
A Computational Model of Bacterial Population Dynamics in Gastrointestinal Yersinia enterocolitica Infections in Mice

 , ,
, ,  ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Generation of Ye Strains Containing Different Antibiotic Selection Markers
2.3. Animal Handling
2.4. Oral Mouse Infection
2.5. Determination of Bacterial Load from Feces
2.6. Calculation of Competitive Indices in Mixed Infections
2.7. 16S rRNA Sequencing from SI Luminal Samples
2.8. Isolation of RNA from Gut Mucosal Scrapings
2.9. Quantification of Immune Parameters by Quantitative Real-Time-PCR (qRT-PCR)
2.10. Determination of the Distribution of Ye along the Mouse GIT
2.11. Systemic Administration of Gentamicin for the Cleansing of a Potential Niche Colonized by Ye
2.12. Determination of GIT Passage of Time
2.13. Determination of the Water Content of the SI Content and Fecal Pellets
2.14. Calculation of the Thickening Factor for SPF and GF Mice
2.15. Alignment of Model Simulation and Lab Observation Time
2.16. Parameter Optimization
3. Results
3.1. Generation of Experimental Datasets to Generate a Dynamic Population Model
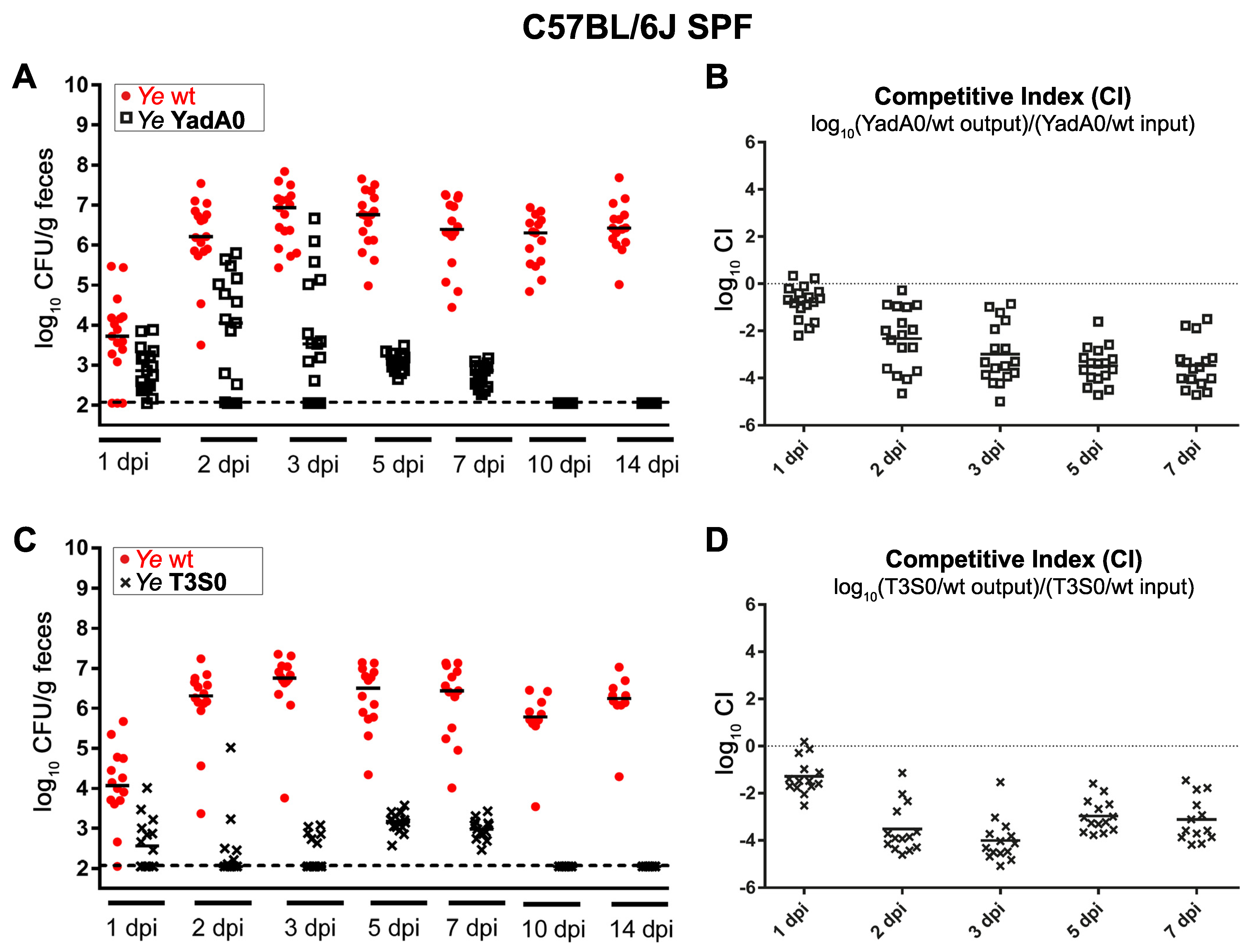
3.1.1. Ye Population Dynamics Are Investigated in the Presence of a Complex Microbiome and an Intact Host Immune Response
3.1.2. The Integration of Experimental Mouse Infection Data, Specific Parameters Determined in Wet-Lab Experiments, and Published Knowledge Are Used to Generate a Conclusive View of Ye Mouse Infection
3.2. Mathematical Description of the Dynamic Population Model
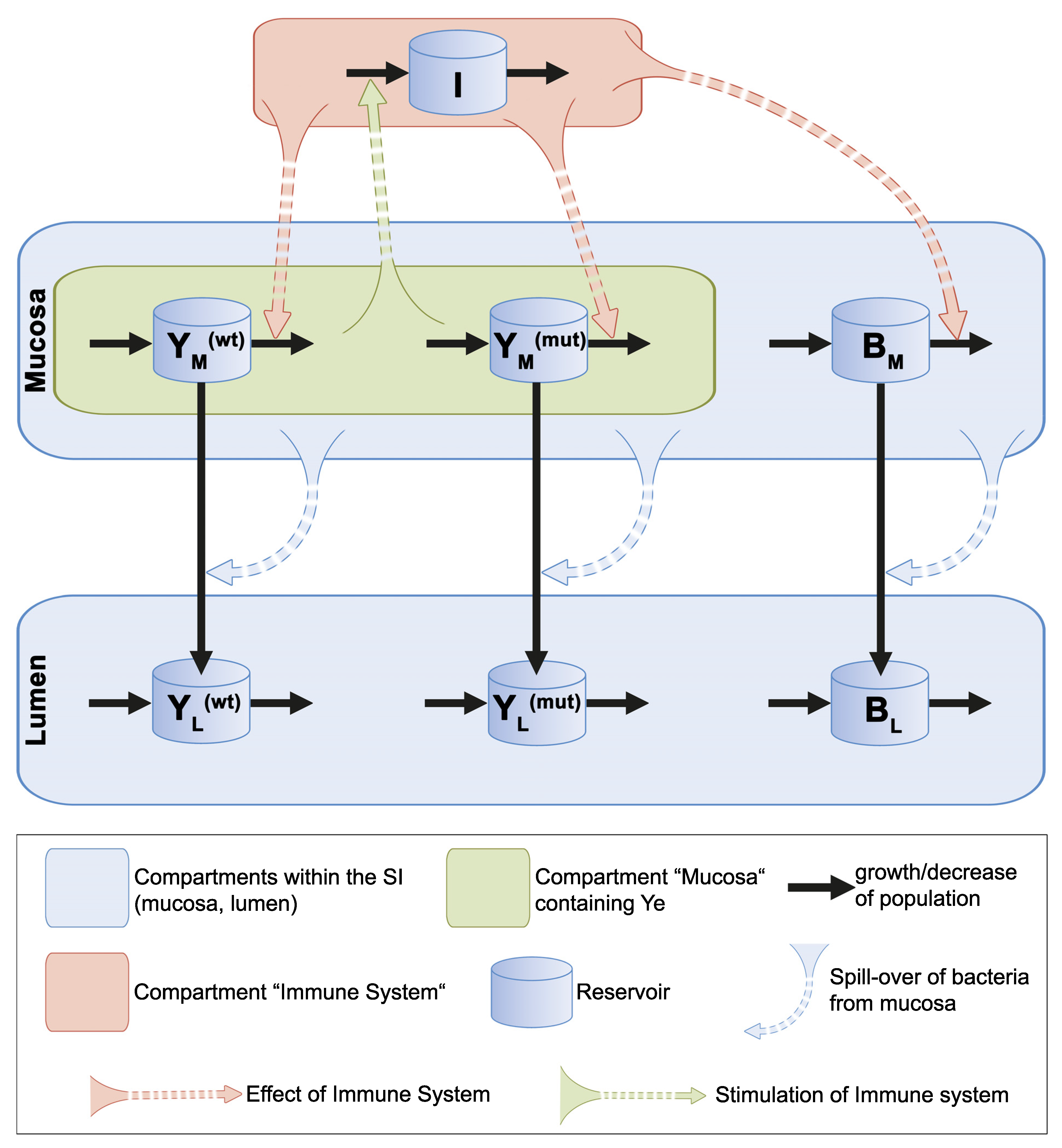
3.2.1. Presumptions Are Made for the Dynamic Population Model
3.2.2. Ordinary Differential Equations Describe the Dynamic Population Model
3.3. Validation of the Dynamic Population Model
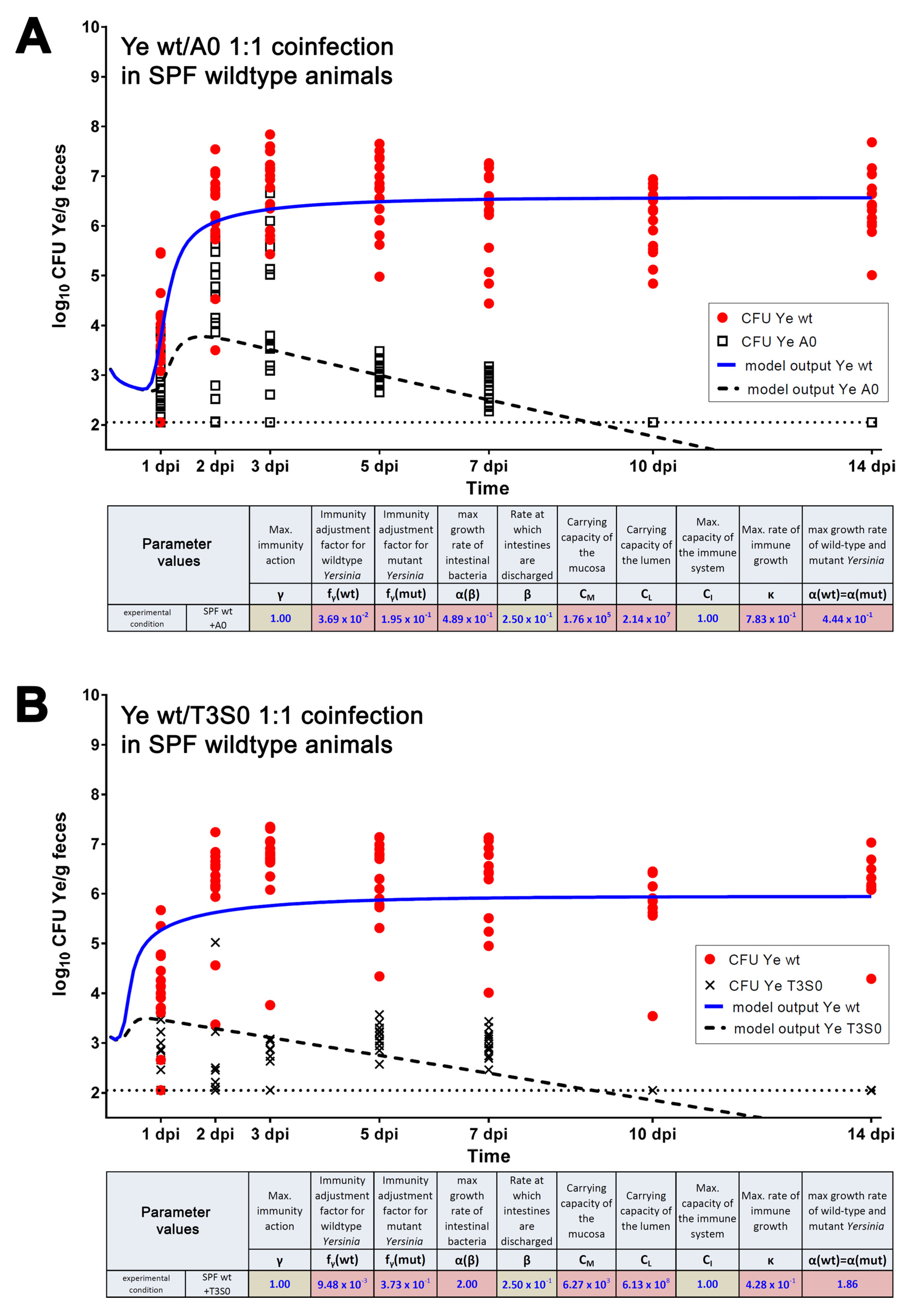
3.3.1. The Dynamic Population Model’s Parameters Were Estimated
3.3.2. Parameters Were Fitted Based on the Coinfection Experiments in SPF Mice
3.3.3. A Sensitivity Analysis of the Estimated Parameters Was Conducted
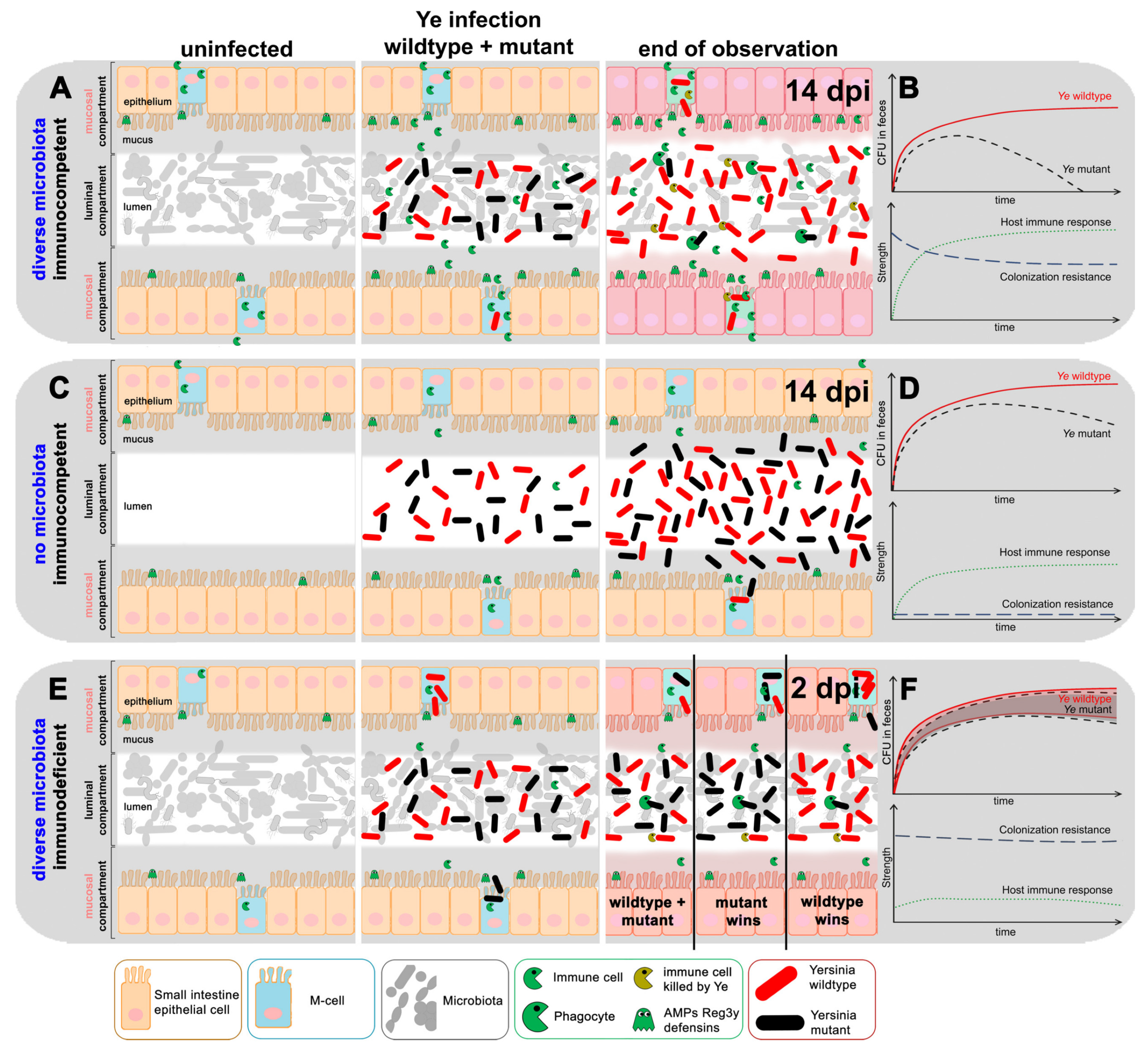
3.4. Refinement of the Dynamic Population Model
3.4.1. The First Model Refinement Was Based on Coinfection Experiments in GF Mice
3.4.2. An Immunocompromised Host Is Mimicked
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Bohnhoff, M.; Rifkind, D. The effect of an antibiotic on the susceptibility of the mouse’s intestinal tract to Salmonella infection. Trans. Am. Clin. Climatol. Assoc. 1956, 68, 51–58. [Google Scholar] [PubMed]
- Bosak, J.; Micenkova, L.; Hrala, M.; Pomorska, K.; Kunova Bosakova, M.; Krejci, P.; Gopfert, E.; Faldyna, M.; Smajs, D. Colicin FY inhibits pathogenic Yersinia enterocolitica in mice. Sci. Rep. 2018, 8, 12242. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Pamer, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Stecher, B.; Hardt, W.D. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 2011, 14, 82–91. [Google Scholar] [CrossRef]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 204. [Google Scholar] [CrossRef]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, e244. [Google Scholar] [CrossRef]
- Moor, K.; Diard, M.; Sellin, M.E.; Felmy, B.; Wotzka, S.Y.; Toska, A.; Bakkeren, E.; Arnoldini, M.; Bansept, F.; Co, A.D.; et al. High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 2017, 544, 498–502. [Google Scholar] [CrossRef]
- Perez-Lopez, A.; Behnsen, J.; Nuccio, S.P.; Raffatellu, M. Mucosal immunity to pathogenic intestinal bacteria. Nat. Rev. Immunol. 2016, 16, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, M.J.; Wekesa, A.N.; Gniadkowski, M.; Antoniadou, A.; Giamarellou, H.; Petrikkos, G.L.; Skiada, A.; Brun-Buisson, C.; Bonten, M.J.; Derde, L.P.; et al. The association between colonization with carbapenemase-producing enterobacteriaceae and overall ICU mortality: An observational cohort study. Crit. Care Med. 2015, 43, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Li, L.; Li, W.; Hou, T.; Ma, H.; Yang, Y.; Wu, A.; Liu, Y.; Wen, J.; Yang, H.; et al. Impact of healthcare-associated infections on length of stay: A study in 68 hospitals in China. Biomed. Res. Int. 2019, 2019, 2590563. [Google Scholar] [CrossRef] [PubMed]
- Dave, M.N.; Silva, J.E.; Eliçabe, R.J.; Jeréz, M.B.; Filippa, V.P.; Gorlino, C.V.; Autenrieth, S.; Autenrieth, I.B.; Di Genaro, M.S. Yersinia enterocolitica YopH-deficient strain activates neutrophil recruitment to Peyer’s patches and promotes clearance of the virulent strain. Infect. Immun. 2016, 84, 3172–3181. [Google Scholar] [CrossRef]
- Deuschle, E.; Keller, B.; Siegfried, A.; Manncke, B.; Spaeth, T.; Köberle, M.; Drechsler-Hake, D.; Reber, J.; Bottcher, R.T.; Autenrieth, S.E.; et al. Role of beta1 integrins and bacterial adhesins for Yop injection into leukocytes in Yersinia enterocolitica systemic mouse infection. Int. J. Med. Microbiol. 2016, 306, 77–88. [Google Scholar] [CrossRef]
- Keller, B.; Mühlenkamp, M.; Deuschle, E.; Siegfried, A.; Mössner, S.; Schade, J.; Griesinger, T.; Katava, N.; Braunsdorf, C.; Fehrenbacher, B.; et al. Yersinia enterocolitica exploits different pathways to accomplish adhesion and toxin injection into host cells. Cell. Microbiol. 2015, 17, 1179–1204. [Google Scholar] [CrossRef]
- Köberle, M.; Klein-Günther, A.; Schütz, M.; Fritz, M.; Berchtold, S.; Tolosa, E.; Autenrieth, I.B.; Bohn, E. Yersinia enterocolitica targets cells of the innate and adaptive immune system by injection of yops in a mouse infection model. PLoS Pathog. 2009, 5, e1000551. [Google Scholar] [CrossRef]
- Lehr, U.; Schütz, M.; Oberhettinger, P.; Ruiz-Perez, F.; Donald, J.W.; Palmer, T.; Linke, D.; Henderson, I.R.; Autenrieth, I.B. C-terminal amino acid residues of the trimeric autotransporter adhesin YadA of Yersinia enterocolitica are decisive for its recognition and assembly by BamA. Mol. Microbiol. 2010, 78, 932–946. [Google Scholar] [CrossRef]
- Pepe, J.C.; Wachtel, M.R.; Wagar, E.; Miller, V.L. Pathogenesis of defined invasion mutants of Yersinia enterocolitica in a BALB/c mouse model of infection. Infect. Immun. 1995, 63, 4837–4848. [Google Scholar] [CrossRef]
- Schindler, M.K.; Schütz, M.S.; Mühlenkamp, M.C.; Rooijakkers, S.H.; Hallström, T.; Zipfel, P.F.; Autenrieth, I.B. Yersinia enterocolitica YadA mediates complement evasion by recruitment and inactivation of C3 products. J. Immunol. 2012, 189, 4900–4908. [Google Scholar] [CrossRef]
- Schütz, M.; Weiss, E.M.; Schindler, M.; Hallström, T.; Zipfel, P.F.; Linke, D.; Autenrieth, I.B. Trimer stability of YadA is critical for virulence of Yersinia enterocolitica. Infect. Immun. 2010, 78, 2677–2690. [Google Scholar] [CrossRef] [PubMed]
- Trülzsch, K.; Sporleder, T.; Igwe, E.I.; Rüssmann, H.; Heesemann, J. Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect. Immun. 2004, 72, 5227–5234. [Google Scholar] [CrossRef] [PubMed]
- Weirich, J.; Bräutigam, C.; Mühlenkamp, M.; Franz-Wachtel, M.; Macek, B.; Meuskens, I.; Skurnik, M.; Leskinen, K.; Bohn, E.; Autenrieth, I.; et al. Identifying components required for OMP biogenesis as novel targets for antiinfective drugs. Virulence 2017, 8, 1170–1188. [Google Scholar] [CrossRef] [PubMed]
- El Tahir, Y.; Skurnik, M. YadA, the multifaceted Yersinia adhesin. Int. J. Med. Microbiol. 2001, 291, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Handley, S.A.; Newberry, R.D.; Miller, V.L. Yersinia enterocolitica invasin-dependent and invasin-independent mechanisms of systemic dissemination. Infect. Immun. 2005, 73, 8453–8455. [Google Scholar] [CrossRef]
- Young, V.B.; Falkow, S.; Schoolnik, G.K. The invasin protein of Yersinia enterocolitica: Internalization of invasin-bearing bacteria by eukaryotic cells is associated with reorganization of the cytoskeleton. J. Cell Biol. 1992, 116, 197–207. [Google Scholar] [CrossRef]
- Mühlenkamp, M.; Oberhettinger, P.; Leo, J.C.; Linke, D.; Schütz, M.S. Yersinia adhesin A (YadA)—Beauty & beast. Int. J. Med. Microbiol. 2015, 305, 252–258. [Google Scholar] [CrossRef]
- Ruckdeschel, K.; Roggenkamp, A.; Schubert, S.; Heesemann, J. Differential contribution of Yersinia enterocolitica virulence factors to evasion of microbicidal action of neutrophils. Infect. Immun. 1996, 64, 724–733. [Google Scholar] [CrossRef]
- Cornelis, G.R. Yersinia type III secretion: Send in the effectors. J. Cell Biol. 2002, 158, 401–408. [Google Scholar] [CrossRef]
- Autenrieth, I.B.; Firsching, R. Penetration of M cells and destruction of Peyer’s patches by Yersinia enterocolitica: An ultrastructural and histological study. J. Med. Microbiol. 1996, 44, 285–294. [Google Scholar] [CrossRef]
- Clark, M.A.; Hirst, B.H.; Jepson, M.A. M-cell surface beta1 integrin expression and invasin-mediated targeting of Yersinia pseudotuberculosis to mouse Peyer’s patch M cells. Infect. Immun. 1998, 66, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.J.; Restif, O.; McKinley, T.J.; Sheppard, M.; Maskell, D.J.; Mastroeni, P. Modelling within-host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol. 2008, 6, e74. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.W.; Carlson, J.M. In silico analysis of antibiotic-induced Clostridium difficile infection: Remediation techniques and biological adaptations. PLoS Comput. Biol. 2018, 14, e1006001. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Regoes, R.R.; Dolowschiak, T.; Wotzka, S.Y.; Lengefeld, J.; Slack, E.; Grant, A.J.; Ackermann, M.; Hardt, W.D. Cecum lymph node dendritic cells harbor slow-growing bacteria phenotypically tolerant to antibiotic treatment. PLoS Biol. 2014, 12, e1001793. [Google Scholar] [CrossRef]
- Kaiser, P.; Slack, E.; Grant, A.J.; Hardt, W.D.; Regoes, R.R. Lymph node colonization dynamics after oral Salmonella Typhimurium infection in mice. PLoS Pathog. 2013, 9, e1003532. [Google Scholar] [CrossRef]
- Leber, A.; Hontecillas, R.; Abedi, V.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Stewart, C.; Bassaganya-Riera, J. Modeling new immunoregulatory therapeutics as antimicrobial alternatives for treating Clostridium difficile infection. Artif. Intell. Med. 2017, 78, 1–13. [Google Scholar] [CrossRef]
- Verma, M.; Bassaganya-Riera, J.; Leber, A.; Tubau-Juni, N.; Hoops, S.; Abedi, V.; Chen, X.; Hontecillas, R. High-resolution computational modeling of immune responses in the gut. Gigascience 2019, 8, giz062. [Google Scholar] [CrossRef]
- Miao, H.; Hollenbaugh, J.A.; Zand, M.S.; Holden-Wiltse, J.; Mosmann, T.R.; Perelson, A.S.; Wu, H.; Topham, D.J. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J. Virol. 2010, 84, 6687–6698. [Google Scholar] [CrossRef]
- Balbas-Martinez, V.; Ruiz-Cerda, L.; Irurzun-Arana, I.; Gonzalez-Garcia, I.; Vermeulen, A.; Gomez-Mantilla, J.D.; Troconiz, I.F. A systems pharmacology model for inflammatory bowel disease. PLoS ONE 2018, 13, e0192949. [Google Scholar] [CrossRef]
- Wendelsdorf, K.; Bassaganya-Riera, J.; Hontecillas, R.; Eubank, S. Model of colonic inflammation: Immune modulatory mechanisms in inflammatory bowel disease. J. Theor. Biol. 2010, 264, 1225–1239. [Google Scholar] [CrossRef]
- Antonenko, V.; Pawlow, V.; Heesemann, J.; Rakin, A. Characterization of a novel unique restriction-modification system from Yersinia enterocolitica O:8 1B. FEMS Microbiol. Lett. 2003, 219, 249–252. [Google Scholar] [CrossRef]
- Miyahara, M.; Maruyama, T.; Wake, A.; Mise, K. Widespread occurrence of the restriction endonuclease YenI, an isoschizomer of PstI, in Yersinia enterocolitica serotype O8. Appl. Environ. Microbiol. 1988, 54, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Dyszel, J.L.; Smith, J.N.; Lucas, D.E.; Soares, J.A.; Swearingen, M.C.; Vross, M.A.; Young, G.M.; Ahmer, B.M. Salmonella enterica serovar Typhimurium can detect acyl homoserine lactone production by Yersinia enterocolitica in mice. J. Bacteriol. 2010, 192, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Godon, J.J.; Zumstein, E.; Dabert, P.; Habouzit, F.; Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997, 63, 2802–2813. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 February 2022).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Herbig, A.; Maixner, F.; Bos, K.I.; Zink, A.; Krause, J.; Huson, D.H. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv, 2016; preprint. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 8 February 2022).
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Gómez-Rubio, V. ggplot2—Elegant graphics for data analysis (2nd edition). J. Stat. Softw. 2017, 77, 1–3. [Google Scholar] [CrossRef]
- Wang, F.; Wang, J.; Liu, D.; Su, Y. Normalizing genes for real-time polymerase chain reaction in epithelial and nonepithelial cells of mouse small intestine. Anal. Biochem. 2010, 399, 211–217. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Van der Waaij, L.A.; Mesander, G.; Limburg, P.C.; van der Waaij, D. Direct flow cytometry of anaerobic bacteria in human feces. Cytometry 1994, 16, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Nocedal, J.S.J.W. Numerical Optimization; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Shi, Z.-J. Convergence of line search methods for unconstrained optimization. Appl. Math. Comput. 2004, 157, 393–405. [Google Scholar] [CrossRef]
- Armijo, L. Minimization of function having Lipschitz continuous first partial derivatives. Pac. J. Math. 1966, 16, 1–3. [Google Scholar] [CrossRef]
- Bertsekas, D.P. On the goldstein-levitin-polyak gradient projection method. IEEE Trans. Autom. Control 1976, 21, 174–184. [Google Scholar] [CrossRef]
- Hinze, M.; Pinnau, R.; Ulbrich, M.; Ulbrich, S. Optimization with PDE Constraints; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Luenberger, D.G. Optimization by Vector Space Methods; John Wiley and Sons, Inc.: New York, NJ, USA, 1969. [Google Scholar]
- Penot, J.P. On the convergence of descent algorithms. Comput. Optim. Appl. 2002, 23, 279–284. [Google Scholar] [CrossRef]
- Kennedy, J.; Eberhart, R. Particle swarm optimization. In Proceedings of the IEEE International Conference on Neural Networks, Perth, WA, Australia, 27 November–1 December 1995; Volume 4, pp. 1942–1948. [Google Scholar]
- Dräger, A.; Kronfeld, M.; Ziller, M.J.; Supper, J.; Planatscher, H.; Magnus, J.B.; Oldiges, M.; Kohlbacher, O.; Zell, A. Modeling metabolic networks in C. glutamicum: A comparison of rate laws in combination with various parameter optimization strategies. BMC Syst. Biol. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Raue, A.; Schilling, M.; Bachmann, J.; Matteson, A.; Schelker, M.; Kaschek, D.; Hug, S.; Kreutz, C.; Harms, B.D.; Theis, F.J.; et al. Lessons learned from quantitative dynamical modeling in systems biology. PLoS ONE 2013, 8, e74335. [Google Scholar] [CrossRef]
- Neath, R.C. On convergence properties of the Monte Carlo EM algorithm. In Advances in Modern Statistical Theory and Applications: A Festschrift in Honor of Morris L. Eaton; Institute of Mathematical Statistics: Washington, DC, USA, 2013; pp. 43–62. [Google Scholar]
- Di Genaro, M.S.; Waidmann, M.; Kramer, U.; Hitziger, N.; Bohn, E.; Autenrieth, I.B. Attenuated Yersinia enterocolitica mutant strains exhibit differential virulence in cytokine-deficient mice: Implications for the development of novel live carrier vaccines. Infect. Immun. 2003, 71, 1804–1812. [Google Scholar] [CrossRef]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Muniz, L.R.; Knosp, C.; Yeretssian, G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front. Immunol. 2012, 3, 310. [Google Scholar] [CrossRef] [PubMed]
- Handley, S.A.; Dube, P.H.; Revell, P.A.; Miller, V.L. Characterization of oral Yersinia enterocolitica infection in three different strains of inbred mice. Infect. Immun. 2004, 72, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.H.; Nielsen, D.S.; Kverka, M.; Zakostelska, Z.; Klimesova, K.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Hansen, A.K. Patterns of early gut colonization shape future immune responses of the host. PLoS ONE 2012, 7, e34043. [Google Scholar] [CrossRef] [PubMed]
- Savidge, T.C.; Smith, M.W.; James, P.S.; Aldred, P. Salmonella-induced M-cell formation in germ-free mouse Peyer’s patch tissue. Am. J. Pathol. 1991, 139, 177–184. [Google Scholar] [PubMed]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, G.; Stahl, M.; Sham, H.P.; Crowley, S.M.; Morampudi, V.; Dalwadi, U.; Ma, C.; Jacobson, K.; Vallance, B.A. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect. Immun. 2014, 82, 3753–3763. [Google Scholar] [CrossRef]
- Friedrich, C.; Mamareli, P.; Thiemann, S.; Kruse, F.; Wang, Z.; Holzmann, B.; Strowig, T.; Sparwasser, T.; Lochner, M. MyD88 signaling in dendritic cells and the intestinal epithelium controls immunity against intestinal infection with C. rodentium. PLoS Pathog. 2017, 13, e1006357. [Google Scholar] [CrossRef]
- Gibson, D.L.; Ma, C.; Bergstrom, K.S.; Huang, J.T.; Man, C.; Vallance, B.A. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell. Microbiol. 2008, 10, 618–631. [Google Scholar] [CrossRef]
- Lebeis, S.L.; Bommarius, B.; Parkos, C.A.; Sherman, M.A.; Kalman, D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J. Immunol. 2007, 179, 566–577. [Google Scholar] [CrossRef]
- Mantle, M.; Rombough, C. Growth in and breakdown of purified rabbit small intestinal mucin by Yersinia enterocolitica. Infect. Immun. 1993, 61, 4131–4138. [Google Scholar] [CrossRef]
- Oellerich, M.F.; Jacobi, C.A.; Freund, S.; Niedung, K.; Bach, A.; Heesemann, J.; Trülzsch, K. Yersinia enterocolitica infection of mice reveals clonal invasion and abscess formation. Infect. Immun. 2007, 75, 3802–3811. [Google Scholar] [CrossRef] [PubMed]
- Mostolizadeh, R.; Dräger, A. Computational model informs effective control interventions against Y. enterocolitica co-infection. Biology 2020, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Hapfelmeier, S.; Stecher, B.; Barthel, M.; Kremer, M.; Müller, A.J.; Heikenwalder, M.; Stallmach, T.; Hensel, M.; Pfeffer, K.; Akira, S.; et al. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 2005, 174, 1675–1685. [Google Scholar] [CrossRef]
- Myhrvold, C.; Kotula, J.W.; Hicks, W.M.; Conway, N.J.; Silver, P.A. A distributed cell division counter reveals growth dynamics in the gut microbiota. Nat. Commun. 2015, 6, 10039. [Google Scholar] [CrossRef]
- Simůnek, J.; Brandysová, V.; Koppová, I.; Simůnek, J., Jr. The antimicrobial action of chitosan, low molar mass chitosan, and chitooligosaccharides on human colonic bacteria. Folia Microbiol. 2012, 57, 341–345. [Google Scholar] [CrossRef]
- Stein, R.R.; Bucci, V.; Toussaint, N.C.; Buffie, C.G.; Ratsch, G.; Pamer, E.G.; Sander, C.; Xavier, J.B. Ecological modeling from time-series inference: Insight into dynamics and stability of intestinal microbiota. PLoS Comput. Biol. 2013, 9, e1003388. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Grosse, J.; Asad, A.B.; Radda, G.K.; Golay, X. Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res. 2013, 3, 60. [Google Scholar] [CrossRef]
- Gordon, H.A.; Pesti, L. The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol. Rev. 1971, 35, 390–429. [Google Scholar] [CrossRef]
- Ge, X.; Ding, C.; Zhao, W.; Xu, L.; Tian, H.; Gong, J.; Zhu, M.; Li, J.; Li, N. Antibiotics-induced depletion of mice microbiota induces changes in host serotonin biosynthesis and intestinal motility. J. Transl. Med. 2017, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Brugiroux, S.; Beutler, M.; Pfann, C.; Garzetti, D.; Ruscheweyh, H.J.; Ring, D.; Diehl, M.; Herp, S.; Lotscher, Y.; Hussain, S.; et al. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol. 2016, 2, 16215. [Google Scholar] [CrossRef] [PubMed]
- Garzetti, D.; Brugiroux, S.; Bunk, B.; Pukall, R.; McCoy, K.D.; Macpherson, A.J.; Stecher, B. High-quality whole-genome sequences of the oligo-mouse-microbiota bacterial community. Genome Announc. 2017, 5, e00758-17. [Google Scholar] [CrossRef]
- Uchimura, Y.; Wyss, M.; Brugiroux, S.; Limenitakis, J.P.; Stecher, B.; McCoy, K.D.; Macpherson, A.J. Complete genome sequences of 12 species of stable defined moderately diverse mouse microbiota 2. Genome Announc. 2016, 4, e00951-16. [Google Scholar] [CrossRef]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef]
- Ackermann, M.; Stecher, B.; Freed, N.E.; Songhet, P.; Hardt, W.D.; Doebeli, M. Self-destructive cooperation mediated by phenotypic noise. Nature 2008, 454, 987–990. [Google Scholar] [CrossRef]
- Carbo, A.; Bassaganya-Riera, J.; Pedragosa, M.; Viladomiu, M.; Marathe, M.; Eubank, S.; Wendelsdorf, K.; Bisset, K.; Hoops, S.; Deng, X.; et al. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PLoS ONE 2013, 8, e73365. [Google Scholar] [CrossRef]
- De Jong, P.; Vissers, M.M.; van der Meer, R.; Bovee-Oudenhoven, I.M. In silico model as a tool for interpretation of intestinal infection studies. Appl. Environ. Microbiol. 2007, 73, 508–515. [Google Scholar] [CrossRef][Green Version]
- Diard, M.; Garcia, V.; Maier, L.; Remus-Emsermann, M.N.; Regoes, R.R.; Ackermann, M.; Hardt, W.D. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 2013, 494, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Leber, A.; Viladomiu, M.; Hontecillas, R.; Abedi, V.; Philipson, C.; Hoops, S.; Howard, B.; Bassaganya-Riera, J. Systems modeling of interactions between mucosal immunity and the gut microbiome during Clostridium difficile infection. PLoS ONE 2015, 10, e0134849. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.E.; Bell, C.S.; McClain, M.S.; Cover, T.L.; Giorgio, T.D. Dynamic computational model of symptomatic bacteremia to inform bacterial separation treatment requirements. PLoS ONE 2016, 11, e0163167. [Google Scholar] [CrossRef] [PubMed]
- Vlazaki, M.; Huber, J.; Restif, O. Integrating mathematical models with experimental data to investigate the within-host dynamics of bacterial infections. Pathog. Dis. 2019, 77, ftaa001. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Bergmann, F.T.; Chaouiya, C.; Dräger, A.; Hoops, S.; Keating, S.M.; Konig, M.; Novère, N.L.; Myers, C.J.; Olivier, B.G.; et al. The systems biology markup language (SBML): Language specification for level 3 version 2 core release 2. J. Integr. Bioinform. 2019, 16, 20190021. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.M.; Waltemath, D.; König, M.; Zhang, F.; Dräger, A.; Chaouiya, C.; Bergmann, F.T.; Finney, A.; Gillespie, C.S.; Helikar, T.; et al. SBML level 3: An extensible format for the exchange and reuse of biological models. Mol. Syst. Biol. 2020, 16, e9110. [Google Scholar] [CrossRef]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; et al. BioModels-15 years of sharing computational models in life science. Nucleic Acids Res. 2020, 48, D407–D415. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
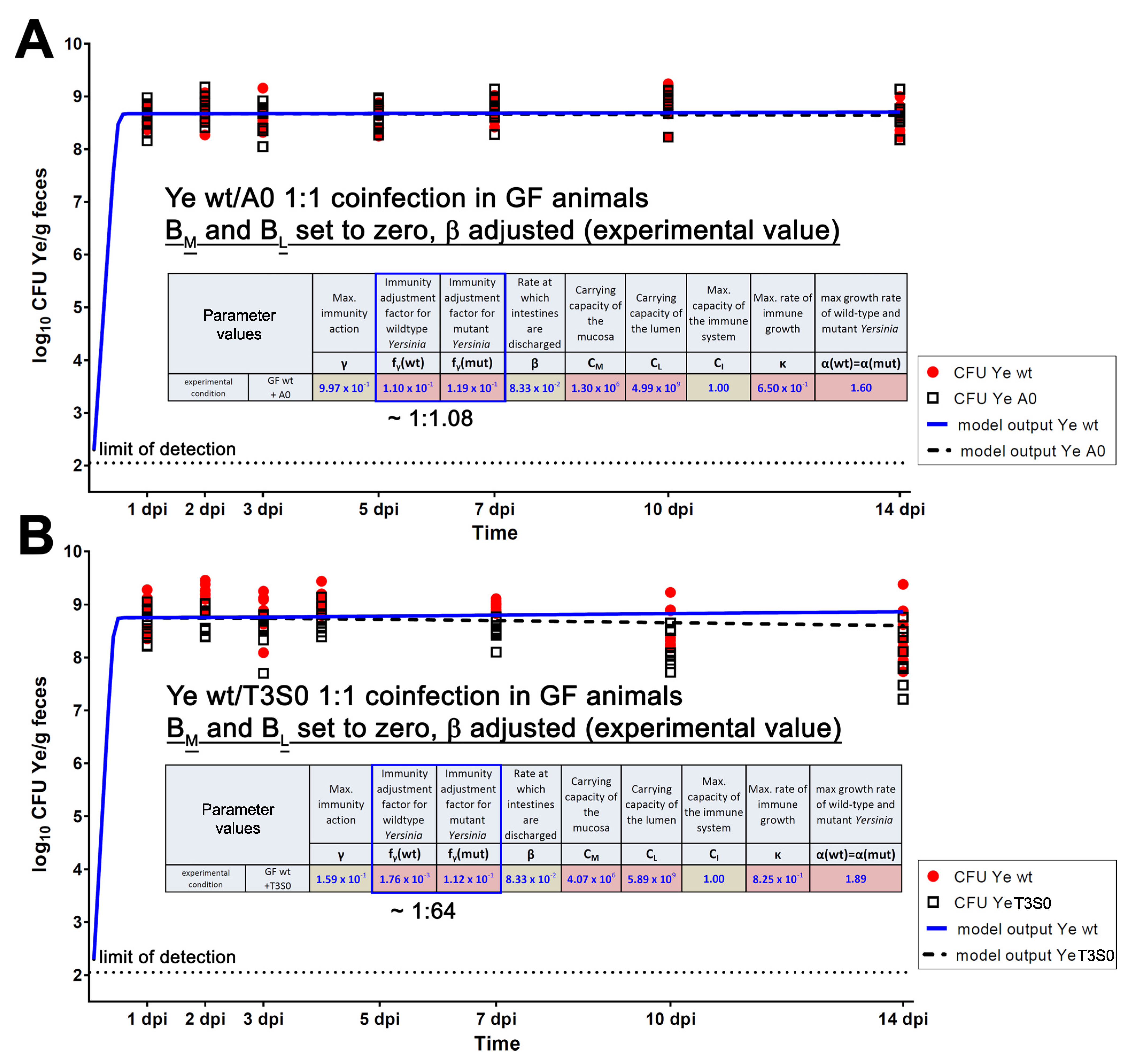{kind=link}
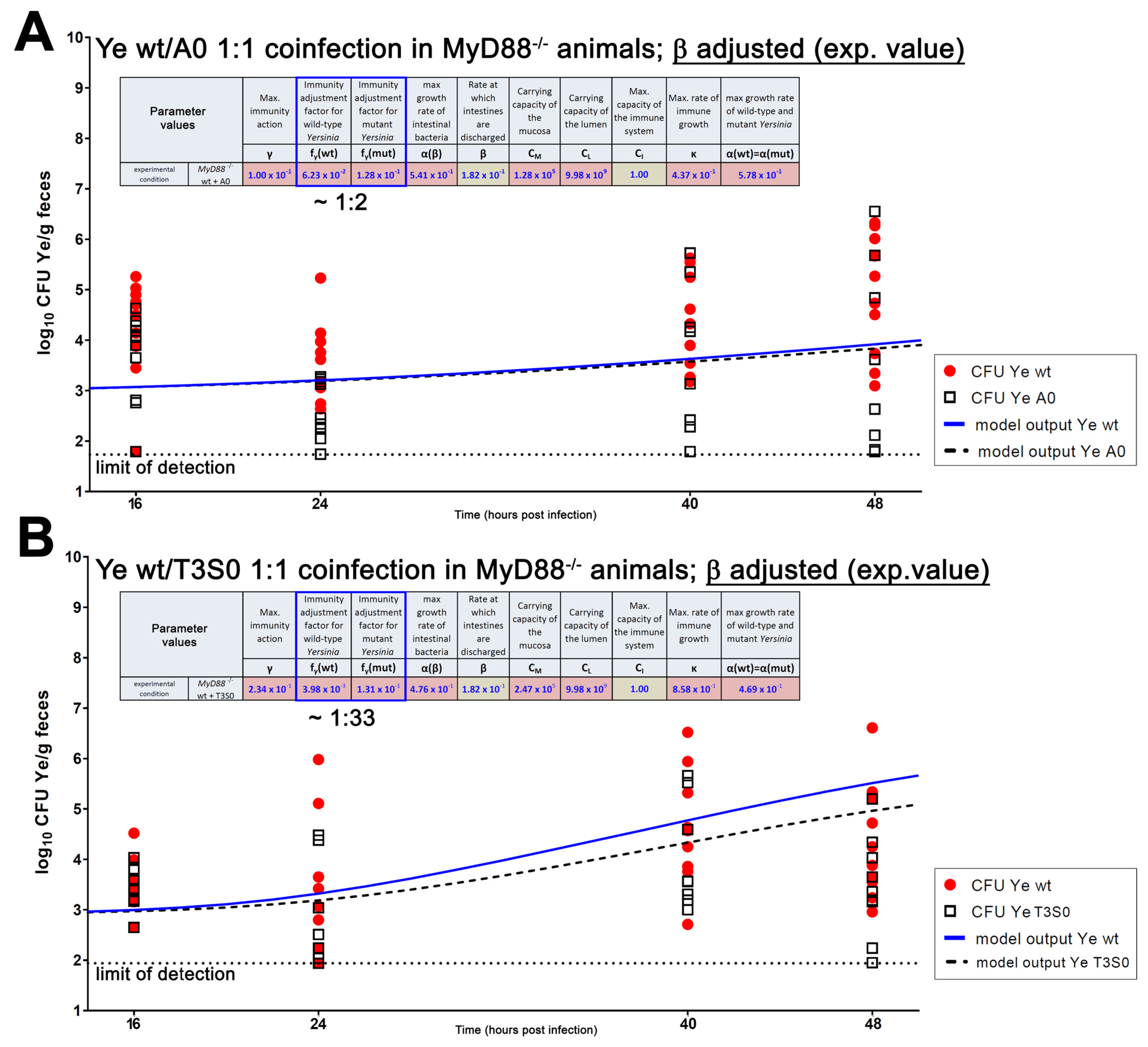{kind=link}
| Parameter | Definition | Source of Parameter Value | Function | Relation to Other/Comment | Preset Boundary/Exact Value | Assumptions Made to Justify the Choice of Preset Boundaries |
|---|---|---|---|---|---|---|
| Growth | ||||||
| α(B) | Growth rate of commensal bacteria | Estimated | Adjustable growth rate of commensal bacteria | Higher compared to growth rate of Ye | 0.4–2.0 | High diversity and different requirements for growth enable overall faster growth compared to Ye. |
| α(wt) | Growth rate of the Ye wt | Estimated | Adjustable growth rate of the Ye wt strain | Same as growth rate α(mut) | 0.4–2.0 | Growth optimum of Ye is at 30 °C; all Ye have the same requirements and compete for nutrients. Therefore, they grow slower compared to the microbiota. |
| α(mut) | Growth rate of the Ye mutant strains | Estimated | By adjustment of the Ye mutant growth rate, the model can account for growth deficiencies. | Same as growth rate α(wt) | 0.4–2.0 | Mutant Ye do not have a growth defect, they just lack a virulence factor dispensable for normal growth; in vitro growth did not reveal a difference in the growth rate of wt and mutant Ye. |
| Discharge | ||||||
| β(SPF) | Discharge rate of intestines | Experimental data (0.22/h) | Adjustable rate accounting for varying GIT passage times in different host models. | Higher as in MyD88−/− and GF | 0.22 | Justified by experimental data. |
| β(GF) | Discharge rate of intestines | Experimental data (0.08/h) | Adjustable rate accounting for varying GIT passage times in different host models | Lower than in SPF and MyD88−/− | 0.08 | Justified by experimental data. |
| β(MyD88−/−) | Discharge rate of intestines | Experimental data (0.18/h) | Adjustable rate accounting for varying GIT passage times in different host models | Lower than in SPF, but higher compared to GF animals | 0.18 | Justified by experimental data. |
| Immunity action related | ||||||
| γ | Immunity action rate | Adjustment factor for the immune action; 1 means 100% activity | Allows adjustment of the global immune action to account for immune deficiencies in a specific host. | Lower in GF and MyD88−/− | 0.1–1.0 | It is known that GF animals have a less developed immune system. MyD88−/− animals suffer from reduced activity of the immune system (see Introduction for references). |
| κ | Rate of immune growth | Estimated | Allows adjusting the rate at which the immune response is activated. | Unknown | 0.004–0.1 | No justification. |
| fγ(wt) | Immunity adjustment factor of the Ye wt | Estimated | Allows adjustment of resistance of the Ye wt strain to immune killing and thereby accounts for immune evasion mechanisms of a pathogen. | Lowest compared to fγ(YadA0) and fγ(T3S0) | 0.001–0.11 | The Ye wt strain is most resistant to killing by the immune system due to its ability to evade the host immune system, e.g., by engaging its T3SS, or by recruiting negative regulators of complement by YadA (see Introduction for references). |
| fγ(YadA0) | Immunity adjustment factor of the Ye YadA0 strain | Estimated | Adjustment allows accounting for an increased (or reduced) susceptibility to immune killing due to mutations affecting Ye immune evasion mechanisms. | Higher compared to fγ(wt) but lower or equal in comparison to fγ(T3S0) | 0.11–0.2 | Ye YadA0 is less resistant to killing by the immune system compared to Ye wt. |
| fγ(T3S0) | Immunity adjustment factor of the Ye T3S0 strain | Estimated | Adjustment allows accounting for an increased (or reduced) susceptibility to immune killing due to mutations affecting Ye immune evasion mechanisms. | Higher compared to fγ(wt) and higher or equal compared to fγ(YadA0) | 0.11–0.2 | Ye T3S0 is less resistant to killing by the immune system compared to Ye wt and less resistant compared to Ye YadA0. |
| Compartment capacities | ||||||
| CI | Capacity of the immune response | Predefined | Caps the maximum activity of the immune system. | CI = 1 means that the immune system is fully operative | ≤1 | Not applicable. |
| CM | Capacity of the mucosal site | Estimated | Caps the replication of the populations within the mucosa to an adjustable maximum capacity. | Lower than CL | 103–107 | Assumed range of commensal bacteria in proximity to the epithelium based on literature [73]. |
| CL | Capacity of the luminal site | Estimated | Caps the replication of populations within the intestinal lumen to an adjustable maximum capacity. | Higher than CM | 106–1010 | The total number of commensal bacteria in the distal small intestine is ~107–1010 per mL. |
| Alignment of experimental data with model output | ||||||
| Thickening factor | Reflects water extraction from fecal material during the colon passage | Experimental data | Allows adjusting experimentally measured CFU in fecal pellets and model-calculated CFU (within intestines). | - | SPF (1.3); MyD88−/−(1.3); GF (0.2) | Justified by experimental data |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geißert, J.K.; Bohn, E.; Mostolizadeh, R.; Dräger, A.; Autenrieth, I.B.; Beier, S.; Deusch, O.; Renz, A.; Eichner, M.; Schütz, M.S. A Computational Model of Bacterial Population Dynamics in Gastrointestinal Yersinia enterocolitica Infections in Mice. Biology 2022, 11, 297. https://doi.org/10.3390/biology11020297
Geißert JK, Bohn E, Mostolizadeh R, Dräger A, Autenrieth IB, Beier S, Deusch O, Renz A, Eichner M, Schütz MS. A Computational Model of Bacterial Population Dynamics in Gastrointestinal Yersinia enterocolitica Infections in Mice. Biology. 2022; 11(2):297. https://doi.org/10.3390/biology11020297
Chicago/Turabian StyleGeißert, Janina K., Erwin Bohn, Reihaneh Mostolizadeh, Andreas Dräger, Ingo B. Autenrieth, Sina Beier, Oliver Deusch, Alina Renz, Martin Eichner, and Monika S. Schütz. 2022. "A Computational Model of Bacterial Population Dynamics in Gastrointestinal Yersinia enterocolitica Infections in Mice" Biology 11, no. 2: 297. https://doi.org/10.3390/biology11020297
APA StyleGeißert, J. K., Bohn, E., Mostolizadeh, R., Dräger, A., Autenrieth, I. B., Beier, S., Deusch, O., Renz, A., Eichner, M., & Schütz, M. S. (2022). A Computational Model of Bacterial Population Dynamics in Gastrointestinal Yersinia enterocolitica Infections in Mice. Biology, 11(2), 297. https://doi.org/10.3390/biology11020297