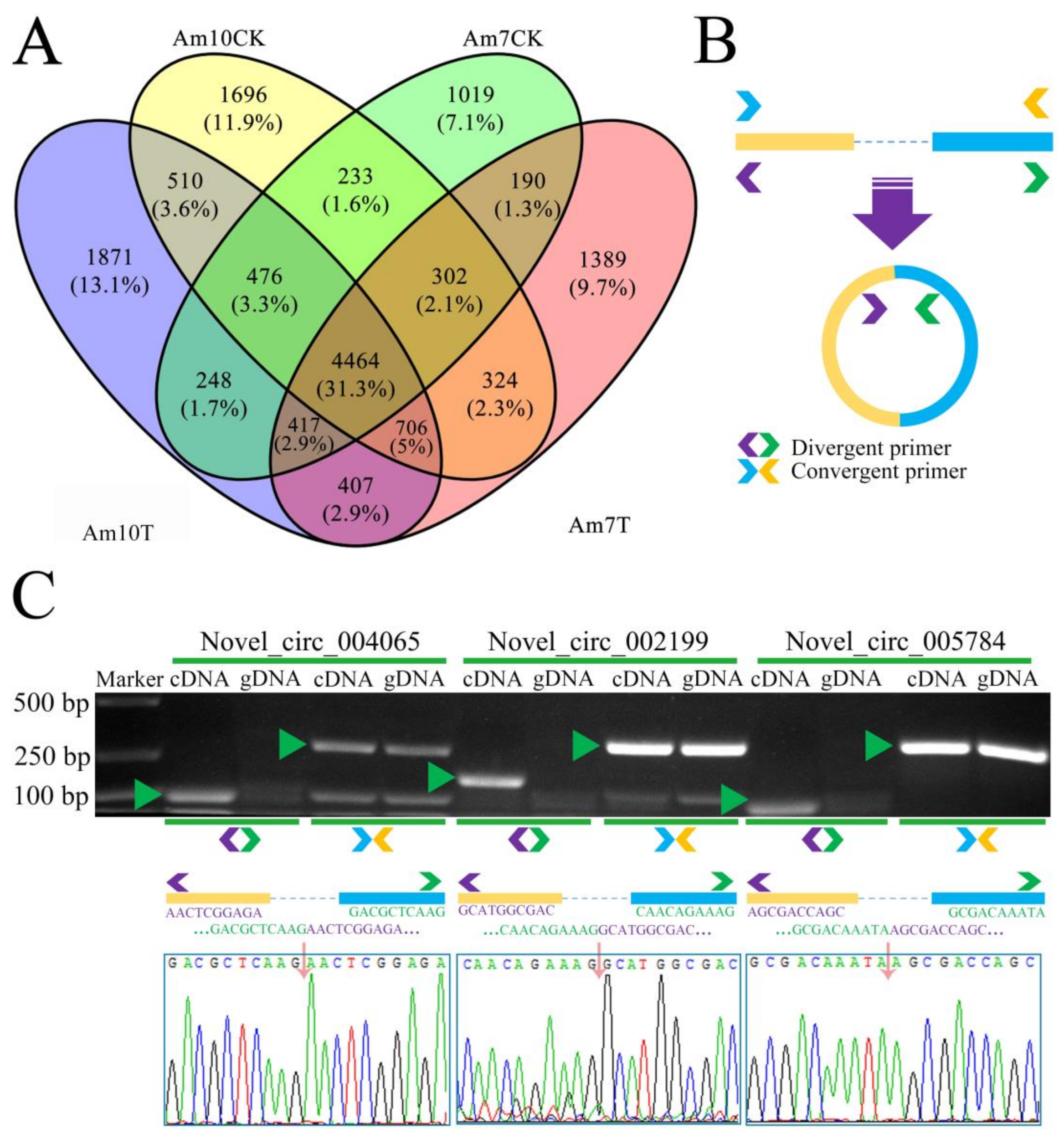
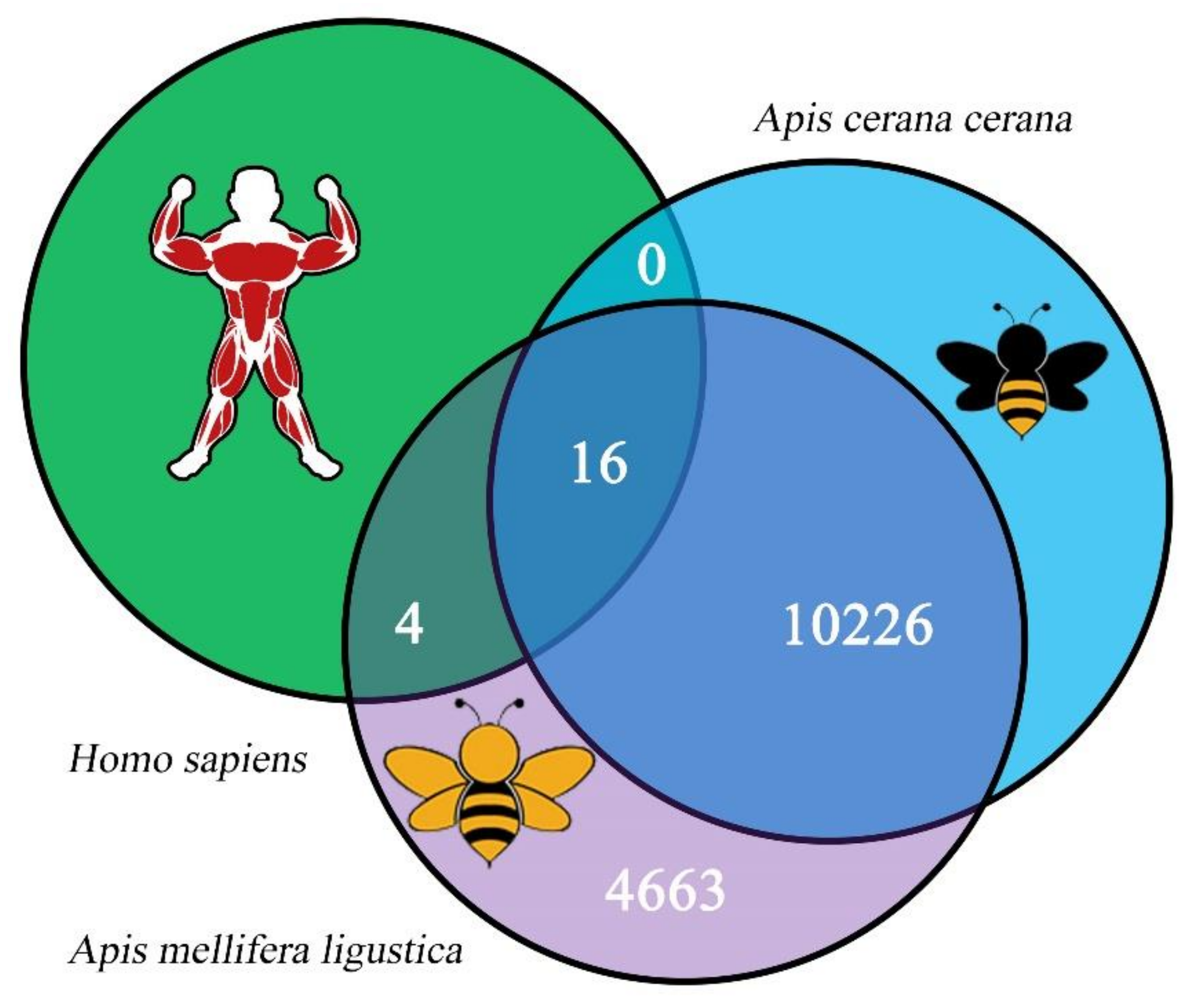
3.2. Quantity, Expression, and Conservation of A. m. ligustica circRNAs
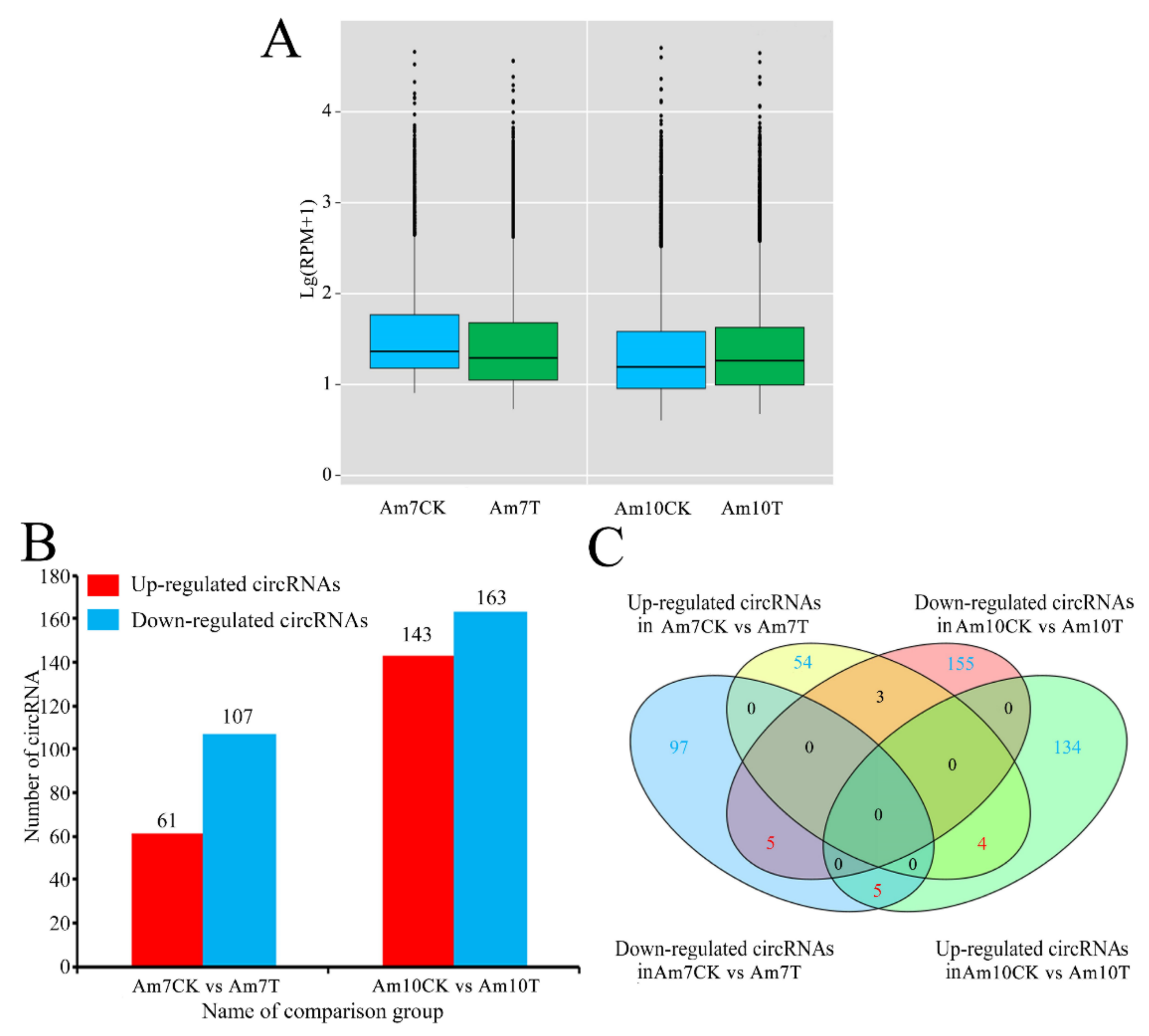
3.3. DEcircRNAs Involved in Response of A. m. ligustica Workers to V. ceranae Infection
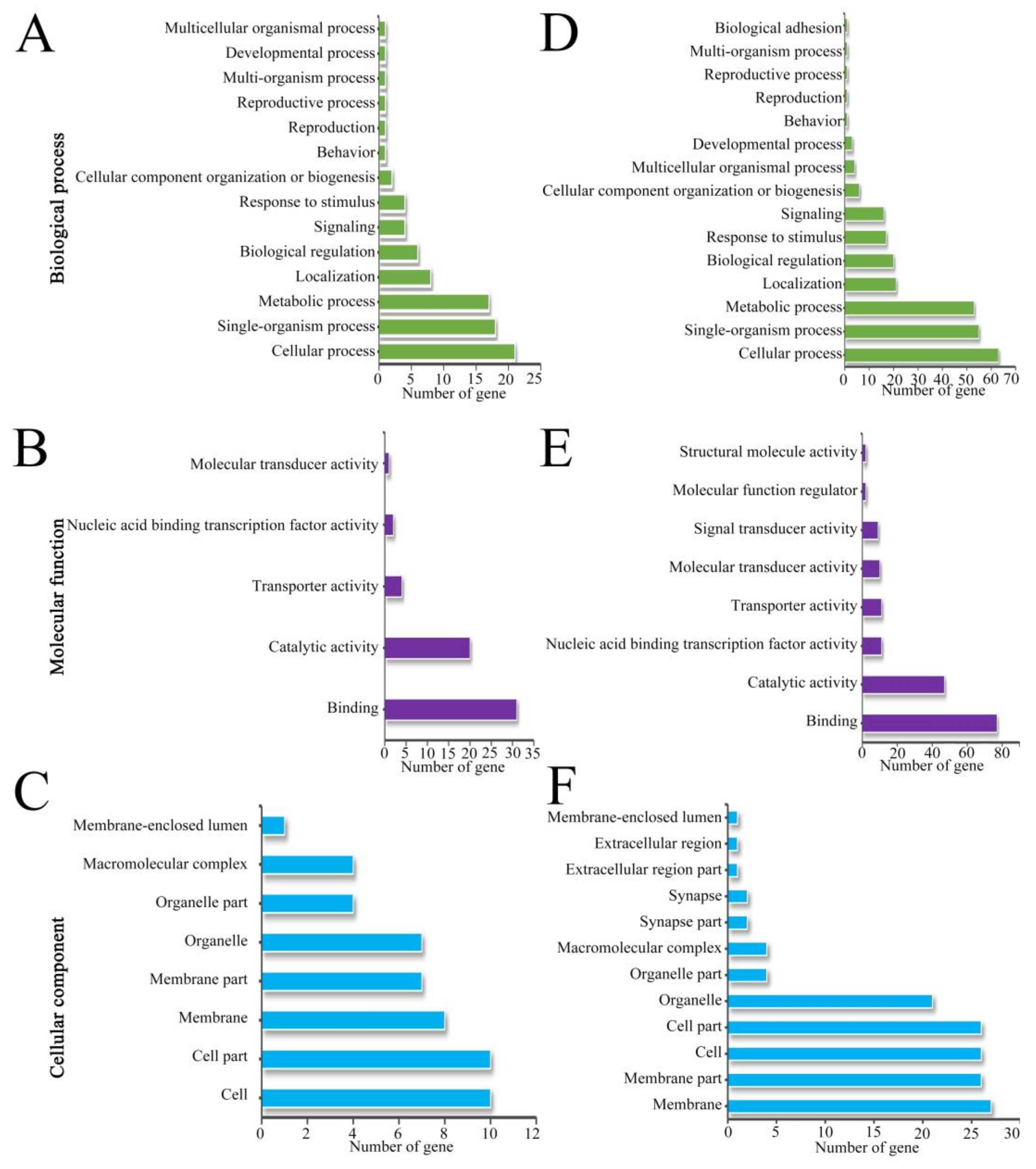
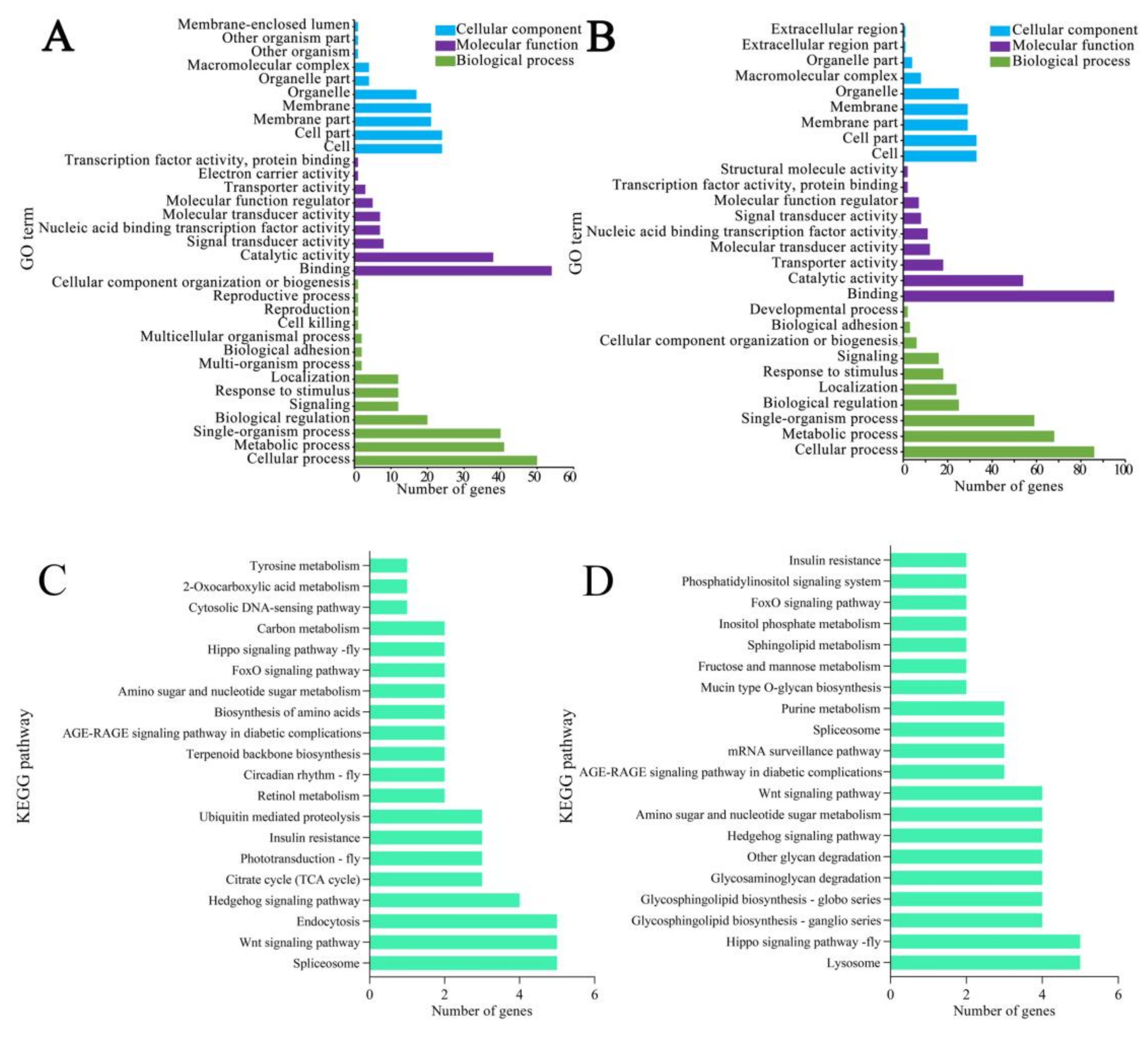
3.4. Functional Annotation of Source Genes of DEcircRNAs
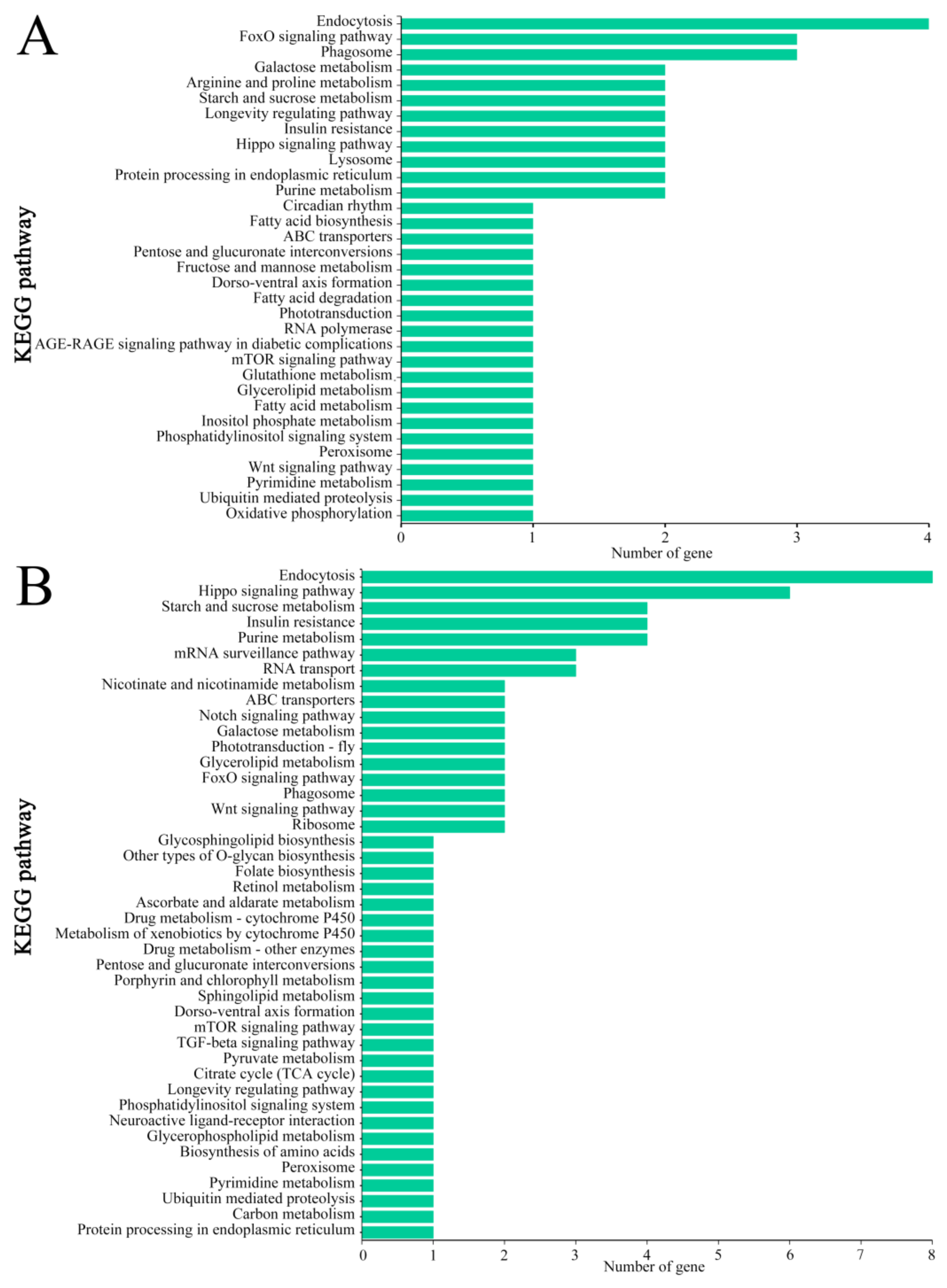
3.5. KEGG Pathway Annotation of Source Genes of DEcircRNAs
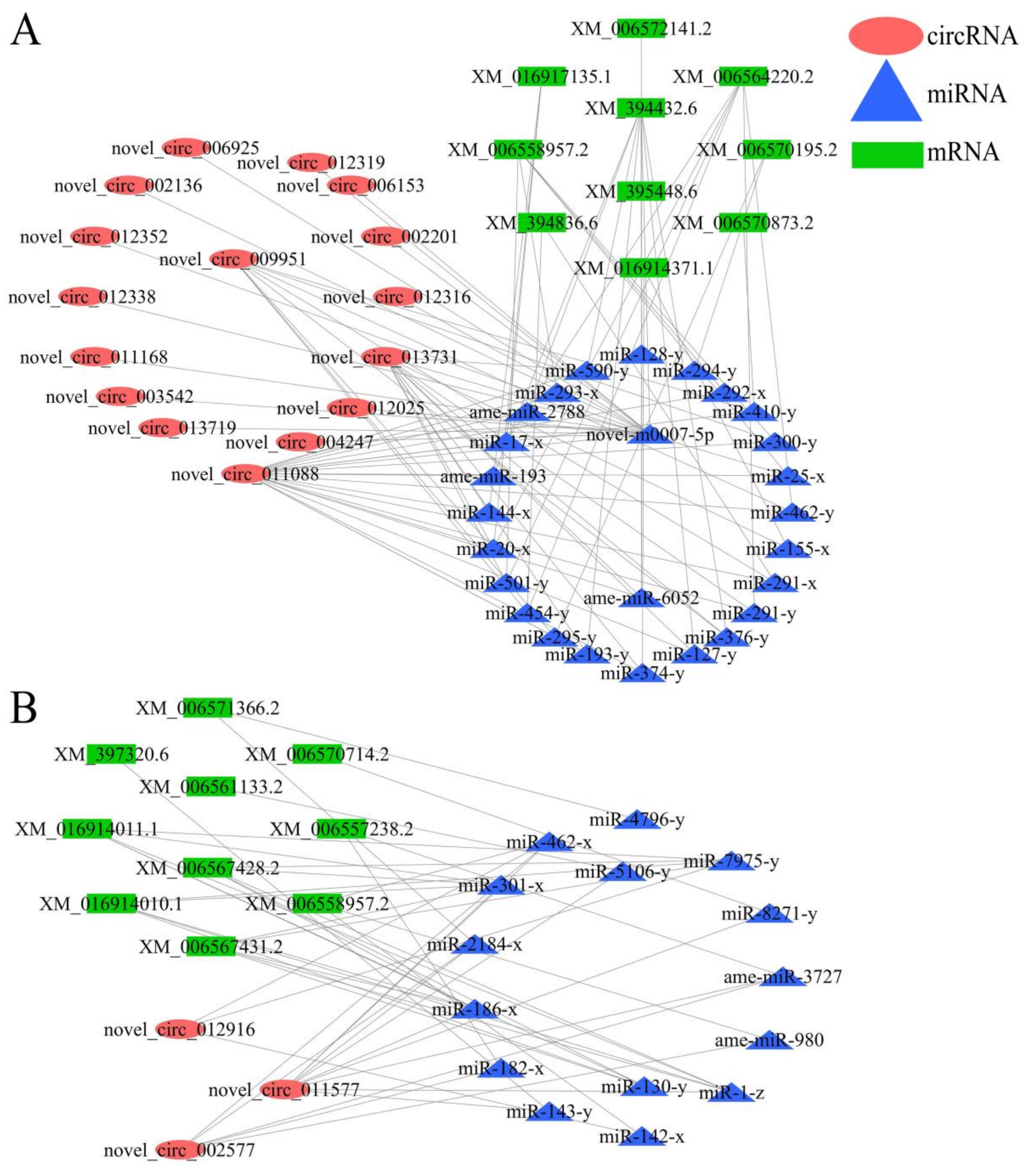
3.6. DEcircRNA-miRNA Regulatory Network Engaged in Response of A. m. ligustica Workers to V. ceranae Invasion
3.7. CeRNA Regulatory Network Associated with V. ceranae Response by A. m. ligustica Workers
4. Discussion
4.1. Number, Conservation, and Expression Pattern of A. m. ligustica circRNAs during V. ceranae Infection
4.2. DEcircRNAs Were Potentially Engaged in the Host Response to V. ceranae Infection by Regulating the Trasncription of Corresponding Source Genes
4.3. DEcircRNAs Were Potentially Involved in the Regulation of Host Immune Defense against V. ceranae Infection via ceRNA Network
4.4. A Working Model of DEcircRNA-Mediated Host Response to V. ceranae Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bromenshenk, J.J.; Henderson, C.B.; Seccomb, R.A.; Welch, P.M.; Debnam, S.E.; Firth, D.R. Bees as biosensors: Chemosensory ability, honey bee monitoring systems, and emergent sensor technologies derived from the pollinator syndrome. Biosensors 2015, 5, 678–711. [Google Scholar] [CrossRef] [PubMed]
- Higes, M.; Garcia-Palencia, P.; Urbieta, A.; Nanetti, A.; Martin-Hernandez, R. Nosema apis and Nosema ceranae tissue tropism in worker honey bees (Apis mellifera). Vet. Pathol. 2020, 57, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Laurianne, P.; Michaël, R.; Bruno, P.; Frédéric, D.; Marie, D. Disruption of oxidative balance in the gut of the western honey bee Apis mellifera exposed to the intracellular parasite Nosema ceranae and to the insecticide fipronil. Microb. Biotechnol. 2017, 10, 1702–1717. [Google Scholar] [CrossRef]
- Antunez, K.; Martin-Hernandez, R.; Prieto, L.; Meana, A.; Zunino, P.; Higes, M. Immune suppression in the honey bee (Apis mellifera) following infection by Nosema ceranae (Microsporidia). Environ. Microbiol. 2009, 11, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Mayack, C.; Naug, D. Energetic stress in the honey bee Apis mellifera from Nosema ceranae infection. J. Invertebr. Pathol. 2009, 100, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Goblirsch, M.; Huang, Z.Y.; Spivak, M. Physiological and behavioral changes in honey bees (Apis mellifera) induced by Nosema ceranae infection. PLoS ONE 2013, 8, e58165. [Google Scholar] [CrossRef]
- Kurze, C.; Le Conte, Y.; Kryger, P.; Lewkowski, O.; Muller, T.; Moritz, R.F.A. Infection dynamics of Nosema ceranae in honey bee midgut and host cell apoptosis. J. Invertebr. Pathol. 2018, 154, 1–4. [Google Scholar] [CrossRef]
- Panek, J.; Paris, L.; Roriz, D.; Mone, A.; Dubuffet, A.; Delbac, F.; Diogon, M.; El Alaoui, H. Impact of the microsporidian Nosema ceranae on the gut epithelium renewal of the honey bee, Apis mellifera. J. Invertebr. Pathol. 2018, 159, 121–128. [Google Scholar] [CrossRef]
- Doublet, V.; Paxton, R.J.; McDonnell, C.M.; Dubois, E.; Nidelet, S.; Moritz, R.F.A.; Alaux, C.; Le Conte, Y. Brain transcriptomes of honey bees (Apis mellifera) experimentally infected by two pathogens: Black queen cell virus and Nosema ceranae. Genom. Data 2016, 10, 79–82. [Google Scholar] [CrossRef]
- Higes, M.; Martin-Hernandez, R.; Garrido-Bailon, E.; Gonzalez-Porto, A.V.; García-Palencia, P.; Mean, A.; Del Nozal, M.J.; Mayo, R.; Bernal, J.L. honey bee colony collapse due to Nosema ceranae in professional apiaries. Env. Microbiol. Rep. 2009, 1, 110–113. [Google Scholar] [CrossRef]
- Currie, R.W.; Pernal, S.F.; Guzma’n-Novoa, E. Honey bee colony losses in Canada. J. Apic. Res. 2010, 49, 104–106. [Google Scholar] [CrossRef]
- Higes, M.; Meana, A.; Bartolomé, C.; Botías, C.; Martín-Hernández, R. Nosema ceranae (Microsporidia), a controversial 21st century honey bee pathogen. Environ. Microbiol. Rep. 2013, 59, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Simeunovic, P.; Stevanovic, J.; Cirkovic, D.; Radojicic, S.; Lakic, N.; Stanisic, L.; Stanimirovic, Z. Nosema ceranae and queen age influence the reproduction and productivity of the honey bee colony. J. Apic. Res. 2014, 53, 545–554. [Google Scholar] [CrossRef]
- Chaimanee, V.; Chantawannakul, P.; Chen, Y.; Evans, J.D.; Pettis, J.S. Differential expression of immune genes of adult honey bee (Apis mellifera) after inoculated by Nosema ceranae. J. Insect Physiol. 2012, 58, 1090–1095. [Google Scholar] [CrossRef]
- Aufauvre, J.; Misme-Aucouturier, B.; Viguès, B.; Texier, C.; Delbac, F.; Blot, N. Transcriptome analyses of the honey bee response to Nosema ceranae and insecticides. PLoS ONE 2014, 9, e91686. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, Y.; Wang, R.W.; Schwarz, R.S.; Evans, J.D. Honey bee microRNAs respond to infection by the microsporidian parasite Nosema ceranae. Sci. Rep. 2015, 5, 17494. [Google Scholar] [CrossRef]
- Badaoui, B.; Fougeroux, A.; Petit, F.; Anselmo, A.; Gorni, C.; Cucurachi, M.; Cersini, A.; Granato, A.; Cardeti, G.; Formato, G.; et al. RNA-sequence analysis of gene expression from honey bees (Apis mellifera) infected with Nosema ceranae. PLoS ONE 2017, 12, e0173438. [Google Scholar] [CrossRef]
- Fu, Z.M.; Chen, H.Z.; Liu, S.Y.; Zhu, Z.W.; Fan, X.X.; Fan, Y.C.; Wan, J.Q.; Zhang, L.; Xiong, C.L.; Xu, G.J.; et al. Immune responses of Apis mellifera ligustia to Nosema ceranae stress. Sci. Agric. Sin. 2019, 52, 3069–3082. (In Chinese) [Google Scholar] [CrossRef]
- Chen, H.Z.; Xiong, C.L.; Zhu, Z.W.; Wang, J.; Fan, X.X.; Jiang, H.B.; Fan, Y.C.; Wan, J.Q.; Lu, J.X.; Zheng, Y.Z.; et al. Unraveling the mechanism underlying the immune responses of Apis mellifera ligustica to Nosema ceranae stress based on small RNA omics analyses. Acta Microbiol. Sin. 2020, 60, 1458–1478. (In Chinese) [Google Scholar] [CrossRef]
- Cocquerelle, C.; Mascrez, B.; Hetuin, D.; Bailleul, B. Mis-splicing yields circular RNA molecules. FASEB J. 1993, 7, 155–160. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xie, Q.; Xu, H.; Li, J.; Li, Y. Circular RNAs and cancer. Cancer Lett. 2017, 396, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Darbani, B.; Noeparvar, S.; Borg, S. Identification of circular RNAs from the parental genes involved in multiple aspects of cellular metabolism in barley. Front. Plant. Sci. 2016, 7, 776. [Google Scholar] [CrossRef]
- Lu, T.; Cui, L.; Zhou, Y.; Zhu, C.; Fan, D.; Gong, H.; Zhao, Q.; Zhou, C.; Zhao, Y.; Lu, D.; et al. Transcriptome-wide investigation of circular RNAs in rice. RNA 2015, 21, 2076–2087. [Google Scholar] [CrossRef]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell. Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef]
- Gan, H.; Feng, T.; Wu, Y.; Liu, C.; Xia, Q.; Cheng, T. Identification of circular RNA in the Bombyx mori silk gland. Insect Biochem. Mol. Biol. 2017, 89, 97–106. [Google Scholar] [CrossRef]
- Chen, X.; Shi, W.; Chen, C. Differential circular RNAs expression in ovary during oviposition in honey bees. Genomics 2019, 111, 598–606. [Google Scholar] [CrossRef]
- Chen, D.; Chen, H.; Du, Y.; Zhu, Z.; Wang, J.; Geng, S.H.; Xiong, C.; Zheng, Y.; Hou, C.; Diao, Q.; et al. Systematic identification of circular RNAs and corresponding regulatory networks unveil their potential roles in the midguts of eastern honey bee workers. Appl. Microbiol. Biotechnol. 2019, 104, 257–276. [Google Scholar] [CrossRef]
- Guo, R.; Chen, D.F.; Chen, H.Z.; Xiong, C.L.; Zheng, Y.Z.; Hou, C.S.; Du, Y.; Geng, S.; Wang, H.; Zhou, D.D.; et al. Genome-wide identification of circular RNAs in fungal parasite Nosema ceranae. Curr. Microbiol. 2018, 75, 1655–1660. [Google Scholar] [CrossRef]
- Guo, R.; Chen, D.; Chen, H.; Fu, Z.; Xiong, C.; Hou, C.; Zheng, Y.; Guo, Y.; Wang, H.; Du, Y.; et al. Systematic investigation of circular RNAs in Ascosphaera apis, a fungal pathogen of honey bee larvae. Gene 2018, 678, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell. 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.D.; Xu, C.D.; Wu, G.; Liu, B.; Wu, D.L. CircRNA-UCK2 increased TET1 inhibits proliferation and invasion of prostate cancer cells via sponge miRNA-767-5p. Open Med. 2019, 14, 833–842. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef]
- Meng, S.; Zhou, H.; Feng, Z.; Xu, Z.; Tang, Y.; Li, P.Y.; Wu, M. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 2017, 16, 94. [Google Scholar] [CrossRef]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of circRNAs. Mol. Cell 2017, 66, 9–21.e7. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef]
- Thölken, C.; Thamm, M.; Erbacher, C.; Lechner, M. Sequence and structural properties of circular RNAs in the brain of nurse and forager honey bees (Apis mellifera). BMC Genom. 2019, 20, 88. [Google Scholar] [CrossRef]
- Xiong, C.; Chen, H.; Chen, D.; Zheng, Y.; Fu, Z.; Xu, G.J.; Du, Y.; Wang, H.P.; Geng, S.H.; Zhou, D.D.; et al. Analysis of circular RNAs and their regulatory networks in the midgut of Apis mellifera ligustica works. Acta Entomol. Sin. 2018, 61, 1363–1375. (In Chinese) [Google Scholar] [CrossRef]
- Hu, X.; Zhu, M.; Zhang, X.; Liu, B.; Liang, Z.; Huang, L.; Xu, J.; Yu, L.; Li, K.; Zar, M.S.; et al. Identification and characterization of circular RNAs in the silkworm midgut following Bombyx mori cytoplasmic polyhedrosis virus infection. RNA Biol. 2018, 15, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhu, M.; Liu, B.; Liang, Z.; Huang, L.; Xu, J.; Yu, L.; Li, K.; Jiang, M.; Xue, R.; et al. Circular RNA alterations in the Bombyx mori midgut following B. mori nucleopolyhedrovirus infection. Mol. Immunol. 2018, 101, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hu, N.; Mo, L.; Zeng, Z.; Sun, J.; Hu, Y. Deep RNA sequencing reveals a repertoire of human fibroblast circular RNAs associated with cellular responses to herpes simplex virus 1 infection. Cell. Physiol. Biochem. 2018, 47, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Qiu, L.; Pan, R.; Bai, H.; Jiang, Y.; Wang, Z.; Bi, Y.; Chen, G.; Chang, G. Expression patterns of novel circular RNAs in chicken cells after avian leukosis virus subgroup J infection. Gene 2019, 701, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chen, H.; Du, Y.; Zhou, D.; Geng, S.; Wang, H.; Wan, J.; Xiong, C.; Zheng, Y.; Guo, R. Genome-wide identification of long non-coding RNAs and their regulatory networks involved in Apis mellifera ligustica response to Nosema ceranae infection. Insects 2019, 10, 245. [Google Scholar] [CrossRef]
- Guo, R.; Chen, D.F.; Xiong, C.L.; Hou, C.S.; Zheng, Y.Z.; Fu, Z.M.; Liang, Q.; Diao, Q.Y.; Zhang, L.; Wang, H.; et al. First identification of long non-coding RNAs in fungal parasite Nosema ceranae. Apidologie 2018, 49, 660–670. [Google Scholar] [CrossRef]
- Guo, R.; Chen, H.; Xiong, C.; Zheng, Y.; Fu, Z.; Xu, G.J.; Du, Y.; Wang, H.P.; Geng, S.H.; Zhou, D.D.; et al. Analysis of differentially expressed circular RNAs and their regulation networks during the developmental process of Apis mellifera ligustica worker’s midgut. Sci. Agric. Sin. 2018, 51, 4575–4590. (In Chinese) [Google Scholar] [CrossRef]
- Chen, H.Z.; Du, Y.; Xiong, C.L.; Zheng, Y.Z.; Chen, D.F.; Guo, R. A comprehensive transcriptome data of normal and Nosema ceranae-stressed midguts of Apis mellifera ligustica workers. Data Brief 2019, 26, 104349. [Google Scholar] [CrossRef]
- The honey bee Genome Sequencing Consortium. Insights into social insects from the genome of the honey bee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID gene functional classification tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H. Stratified Fisher’s exact test and its sample size calculation. Biom. J. 2014, 56, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Du, J.L.; Yuan, Z.F.; Ma, Z.W.; Song, J.Z.; Xie, X.L.; Chen, Y.L. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. Biosyst. 2014, 10, 2441–2447. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Lai, X.; Bazin, J.; Webb, S.; Crespi, M.; Zubieta, C.; Conn, S.J. CircRNAs in Plants. Adv. Exp. Med. Biol. 2018, 1087, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Agarwal, V.; Guo, H.; Bartel, D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Li, D.; Li, L.; Zhang, Q.; Wang, S.; Huang, H. Identification of circular RNAs in kiwifruit and their species-specific response to bacterial canker pathogen invasion. Front. Plant Sci. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Paris, L.; El Alaoui, H.; Delbac, F.; Diogon, M. Effects of the gut parasite Nosema ceranae on honey bee physiology and behavior. Curr. Opin. Insect Sci. 2018, 26, 149–154. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Chakrabarti, S.; Lemaitre, B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev. 2009, 23, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Xu, N.; Xi, R. Paracrine Wingless signalling controls self-renewal of Drosophila intestinal stem cells. Nature 2008, 455, 1119–1123. [Google Scholar] [CrossRef]
- Chen, Y.P.; Pettis, J.S.; Zhao, Y.; Liu, X.Y.; Tallon, L.J.; Sadzewicz, L.D.; Li, R.; Zheng, H.; Huang, S.; Zhang, X.; et al. Genome sequencing and comparative genomics of honey bee microsporidia, Nosema apis reveal novel insights into host-parasite interactions. BMC Genom. 2013, 14, 451. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, R.; Liu, S.; Donath, A.; Peters, R.S.; Ware, J.; Misof, B.; Niehuis, O.; Pfrender, M.E.; Zhou, X. The molecular evolutionary dynamics of oxidative phosphorylation (OXPHOS) genes in Hymenoptera. BMC Evol. Biol. 2017, 17, 269. [Google Scholar] [CrossRef]
- Lavine, M.D.; Strand, M.R. Insect hemocytes and their role in immunity. Insect Biochem. Molec. 2002, 32, 1295–1309. [Google Scholar] [CrossRef]
- Aronstein, K.A.; Murray, K.D. Chalkbrood disease in honey bees. J. Invertebr. Pathol. 2010, 103 (Suppl. 1), S20–S29. [Google Scholar] [CrossRef]
- Samaranayake, Y.H.; Samaranayake, L.P.; Pow, E.H.; Beena, V.T.; Yeung, K.W. Antifungal effects of lysozyme and lactoferrin against genetically similar, sequential Candida albicans isolates from a human immunodeficiency virus-infected southern Chinese cohort. J. Clin. Microbiol. 2001, 39, 3296–3302. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.H.; Iwamoto, K.S.; Syljuasen, R.; Pervan, M.; Pajonk, F. The role of the ubiquitin/proteasome system in cellular responses to radiation. Oncogene 2003, 22, 5755–5773. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.Z.; Wang, J.; Fan, X.X.; Long, Q.; Chen, H.Z.; Ye, Y.P.; Zhang, K.Y.; Ren, Z.M.; Zhang, Y.; Niu, Q.S.; et al. CircRNA-regulated immune response of Asian honey bee workers to microsporidian infection. bioRxiv 2022, 498258. [Google Scholar] [CrossRef]
- Scott, J.G.; Liu, N.; Wen, Z. Insect cytochromes P450: Diversity, insecticide resistance and tolerance to plant toxins. Comp. Biochem. Physiol. 1998, 121, 147–155. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Dijkers, P.F.; Medema, R.H.; Lammers, J.W.; Koenderman, L.; Coffer, P.J. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 2000, 10, 1201–1204. [Google Scholar] [CrossRef]
- Evans, J.D.; Aronstein, K.; Chen, Y.P.; Hetru, C.; Imler, J.L.; Jiang, H.; Kanost, M.; Thompson, G.J.; Zou, Z.; Hultmark, D. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol. Biol. 2006, 15, 645–656. [Google Scholar] [CrossRef]
- Huang, R.; Zhang, Y.; Han, B.; Bai, Y.; Zhou, R.; Yao, H. Circular RNA HIPK2 regulates astrocyte activation via cooperation of autophagy and ER stress by targeting MIR124-2HG. Autophagy 2017, 13, 1722–1741. [Google Scholar] [CrossRef]
- Li, R.-C.; Ke, S.; Meng, F.-K.; Lu, J.; Zou, X.-J.; He, Z.-G.; Wang, W.-F.; Fang, M.-H. CiRS-7 promotes growth and metastasis of esophageal squamous cell carcinoma via regulation of miR-7/HOXB13. Cell Death Dis. 2018, 9, 838. [Google Scholar] [CrossRef] [Green Version]
- Hori, S.; Kaneko, K.; Saito, T.H.; Takeuchi, H.; Kubo, T. Expression of two microRNAs, ame-mir-276 and-1000, in the adult honey bee (Apis mellifera) brain. Apidologie 2011, 42, 89–102. [Google Scholar] [CrossRef]
- Liu, F.; Peng, W.; Li, Z.; Li, W.; Li, L.; Pan, J.; Zhang, S.; Miao, Y.; Chen, S.; Su, S. Next-generation small RNA sequencing for microRNAs profiling in Apis mellifera: Comparison between nurses and foragers. Insect Mol. Biol. 2012, 21, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Y.; Zheng, H.J.; Pan, Q.Z.; Wang, Z.L.; Zeng, Z.J. Differentially expressed microRNAs between queen and worker larvae of the honey bee (Apis mellifera). Apidologie 2015, 46, 35–45. [Google Scholar] [CrossRef]
- Fullaondo, A.; Lee, S.Y. Identification of putative miRNA involved in Drosophila melanogaster immune response. Dev. Comp. Immunol. 2012, 36, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Shrinet, J.; Jain, S.; Jain, J.; Bhatnagar, R.K.; Sunil, S. Next generation sequencing reveals regulation of distinct Aedes microRNAs during chikungunya virus development. PLoS. Negl. Trop. Dis. 2014, 8, e2616. [Google Scholar] [CrossRef]
- Hua, Y.; Zhao, K.; Tao, G.; Dai, C.; Su, Y. miR-25 promotes metastasis via targeting FBXW7 in esophageal squamous cell carcinoma. Oncol. Rep. 2017, 38, 3030–3038. [Google Scholar] [CrossRef]
- Wang, L.; Wu, J.; Xie, C. miR-92a promotes hepatocellular carcinoma cells proliferation and invasion by FOXA2 targeting. Iran. J. Basic Med. Sci. 2017, 20, 783–790. [Google Scholar] [CrossRef]
- Xie, M.; Qin, H.; Luo, Q.; Huang, Q.; He, X.; Yang, Z.; Lan, P.; Lian, L. MicroRNA-30a regulates cell proliferation and tumor growth of colorectal cancer by targeting CD73. BMC Cancer 2017, 17, 305. [Google Scholar] [CrossRef]
- Steiner, H. Peptidoglycan recognition proteins: On and off switches for innate immunity. Immunol. Rev. 2004, 198, 83–96. [Google Scholar] [CrossRef]
- Kaneko, T.; Silverman, N. Bacterial recognition and signalling by the Drosophila IMD pathway. Cell Microbiol. 2005, 7, 461–469. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Anchor Reads | Mapped Anchor Reads | Data Source |
|---|---|---|---|
| Am7T1 | 220,977,612 | 19,786,662 (8.95%) | This study |
| Am7T2 | 302,675,036 | 26,173,216 (8.65%) | This study |
| Am7T3 | 184,550,634 | 16,349,641 (8.86%) | This study |
| Am10T1 | 389,439,122 | 26,881,797 (6.90%) | This study |
| Am10T2 | 327,093,752 | 22,105,363 (6.76%) | This study |
| Am10T3 | 264,237,670 | 18,445,112 (6.98%) | This study |
| Am7CK1 | 236,400,908 | 19,006,601 (8.04%) | [40] |
| Am7CK2 | 168,420,472 | 19,873,468 (11.80%) | [40] |
| Am7CK3 | 136,779,122 | 17,502,228 (12.80%) | [40] |
| Am10CK1 | 195,020,440 | 22,441,139 (11.51%) | [40] |
| Am10CK2 | 184,091,234 | 21,109,367 (11.47%) | [40] |
| Am10CK3 | 168,790,774 | 17,765,330 (10.53%) | [40] |
| Immune Pathway | Number of Source Genes in Am7CK vs. Am7T | Number of Source Genes in Am10CK vs. Am10T | Ko Number |
|---|---|---|---|
| Endocytosis | 4 | 8 | ko04144 |
| Phagosome | 3 | 2 | ko04145 |
| Lysosome | 2 | 0 | ko04142 |
| Ubiquitin-mediated proteolysis | 0 | 1 | ko04120 |
| Metabolism of xenobiotics by cytochrome P450 | 0 | 1 | ko00980 |
| FoxO signaling pathway | 3 | 2 | ko04068 |
| Pathway ID | Number of DEcircRNAs in Am7CK vs. Am7T | DEcircRNA ID | Number of DEcircRNAs in Am10CK vs. Am10T | DEcircRNA ID |
|---|---|---|---|---|
| Cellular immune-related pathway | ||||
| Endocytosis | 3 | novel_circ_011088, novel_circ_013731, novel_circ_009951 | 1 | novel_circ_011577 |
| Phagosome | 2 | novel_circ_011088, novel_circ_009951 | 0 | |
| Ubiquitin-mediated proteolysis | 3 | novel_circ_011088, novel_circ_013731, novel_circ_009951 | 1 | novel_circ_011577 |
| Lysosome | 0 | 2 | novel_circ_011577, novel_circ_012916 | |
| Humoral immune-related pathway | ||||
| FoxO signaling pathway | 16 | novel_circ_011088, novel_circ_013731, novel_circ_009951, novel_circ_013719, novel_circ_012352, novel_circ_012338, novel_circ_012319, novel_circ_012316, novel_circ_012025, novel_circ_011168, novel_circ_006925, novel_circ_006153, novel_circ_004247, novel_circ_003542, novel_circ_002201, novel_circ_002136 | 3 | novel_circ_011577, novel_circ_002577, novel_circ_012916 |
| MAPK signaling pathway | 2 | novel_circ_013731, novel_circ_011088 | 2 | novel_circ_011577, novel_circ_002577 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Fan, X.; Zhang, W.; Ye, Y.; Cai, Z.; Zhang, K.; Zhang, K.; Fu, Z.; Chen, D.; Guo, R. Deciphering the CircRNA-Regulated Response of Western Honey Bee (Apis mellifera) Workers to Microsporidian Invasion. Biology 2022, 11, 1285. https://doi.org/10.3390/biology11091285
Chen H, Fan X, Zhang W, Ye Y, Cai Z, Zhang K, Zhang K, Fu Z, Chen D, Guo R. Deciphering the CircRNA-Regulated Response of Western Honey Bee (Apis mellifera) Workers to Microsporidian Invasion. Biology. 2022; 11(9):1285. https://doi.org/10.3390/biology11091285
Chicago/Turabian StyleChen, Huazhi, Xiaoxue Fan, Wende Zhang, Yaping Ye, Zongbing Cai, Kaiyao Zhang, Kuihao Zhang, Zhongmin Fu, Dafu Chen, and Rui Guo. 2022. "Deciphering the CircRNA-Regulated Response of Western Honey Bee (Apis mellifera) Workers to Microsporidian Invasion" Biology 11, no. 9: 1285. https://doi.org/10.3390/biology11091285
APA StyleChen, H., Fan, X., Zhang, W., Ye, Y., Cai, Z., Zhang, K., Zhang, K., Fu, Z., Chen, D., & Guo, R. (2022). Deciphering the CircRNA-Regulated Response of Western Honey Bee (Apis mellifera) Workers to Microsporidian Invasion. Biology, 11(9), 1285. https://doi.org/10.3390/biology11091285