
To Stick or Not to Stick: Adhesions in Orofacial Clefts



Abstract
Simple Summary
Abstract
1. Introduction
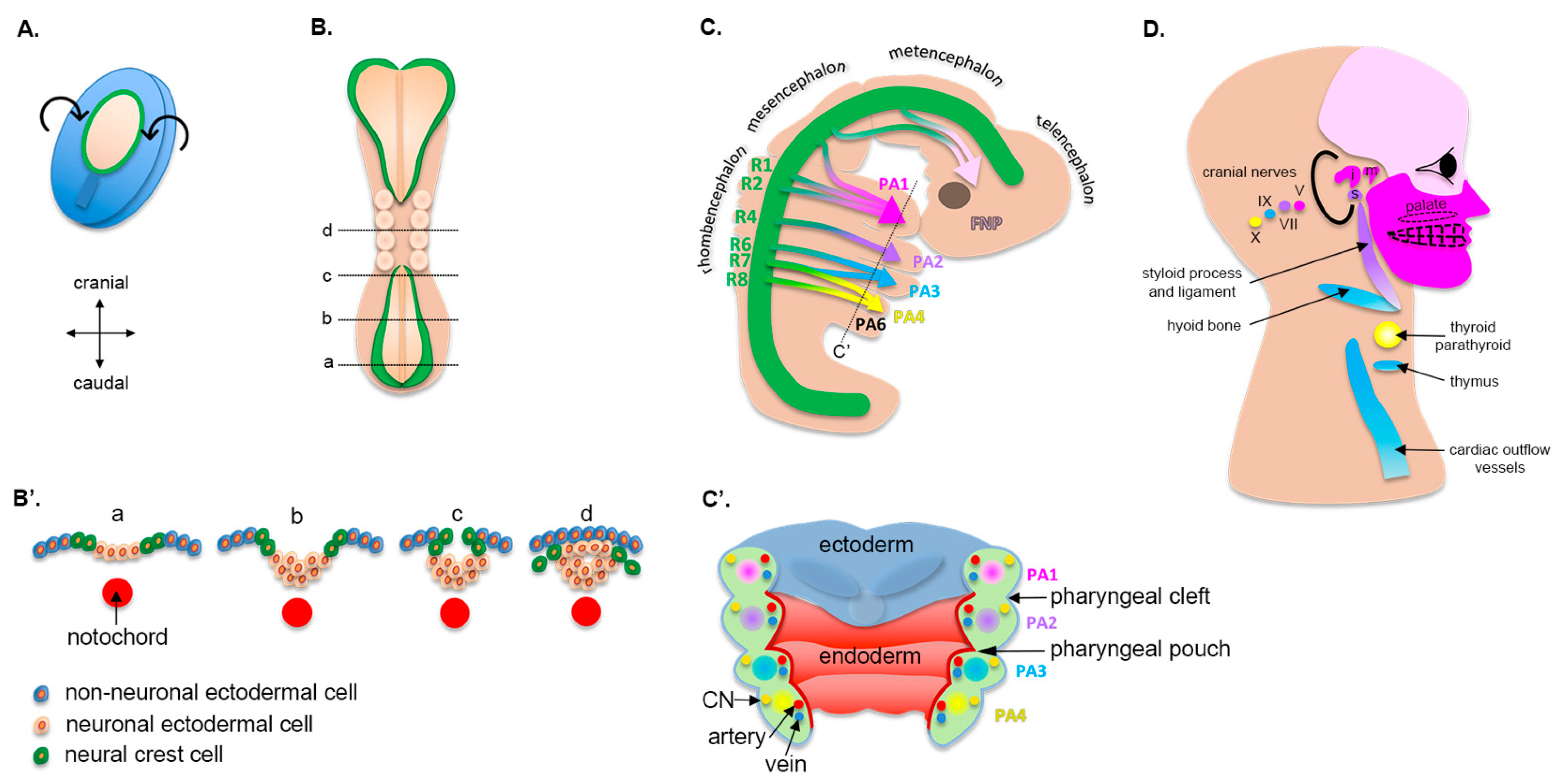
2. Craniofacial Morphogenesis
2.1. Early Embryogenesis
2.2. Cranial Neural Crest Cells
2.3. Pharyngeal Arches
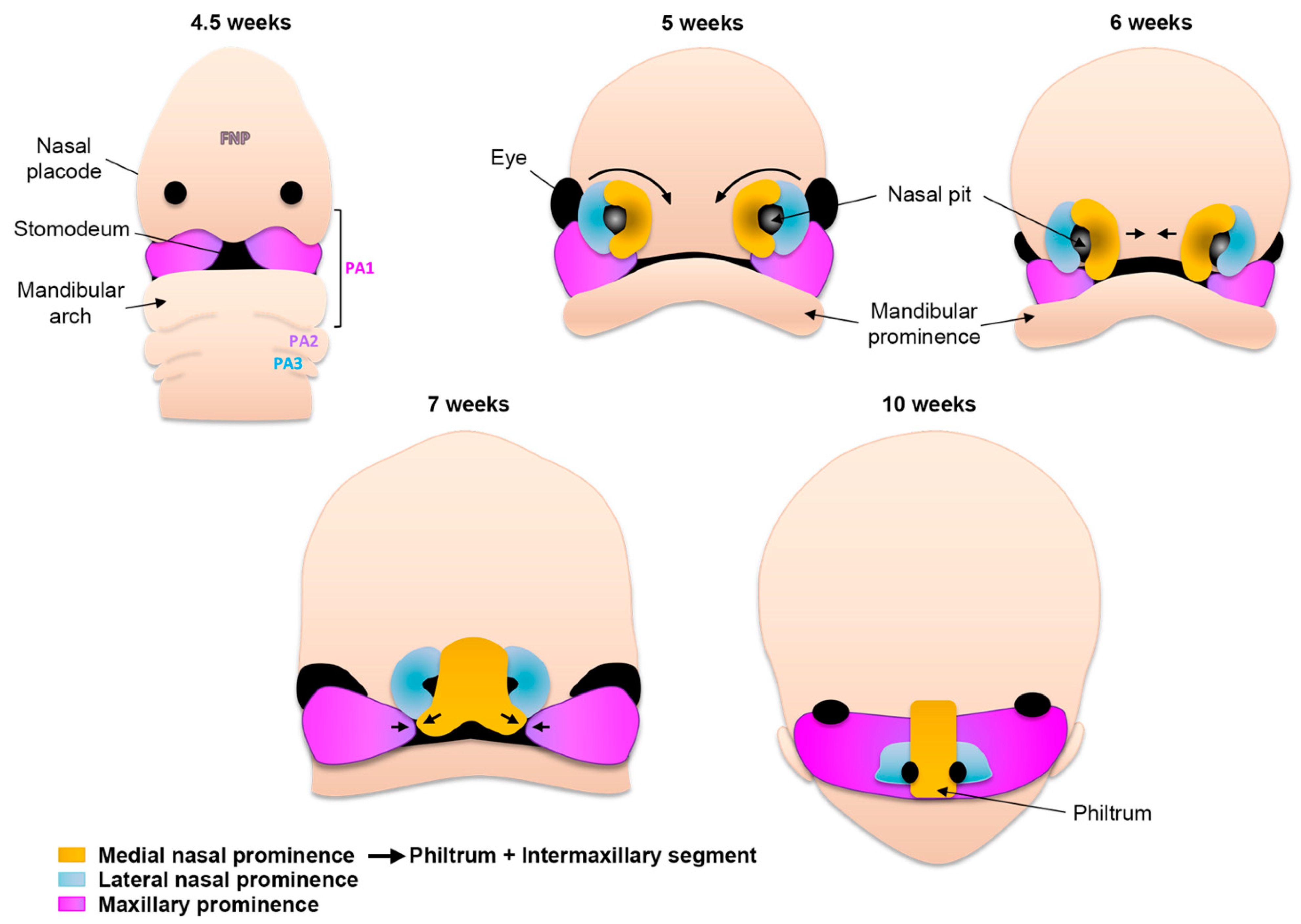
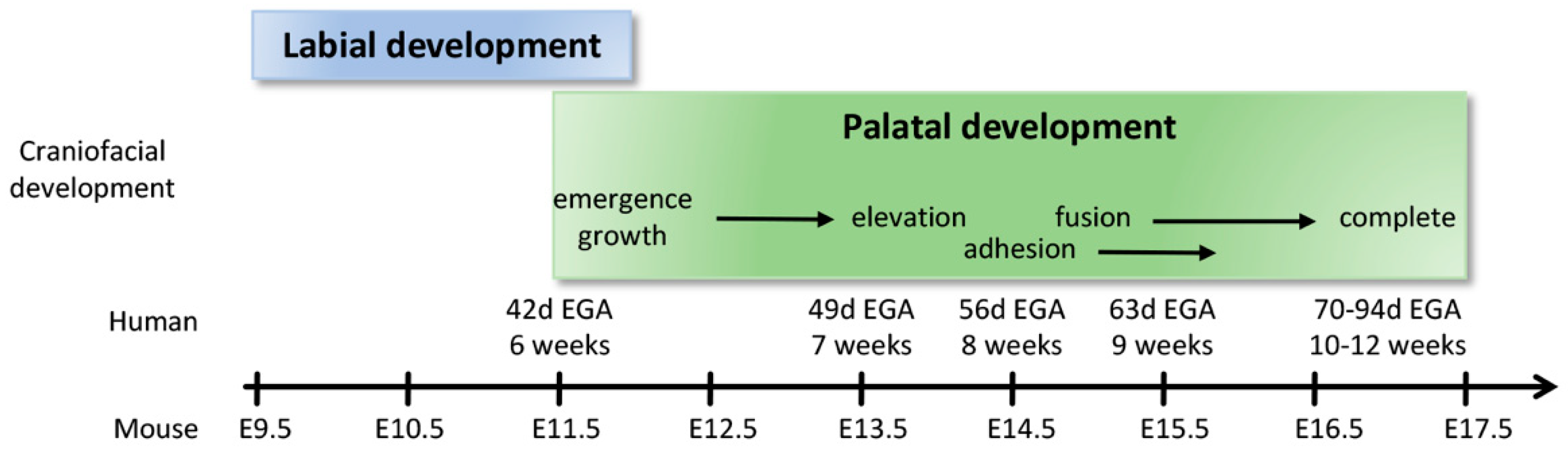
2.4. Formation of the Upper Lip, Nose, and Primary Palate
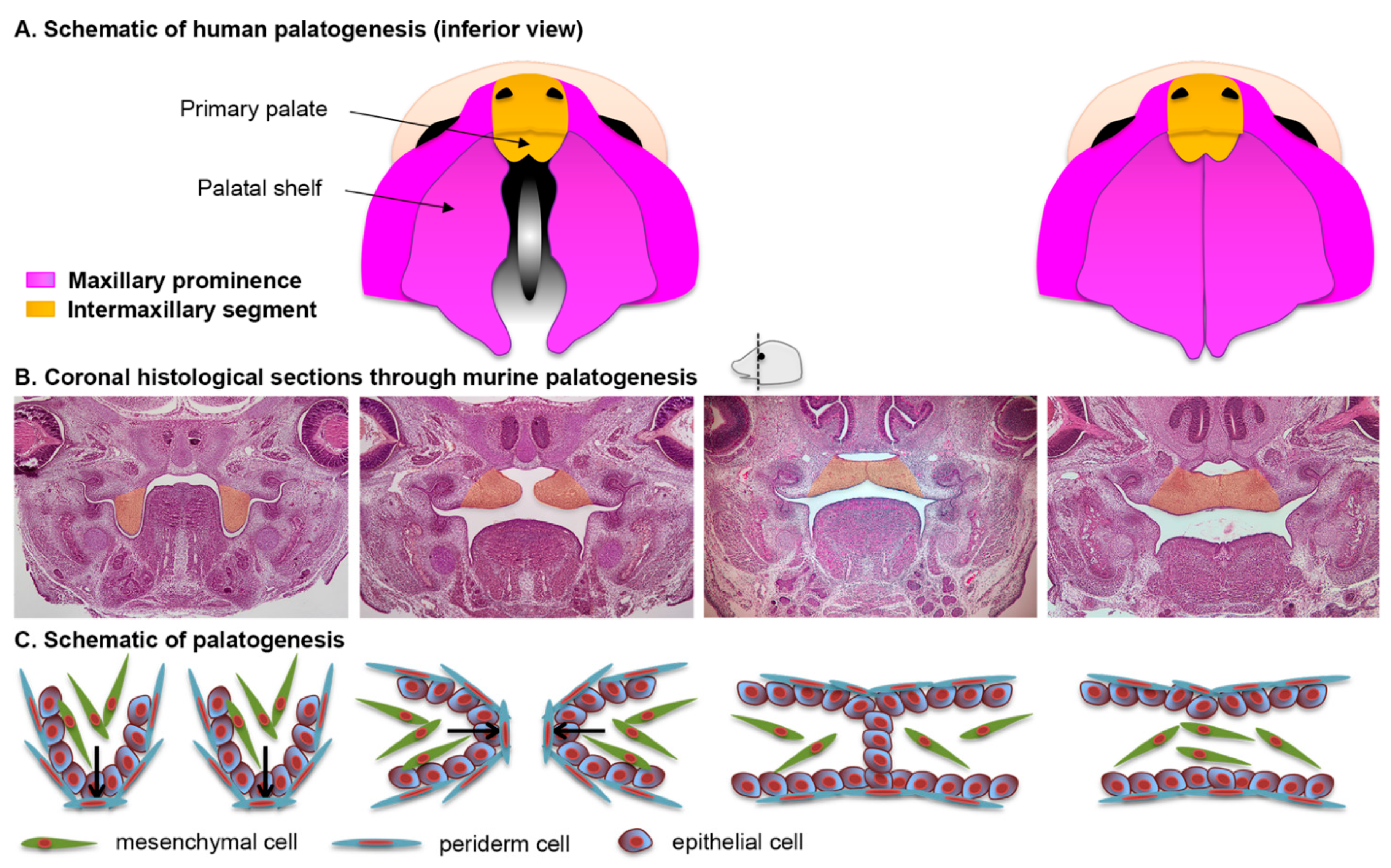
2.5. Formation of the Secondary Palate
2.6. The Periderm
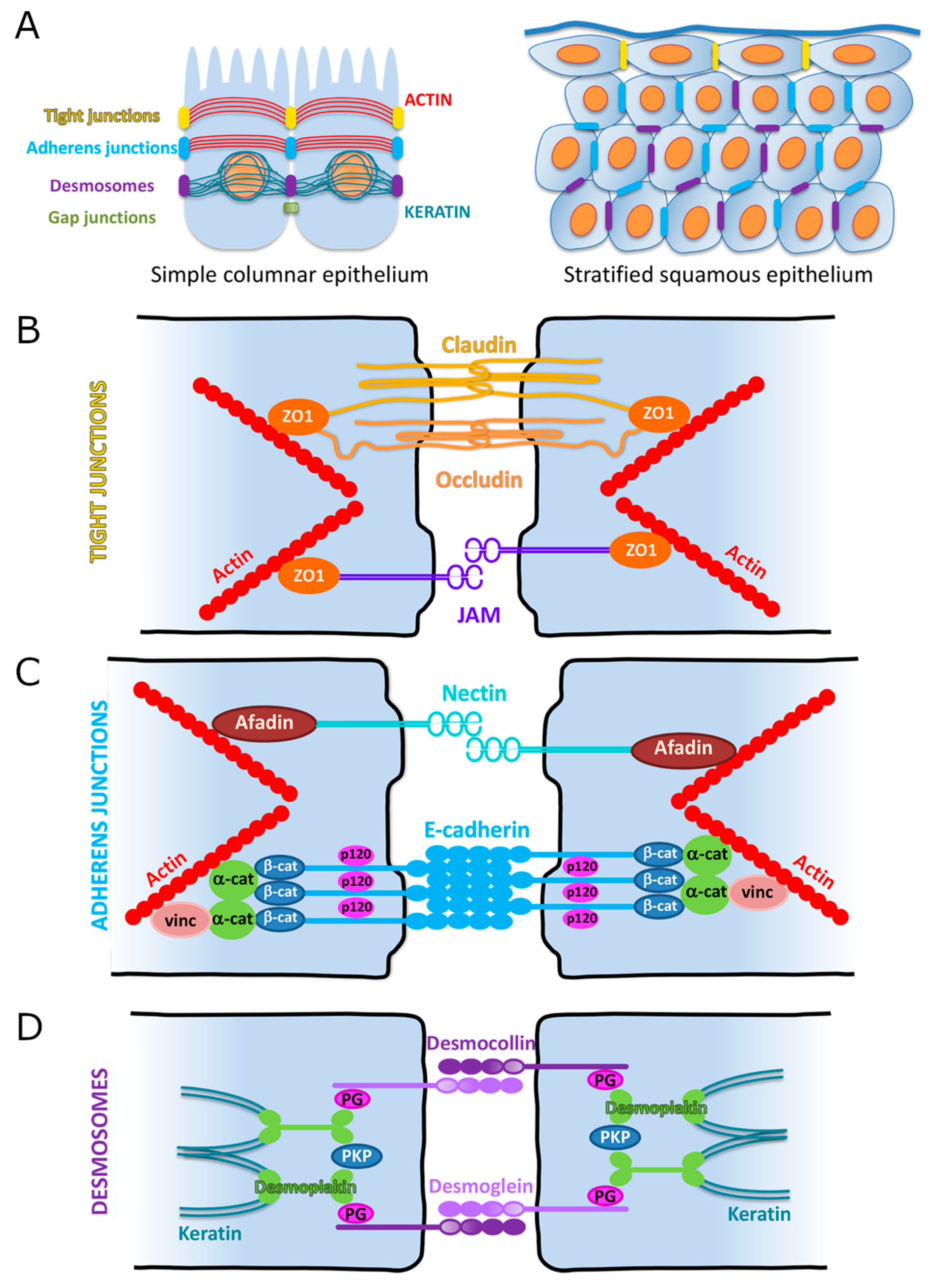
3. Cellular Adhesions
3.1. Overview
3.2. Adherens Junctions (AJs)
3.2.1. Cadherin-Based Adhesions
3.2.2. Nectin-Based Adhesions
3.3. Tight Junctions (TJs)
3.3.1. Claudins
3.3.2. Occludin
3.3.3. Zonula Occludens
3.3.4. Junctional Adhesion Molecules (JAMs)
3.4. Desmosomes
3.5. Desmosomal Cadherins (Desmogleins, Desmocollins)
3.6. Desmosomal Armadillo Proteins (Plakophilin, Plakoglobin)
3.7. Desmoplakin
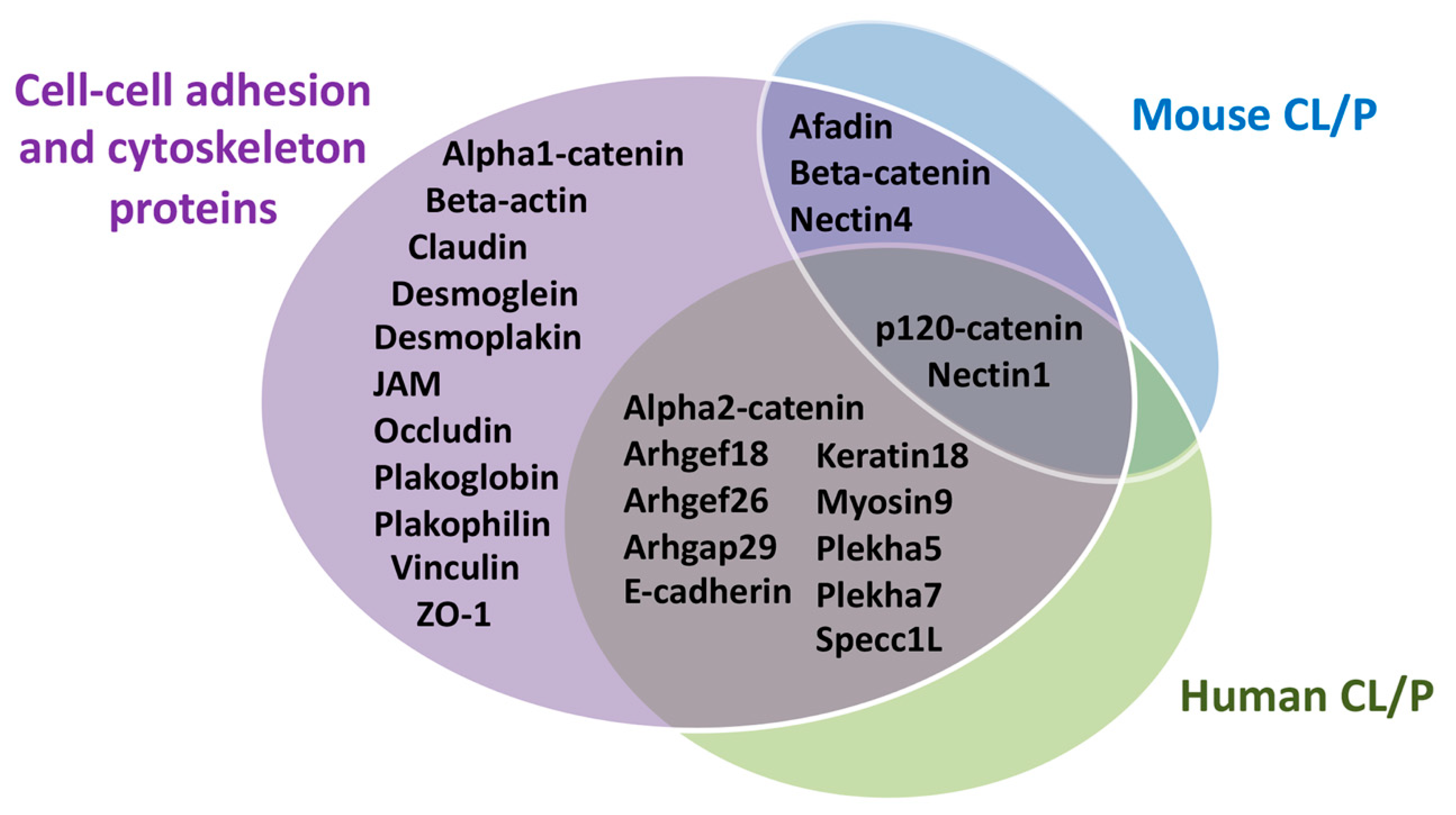
4. Contribution of Cellular Adhesion to Craniofacial Morphogenesis
4.1. Cytoskeleton in Craniofacial Morphogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Protein) | Type of Human Craniofacial Clefts | Human Variants Associated with the CL/P Phenotype | Mouse Knockouts Craniofacial Phenotype |
|---|---|---|---|
| AFDN (Afadin) | No CL/P | KO embryonic lethal [99] K14-Cre cKO no CP [100] In utero cKO CP and oral adhesions [100] | |
| CTNNA2 (Alpha2-catenin) | NSCL/P [101] | g.82025185 | None |
| ARHGEF18 (Arhgef18) | NSCL/P [102] | c.1484G>A p.Arg495Gln | None |
| ARHGEF26 (Arhgef26) | NSCL/P [103] | g.153840512:A>T g.153943770:C>G | None |
| ARHGAP29 (Arhgap29) | NSCL/P [104,105,106,107] | c.62_63delCT p.Ser21Tyrfs*20 c.91C>T p.Leu31Phe c.698-1G>C c.888G>C p.Arg296Ser c.976A>T p.Lys326* c.1252G>A p.Val418Ile c.1475C>A p.Ser492* c.1576+1G>A c.1654T>C p.Ser552Pro c.1847G>A p.Arg616His c.1865C>T p.Thr622Met c.2017T>G p.Phe673Val c.2109+1G>A c.2393G>A p.Arg798Gln c.2533A>G p.Ile845Val c.2617C>T p.Arg873Cys c.3023G>A. p.(Arg1008Lys) c.3326_3328delCAA p.Thr1109del c.3339T>G p.Ile1113Met g.94545160:T>C g.94547883:C>G g.94547889:G>A | KO embryonic lethal [108] Heterozygote no CP but oral adhesion [108] |
| CTNNB1 (Beta-catenin) | No CL/P [109] | cKO CP [110] | |
| CTNND1 (p120-catenin) | Syndromic CL/P [111] NSCL/P [112,113] | c.606_627del p.Pro203Leufs*25 c.1093C>T p.Gln365* c.2098C>T p.Arg700* g.57559005:C>G p.Gln19Glu g.57564445_5756446del p.Asp313Profs*9 g.57569255:G>A p.Trp336* g.57571168:A>G p.Asp499Gly g.57572202:C>T p.Leu558Phe g.57573381:C>T p.Arg584Trp g57575761:G>T p.Try690Cys g.57576939:G>T c.2417+1G>T g.57578892:C>T p.Arg852* c.1381C>T p.Arg461* c1481_1485del p.Leu494Argfs*5 c.2598_2601dupTGAT p.Ser868* c.2737dupC p.His913Profs*3 | CreCT cKO 47% CP [112] |
| CDH1 (E-cadherin) | Syndromic CL/P [111] NSCL/P [112,114,115,116,117,118] | c.760G>T p.Asp254Tyr c.770A>T p.Asp257Val c.1320G>T p. ? c.1320+1G>C p. ? c.1361_1363del p.Val454del c.387+5G>A p.? c.468G>C p.Trp156Cys c.752C>T p.Thr251Met c.760G>A p.Asp254Asn c.768T>A p.Asn256Lys c.1023T>G p.Tyr341* c.1489G>A p.Glu497Lys c.1766A>T p.Asn589Ile c.2351G>A p.Arg784His c.2426_2427del p.Asn809Ilefs*3 | KO embryonic lethal [119] Wnt1-Cre cKO no cleft [120] K14-Cre cKO no cleft [121] |
| KRT18 (Keratin18) | NSCL/P [97] | g.53344318:G>T | None |
| MYOSIN9 (Myosin9) | NSCL/P [94,95,96] | g.35044605:C>T g.35048804:C>T g.35007860:T>C | None |
| PVRL1 (Nectin1) | Syndromic CL/P [122] | p.Tryp185X | KO no CL/P [123,124] In utero cKO CP [100] |
| PVRL4 (Nectin4) | No CL/P [125] | KO 11–40% CP [100] | |
| PLEKHA5 (Plekha5) | NSCL/P [112] | g.19440414:A>G p.Tyr590Cys | None |
| PLEKHA7 (Plekha7) | NSCL/P [112] | g.16838582:C>T p.Gly544Asp g.16838676:G>A p.Arg513Trp g.16834682:T>C p.Asp662Gly | None |
| SPECC1L (Specc1L) | Syndromic CL/P [126] NSCL/P [127] | c.569C>T p.Thr190Met c.1244A>C p.Gln415Pro c.273G>A p.Met91Iso c.256G>A p.Ala86Thr c.895A>G p.Thr299Ala c.1637G>A p.Arg546Gln | KO embryonic lethal [127] In-frame deletion CP [128] |
4.2. Role of Adherens Junctions in Facial Morphogenesis
4.2.1. Cadherin-Based Adhesions: Lessons from Patients
4.2.2. Cadherin-Based Adhesions: Lessons from Murine Models
4.2.3. Nectin-Based Adhesions: Lessons from Patients
4.2.4. Nectin-Based Adhesions: Lessons from Murine Models
4.3. Role of Other Adhesion Components in Facial Morphogenesis
4.3.1. Lessons from Patients
4.3.2. Lessons from Murine Models
4.4. Other Molecules Modulating Cell–Cell Junctions Critical for Facial Morphogenesis
4.4.1. SPECC1L and Pleckstrin
4.4.2. IRF6 and the Rho Pathway
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chuai, M.; Weijer, C.J. The mechanisms underlying primitive streak formation in the chick embryo. Curr. Top. Dev. Biol. 2008, 81, 135–156. [Google Scholar]
- Kousa, Y.A.; Mansour, T.A.; Seada, H.; Matoo, S.; Schutte, B.C. Shared molecular networks in orofacial and neural tube development. Birth Defects Res. 2017, 109, 169–179. [Google Scholar] [CrossRef]
- Munoz, W.A.; Trainor, P.A. Neural crest cell evolution: How and when did a neural crest cell become a neural crest cell. Curr. Top. Dev. Biol. 2015, 111, 3–26. [Google Scholar]
- Minoux, M.; Rijli, F.M. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef]
- Simoes-Costa, M.; Bronner, M.E. Establishing neural crest identity: A gene regulatory recipe. Development 2015, 142, 242–257. [Google Scholar] [CrossRef]
- Santagati, F.; Rijli, F.M. Cranial neural crest and the building of the vertebrate head. Nat. Rev. Neurosci. 2003, 4, 806–818. [Google Scholar] [CrossRef]
- Frisdal, A.; Trainor, P.A. Development and evolution of the pharyngeal apparatus. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 403–418. [Google Scholar] [CrossRef]
- Guthrie, S. Patterning and axon guidance of cranial motor neurons. Nat. Rev. Neurosci. 2007, 8, 859–871. [Google Scholar] [CrossRef]
- Diewert, V.M. A morphometric analysis of craniofacial growth and changes in spatial relations during secondary palatal development in human embryos and fetuses. Am. J. Anat. 1983, 167, 495–522. [Google Scholar] [CrossRef]
- Bush, J.O.; Jiang, R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 231–243. [Google Scholar] [CrossRef]
- Kim, S.; Lewis, A.E.; Singh, V.; Ma, X.; Adelstein, R.; Bush, J.O. Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol. 2015, 13, e1002122. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.J.; Ammond, N.L.; Coulombe, P.A.; Saloranta, C.; Nousiainen, H.O.; Salonen, R.; Berry, A.; Hanley, N.; Headon, D.; Karikoski, R.; et al. Periderm prevents pathological epithelial adhesions during embryogenesis. J. Clin. Investig. 2014, 124, 3891–3900. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, K.A. Structure and Function of the Developing Human Skin. In Physiology, Biochemistry, and Molecular Biology of the Skin; Goldsmith, L.A., Ed.; Oxford University Press: New York, NY, USA, 1991; pp. 63–110. [Google Scholar]
- McGowan, K.M.; Coulombe, P.A. Onset of keratin 17 expression coincides with the definition of major epithelial lineages during skin development. J. Cell Biol. 1998, 143, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, J.; Li, Z.; Ozturk, F.; Gurumurthy, C.; Romano, R.A.; Sinha, S.; Nawshad, A. TGFbeta3 regulates periderm removal through DeltaNp63 in the developing palate. J. Cell. Physiol. 2015, 230, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, A.; Takeichi, M. Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO J. 1988, 7, 3679–3684. [Google Scholar] [CrossRef]
- Danjo, Y.; Gipson, I.K. Actin ‘purse string’ filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J. Cell Sci. 1998, 111, 3323–3332. [Google Scholar] [CrossRef]
- Yulis, M.; Kusters, D.H.M.; Nusrat, A. Cadherins: Cellular adhesive molecules serving as signalling mediators. J. Physiol. 2018, 596, 3883–3898. [Google Scholar] [CrossRef]
- McCrea, P.D.; Gumbiner, B.M. Purification of a 92-kDa cytoplasmic protein tightly associated with the cell-cell adhesion molecule E-cadherin (uvomorulin). Characterization and extractability of the protein complex from the cell cytostructure. J. Biol. Chem. 1991, 266, 4514–4520. [Google Scholar] [CrossRef]
- Stappert, J.; Kemler, R. A short core region of E-cadherin is essential for catenin binding and is highly phosphorylated. Cell Adhes. Commun. 1994, 2, 319–327. [Google Scholar] [CrossRef]
- Su, W.; Kowalczyk, A.P. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol. Biol. Cell 2017, 28, 76–84. [Google Scholar] [CrossRef]
- Brieher, W.M.; Yap, A.S.; Gumbiner, B.M. Lateral dimerization is required for the homophilic binding activity of C-cadherin. J. Cell Biol. 1996, 135, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Nagar, B.; Overduin, M.; Ikura, M.; Rini, J.M. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature 1996, 380, 360–364. [Google Scholar] [CrossRef]
- Shapiro, L.; Kwong, P.D.; Fannon, A.M.; Colman, D.R.; Hendrickson, W.A. Considerations on the folding topology and evolutionary origin of cadherin domains. Proc. Natl. Acad. Sci. USA 1995, 92, 6793–6797. [Google Scholar] [CrossRef]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef]
- Yamada, S.; Pokutta, S.; Drees, F.; Weis, W.I.; Nelson, W.J. Deconstructing the cadherin-catenin-actin complex. Cell 2005, 123, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Ireton, R.C.; Reynolds, A.B. A core function for p120-catenin in cadherin turnover. J. Cell Biol. 2003, 163, 525–534. [Google Scholar] [CrossRef]
- Shibamoto, S.; Hayakawa, M.; Takeuchi, K.; Hori, T.; Miyazawa, K.; Kitamura, N.; Johnson, K.R.; Wheelock, M.J.; Matsuyoshi, N.; Takeichi, M.; et al. Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes. J. Cell Biol. 1995, 128, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Sheth, B.; Fesenko, I. Cell adhesion in the preimplantation mammalian embryo and its role in trophectoderm differentiation and blastocyst morphogenesis. Front. Biosci. 2001, 6, 1000–1007. [Google Scholar] [CrossRef]
- Larue, L.; Ohsugi, M.; Hirchenhain, J.; Kemler, R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc. Natl. Acad. Sci. USA 1994, 91, 8263–8267. [Google Scholar] [CrossRef]
- Perez-Moreno, M.; Davis, M.A.; Wong, E.; Pasolli, H.A.; Reynolds, A.B. p120-catenin mediates inflammatory responses in the skin. Cell 2006, 124, 631–644. [Google Scholar] [CrossRef]
- Batsche, E.; Cremisi, C. Opposite transcriptional activity between the wild type c-myc gene coding for c-Myc1 and c-Myc2 proteins and c-Myc1 and c-Myc2 separately. Oncogene 1999, 18, 5662–5671. [Google Scholar] [CrossRef][Green Version]
- Gumbiner, B.M. Regulation of cadherin adhesive activity. J. Cell Biol. 2000, 148, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.M.; Reynolds, A.B. Tyrosine phosphorylation and cadherin/catenin function. Bioessays 1997, 19, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.E.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef]
- Lickert, H.; Bauer, A.; Kemler, R.; Stappert, J. Casein kinase II phosphorylation of E-cadherin increases E-cadherin/beta-catenin interaction and strengthens cell-cell adhesion. J. Biol. Chem. 2000, 275, 5090–5095. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Behrens, J.; Vakaet, L.; Friis, R.; Winterhager, E.; Van Roy, F.; Mareel, M.M.; Birchmeier, W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 1993, 120, 757–766. [Google Scholar] [CrossRef]
- Hazan, R.B.; Norton, L. The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J. Biol. Chem. 1998, 273, 9078–9084. [Google Scholar] [CrossRef] [PubMed]
- Kypta, R.M.; Su, H.; Reichardt, L.F. Association between a transmembrane protein tyrosine phosphatase and the cadherin-catenin complex. J. Cell Biol. 1996, 134, 1519–1529. [Google Scholar] [CrossRef]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- Braga, V.M.; Machesky, L.M.; Hall, A.; Hotchin, N.A. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J. Cell Biol. 1997, 137, 1421–1431. [Google Scholar] [CrossRef]
- Morrison, M.E.; Racaniello, V.R. Molecular cloning and expression of a murine homolog of the human poliovirus receptor gene. J. Virol. 1992, 66, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, F.; Menotti, L.; Mirandola, P.; Lopez, M.; Campadelli-Fiume, G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998, 72, 9992–10002. [Google Scholar] [CrossRef]
- Satoh-Horikawa, K.; Nakanishi, H.; Takahashi, K.; Miyahara, M.; Nishimura, M.; Tachibana, K.; Mizoguchi, A.; Takai, Y. Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J. Biol. Chem. 2000, 275, 10291–10299. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaïde, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef] [PubMed]
- Sakisaka, T.; Nakanishi, H.; Takahashi, K.; Mandai, K.; Miyahara, M.; Satoh, A.; Takaishi, K.; Takai, Y. Different behavior of l-afadin and neurabin-II during the formation and destruction of cell-cell adherens junction. Oncogene 1999, 18, 1609–1617. [Google Scholar] [CrossRef][Green Version]
- Takahashi, K.; Nakanishi, H.; Miyahara, M.; Mandai, K.; Satoh, K.; Satoh, A.; Nishioka, H.; Aoki, J.; Nomoto, A.; Mizoguchi, A. Nectin/PRR: An immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 1999, 145, 539–549. [Google Scholar] [CrossRef]
- Rajasekaran, A.K.; Hojo, M.; Huima, T.; Rodriguez-Boulan, E. Catenins and zonula occludens-1 form a complex during early stages in the assembly of tight junctions. J. Cell Biol. 1996, 132, 451–463. [Google Scholar] [CrossRef]
- Morita, K.; Itoh, M.; Saitou, M.; Ando-Akatsuka, Y.; Furuse, M.; Yoneda, K.; Imamura, S.; Fujimoto, K.; Tsukita, S. Subcellular distribution of tight junction-associated proteins (occludin, ZO-1, ZO-2) in rodent skin. J. Investig. Derm. 1998, 110, 862–866. [Google Scholar] [CrossRef]
- Ooshio, T.; Kobayashi, R.; Ikeda, W.; Miyata, M.; Fukumoto, Y.; Matsuzawa, N.; Ogita, H.; Takai, Y. Involvement of the interaction of afadin with ZO-1 in the formation of tight junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 2010, 285, 5003–5012. [Google Scholar] [CrossRef]
- Anderson, J.M. Molecular structure of tight junctions and their role in epithelial transport. Physiology 2001, 16, 126–130. [Google Scholar] [CrossRef]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Van Itallie, C.M.; Anderson, J.M. Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol. Biol. Cell 2012, 23, 577–590. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Itoh, M.; Nagafuchi, A.; Moroi, S.; Tsukita, S. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J. Cell Biol. 1997, 138, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Matlis, S.; Ullmer, C.; Scherer, S.S.; Peles, E. Distinct claudins and associated PDZ proteins form different autotypic tight junctions in myelinating Schwann cells. J. Cell Biol. 2002, 159, 361–372. [Google Scholar] [CrossRef]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20, quiz 21–2. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Anderson, J.M. Two classes of tight junctions are revealed by ZO-1 isoforms. Am. J. Physiol. 1993, 264, C918–C924. [Google Scholar] [CrossRef]
- Haskins, J.; Gu, L.; Wittchen, E.S.; Hibbard, J.; Stevenson, B.R. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J. Cell Biol. 1998, 141, 199–208. [Google Scholar] [CrossRef]
- Rodgers, L.S.; Beam, M.T.; Anderson, J.M.; Fanning, A.S. Epithelial barrier assembly requires coordinated activity of multiple domains of the tight junction protein ZO-1. J. Cell Sci. 2013, 126, 1565–1575. [Google Scholar] [CrossRef]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 1986, 103, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Mandell, K.J.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J. Biol. Chem. 2005, 280, 11665–11674. [Google Scholar] [CrossRef]
- Kostrewa, D.; Brockhaus, M.; D’Arcy, A.; Dale, G.E.; Nelboeck, P.; Schmid, G.; Mueller, F.; Bazzoni, G.; Dejana, E.; Bartfai, T.; et al. X-ray structure of junctional adhesion molecule: Structural basis for homophilic adhesion via a novel dimerization motif. EMBO J. 2001, 20, 4391–4398. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K.; Suzuki, A.; Horikoshi, Y.; Hirose, T.; Meyer Zu Brickwedde, M.K.; Ohno, S.; Vestweber, D. The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J. 2001, 20, 3738–3748. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Bazzoni, G.; Martinez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, S.A.; Rodriguez, J.M.; Arrate, M.P.; Tran, T.M.; Brock, T.A. JAM2 interacts with alpha4beta1. Facilitation by JAM3. J. Biol. Chem. 2002, 277, 27589–27592. [Google Scholar] [CrossRef]
- Liang, T.W.; Chiu, H.H.; Gurney, A.; Sidle, A.; Tumas, D.B.; Schow, P.; Foster, J.; Klassen, T.; Dennis, K.; DeMarco, R.A.; et al. Vascular endothelial-junctional adhesion molecule (VE-JAM)/JAM 2 interacts with T, NK, and dendritic cells through JAM 3. J. Immunol. 2002, 168, 1618–1626. [Google Scholar] [CrossRef]
- Franke, W.W.; Schmid, E.; Winter, S.; Osborn, M.; Weber, K. Widespread occurrence of intermediate-sized filaments of the vimentin-type in cultured cells from diverse vertebrates. Exp. Cell Res. 1979, 123, 25–46. [Google Scholar] [CrossRef]
- Troyanovsky, S.M.; Eshkind, L.G.; Troyanovsky, R.B.; Leube, R.E.; Franke, W.W. Contributions of cytoplasmic domains of desmosomal cadherins to desmosome assembly and intermediate filament anchorage. Cell 1993, 72, 561–574. [Google Scholar] [CrossRef]
- Kowalczyk, A.P.; Bornslaeger, E.A.; Norvell, S.M.; Palka, H.L.; Green, K.J. Desmosomes: Intercellular adhesive junctions specialized for attachment of intermediate filaments. Int. Rev. Cytol. 1999, 185, 237–302. [Google Scholar]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Mattey, D.L.; Garrod, D.R. Calcium-induced reorganization of desmosomal components in cultured human keratinocytes. J. Cell Biol. 1984, 99, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Stuart, R.O.; Sun, A.; Bush, K.T.; Nigam, S.K. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J. Biol. Chem. 1996, 271, 13636–13641. [Google Scholar] [CrossRef]
- Kimura, T.E.; Merritt, A.J.; Garrod, D.R. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J. Investig. Dermatol. 2007, 127, 775–781. [Google Scholar] [CrossRef]
- Boggon, T.J.; Murray, J.; Chappuis-Flament, S.; Wong, E.; Gumbiner, B.M.; Shapiro, L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 2002, 296, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Hulpiau, P.; van Roy, F. Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol. 2009, 41, 349–369. [Google Scholar] [CrossRef]
- Hatzfeld, M. Plakophilins: Multifunctional proteins or just regulators of desmosomal adhesion? Biochim. Biophys. Acta 2007, 1773, 69–77. [Google Scholar] [CrossRef]
- Mahoney, M.G.; Hu, Y.; Brennan, D.; Bazzi, H.; Christiano, A.M.; Wahl, J.K., 3rd. Delineation of diversified desmoglein distribution in stratified squamous epithelia: Implications in diseases. Exp. Derm. 2006, 15, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Rubsam, M.; Broussard, J.A.; Wickström, S.A.; Nekrasova, O.; Green, K.J.; Niessen, C.M. Adherens Junctions and Desmosomes Coordinate Mechanics and Signaling to Orchestrate Tissue Morphogenesis and Function: An Evolutionary Perspective. Cold Spring Harb. Perspect. Biol. 2018, 10, a029207. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Sweeton, D.; Casey, M.; Wieschaus, E. Wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development 1994, 120, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Hatzfeld, M.; Nachtsheim, C. Cloning and characterization of a new armadillo family member, p0071, associated with the junctional plaque: Evidence for a subfamily of closely related proteins. J. Cell Sci. 1996, 109, 2767–2778. [Google Scholar] [CrossRef]
- Choi, H.J.; Weis, W.I. Structure of the armadillo repeat domain of plakophilin 1. J. Mol. Biol. 2005, 346, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.; Franke, W.W. Biochemical and immunological characterization of desmoplakins I and II, the major polypeptides of the desmosomal plaque. J. Mol. Biol. 1983, 163, 647–671. [Google Scholar] [CrossRef]
- Smith, E.A.; Fuchs, E. Defining the interactions between intermediate filaments and desmosomes. J. Cell Biol. 1998, 141, 1229–1241. [Google Scholar] [CrossRef]
- Liovic, M.; D’Alessandro, M.; Tomic-Canic, M.; Bolshakov, V.N.; Coats, S.E.; Lane, E.B. Severe keratin 5 and 14 mutations induce down-regulation of junction proteins in keratinocytes. Exp. Cell Res. 2009, 315, 2995–3003. [Google Scholar] [CrossRef]
- Amar, L.S.; Shabana, A.H.; Oboeuf, M.; Martin, N.; Forest, N. Involvement of desmoplakin phosphorylation in the regulation of desmosomes by protein kinase C, in HeLa cells. Cell Adhes. Commun. 1999, 7, 125–138. [Google Scholar] [CrossRef]
- Bornslaeger, E.A.; Corcoran, C.M.; Stappenbeck, T.S.; Green, K.J. Breaking the connection: Displacement of the desmosomal plaque protein desmoplakin from cell-cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J. Cell Biol. 1996, 134, 985–1001. [Google Scholar] [CrossRef]
- Lessard, J.L.; Wee, E.L.; Zimmerman, E.F. Presence of contractile proteins in mouse fetal palate prior to shelf elevation. Teratology 1974, 9, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, M.; Blumer, S.; Angelini, M.; Mitsiadis, T.A.; Katsaros, C. Mesenchymal Remodeling during Palatal Shelf Elevation Revealed by Extracellular Matrix and F-Actin Expression Patterns. Front. Physiol. 2016, 7, 392. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.; Reutter, H.; Mende, M.; de Assis, N.A.; Diaz-Lacava, A.; Herms, S.; Scheer, M.; Lauster, C.; Braumann, B.; Schmidt, G.; et al. Further evidence for the involvement of MYH9 in the etiology of non-syndromic cleft lip with or without cleft palate. Eur. J. Oral Sci. 2009, 117, 200–203. [Google Scholar] [CrossRef]
- Chiquet, B.T.; Hashmi, S.S.; Henry, R.; Burt, A.; Mulliken, J.B.; Stal, S.; Bray, M.; Blanton, S.H.; Hecht, J.T. Genomic screening identifies novel linkages and provides further evidence for a role of MYH9 in nonsyndromic cleft lip and palate. Eur. J. Hum. Genet. 2009, 17, 195–204. [Google Scholar] [CrossRef][Green Version]
- Moreno Uribe, L.M.; Fomina, T.; Munger, R.G.; Romitti, P.A.; Jenkins, M.M.; Gjessing, H.K.; Gjerdevik, M.; Christensen, K.; Wilcox, A.J.; Murray, J.C.; et al. A Population-Based Study of Effects of Genetic Loci on Orofacial Clefts. J. Dent. Res. 2017, 96, 1322–1329. [Google Scholar] [CrossRef]
- Liu, H.; Duncan, K.; Helverson, A.; Kumari, P.; Mumm, C.; Xiao, Y.; Carlson, J.C.; Darbellay, F.; Visel, A.; Leslie, E.; et al. Analysis of zebrafish periderm enhancers facilitates identification of a regulatory variant near human KRT8/18. eLife 2020, 9, e51325. [Google Scholar] [CrossRef]
- Lu, H.; Hesse, M.; Peters, B.; Magin, T.M. Type II keratins precede type I keratins during early embryonic development. Eur. J. Cell Biol. 2005, 84, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, W.; Nakanishi, H.; Miyoshi, J.; Mandai, K.; Ishizaki, H.; Tanaka, M.; Togawa, A.; Takahashi, K.; Nishioka, H.; Yoshida, H.; et al. Afadin: A key molecule essential for structural organization of cell-cell junctions of polarized epithelia during embryogenesis. J. Cell Biol. 1999, 146, 1117–1132. [Google Scholar] [CrossRef]
- Lough, K.J.; Spitzer, D.C.; Bergman, A.J.; Wu, J.J.; Byrd, K.M.; Williams, S.E. Disruption of the nectin-afadin complex recapitulates features of the human cleft lip/palate syndrome CLPED1. Development 2020, 147, dev189241. [Google Scholar] [CrossRef]
- Camargo, M.; Rivera, D.; Moreno, L.; Lidral, A.C.; Harper, U.; Jones, M.; Solomon, B.D.; Roessler, E.; Vélez, J.I.; Martinez, A.F. GWAS reveals new recessive loci associated with non-syndromic facial clefting. Eur. J. Med. Genet. 2012, 55, 510–514. [Google Scholar] [CrossRef]
- El-Sibai, M.; El Hajj, J.; Al Haddad, M.; El Baba, N.; Al Saneh, M.; Daoud Khatoun, W.; Helaers, R.; Vikkula, M.; El Atat, O.; Sabbagh, J.; et al. Dysregulation of Rho GTPases in orofacial cleft patients-derived primary cells leads to impaired cell migration, a potential cause of cleft/lip palate development. Cells Dev. 2021, 165, 203656. [Google Scholar] [CrossRef] [PubMed]
- Hoebel, A.K.; Drichel, D.; van de Vorst, M.; Böhmer, A.C.; Sivalingam, S.; Ishorst, N.; Klamt, J.; Gölz, L.; Alblas, M.; Maaser, A.; et al. Candidate Genes for Nonsyndromic Cleft Palate Detected by Exome Sequencing. J. Dent. Res. 2017, 96, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Mansilla, M.A.; Biggs, L.C.; Schuette, K.; Bullard, S.; Cooper, M.; Dunnwald, M.; Lidral, A.C.; Marazita, M.L.; Beaty, T.H.; et al. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Busch, T.; Eliason, S.; Anand, D.; Bullard, S.; Gowans, L.J.J.; Nidey, N.; Petrin, A.; Augustine-Akpan, E.A.; Saadi, I.; et al. Exome sequencing provides additional evidence for the involvement of ARHGAP29 in Mendelian orofacial clefting and extends the phenotypic spectrum to isolated cleft palate. Birth Defects Res. 2017, 109, 27–37. [Google Scholar] [CrossRef]
- Liu, H.; Leslie, E.J.; Carlson, J.C.; Beaty, T.H.; Marazita, M.L.; Lidral, A.C.; Cornell, R.A. Identification of common non-coding variants at 1p22 that are functional for non-syndromic orofacial clefting. Nat. Commun. 2017, 8, 14759. [Google Scholar] [CrossRef]
- Savastano, C.P.; Brito, L.A.; Faria, Á.C.; Setó-Salvia, N.; Peskett, E.; Musso, C.M.; Alvizi, L.; Ezquina, S.A.; James, C. Impact of rare variants in ARHGAP29 to the etiology of oral clefts: Role of loss-of-function vs. missense variants. Clin. Genet. 2017, 91, 683–689. [Google Scholar] [CrossRef]
- Paul, B.J.; Palmer, K.; Sharp, J.C.; Pratt, C.H.; Murray, S.A.; Dunnwald, M. ARHGAP29 Mutation Is Associated with Abnormal Oral Epithelial Adhesions. J. Dent. Res. 2017, 96, 1298–1305. [Google Scholar] [CrossRef]
- Tucci, V.; Kleefstra, T.; Hardy, A.; Heise, I.; Maggi, S.; Willemsen, M.H.; Hilton, H.; Esapa, C.; Simon, M.; Buenavista, M.T.; et al. Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Investig. 2014, 124, 1468–1482. [Google Scholar] [CrossRef]
- He, F.; Xiong, W.; Wang, Y.; Li, L.; Liu, C.; Yamagami, T.; Taketo, M.M.; Zhou, C.; Chen, Y. Epithelial Wnt/beta-catenin signaling regulates palatal shelf fusion through regulation of Tgfbeta3 expression. Dev. Biol. 2011, 350, 511–519. [Google Scholar] [CrossRef][Green Version]
- Ghoumid, J.; Stichelbout, M.; Jourdain, A.S.; Frenois, F.; Lejeune-Dumoulin, S.; Alex-Cordier, M.P.; Lebrun, M.; Guerreschi, P.; Duquennoy-Martinot, V.; Vinchon, M.; et al. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet. Med. 2017, 19, 1013–1021. [Google Scholar] [CrossRef]
- Cox, L.L.; Cox, T.C.; Moreno Uribe, L.M.; Zhu, Y.; Richter, C.T.; Nidey, N.; Standley, J.M.; Deng, M.; Blue, E.; Chong, J.X.; et al. Mutations in the Epithelial Cadherin-p120-Catenin Complex Cause Mendelian Non-Syndromic Cleft Lip with or without Cleft Palate. Am. J. Hum. Genet. 2018, 102, 1143–1157. [Google Scholar] [CrossRef]
- Alharatani, R.; Ververi, A.; Beleza-Meireles, A.; Ji, W.; Mis, E.; Patterson, Q.T.; Griffin, J.N.; Bhujel, N.; Chang, C.A.; Dixit, A.; et al. Novel truncating mutations in CTNND1 cause a dominant craniofacial and cardiac syndrome. Hum. Mol. Genet. 2020, 29, 1900–1921. [Google Scholar] [CrossRef]
- Brito, L.A.; Yamamoto, G.L.; Melo, S.; Malcher, C.; Ferreira, S.G.; Figueiredo, J.; Alvizi, L.; Kobayashi, G.S.; Naslavsky, M.S.; Alonso, N.; et al. Rare Variants in the Epithelial Cadherin Gene Underlying the Genetic Etiology of Nonsyndromic Cleft Lip with or without Cleft Palate. Hum. Mutat. 2015, 36, 1029–1033. [Google Scholar] [CrossRef]
- Bureau, A.; Parker, M.M.; Ruczinski, I.; Taub, M.A.; Marazita, M.L.; Murray, J.C.; Mangold, E.; Noethen, M.M.; Ludwig, K.U.; Hetmanski, J.B.; et al. Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics 2014, 197, 1039–1044. [Google Scholar] [CrossRef]
- Du, S.; Yang, Y.; Yi, P.; Luo, J.; Liu, T.; Chen, R.; Liu, C.J.; Ma, T.; Li, Y.; Wang, C.; et al. A Novel CDH1 Mutation Causing Reduced E-Cadherin Dimerization Is Associated with Nonsyndromic Cleft Lip With or Without Cleft Palate. Genet. Test. Mol. Biomark. 2019, 23, 759–765. [Google Scholar] [CrossRef]
- Vogelaar, I.P.; Figueiredo, J.; van Rooij, I.A.; Simões-Correia, J.; van der Post, R.S.; Melo, S.; Seruca, R.; Carels, C.E.; Ligtenberg, M.J.; Hoogerbrugge, N. Identification of germline mutations in the cancer predisposing gene CDH1 in patients with orofacial clefts. Hum. Mol. Genet. 2013, 22, 919–926. [Google Scholar] [CrossRef]
- Selvanathan, A.; Nixon, C.Y.; Zhu, Y.; Scietti, L.; Forneris, F.; Uribe, L.M.M.; Lidral, A.C.; Jezewski, P.A.; Mulliken, J.B.; Murray, J.C.; et al. CDH1 Mutation Distribution and Type Suggests Genetic Differences between the Etiology of Orofacial Clefting and Gastric Cancer. Genes 2020, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Higuera, I.; Manchado, E.; Dubus, P.; Cañamero, M.; Méndez, J.; Moreno, S.; Malumbres, M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008, 10, 802–811. [Google Scholar] [CrossRef]
- Shao, R.; Liu, J.; Yan, G.; Zhang, J.; Han, Y.; Guo, J.; Xu, Z.; Yuan, Z.; Liu, J.; Malumbres, M.; et al. Cdh1 regulates craniofacial development via APC-dependent ubiquitination and activation of Goosecoid. Cell Res. 2016, 26, 699–712. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tunggal, J.A.; Helfrich, I.; Schmitz, A.; Schwarz, H.; Günzel, D.; Fromm, M.; Kemler, R.; Krieg, T.; Niessen, C.M. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005, 24, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Hu, D.; Bustos, T.; Zlotogora, J.; Richieri-Costa, A.; Helms, J.A.; Spritz, R.A. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat. Genet. 2000, 25, 427–430. [Google Scholar] [CrossRef]
- Barron, M.J.; Brookes, S.J.; Draper, C.E.; Garrod, D.; Kirkham, J.; Shore, R.C.; Dixon, M.J. The cell adhesion molecule nectin-1 is critical for normal enamel formation in mice. Hum. Mol. Genet. 2008, 17, 3509–3520. [Google Scholar] [CrossRef][Green Version]
- Yoshida, T.; Iwata, T.; Takai, Y.; Birchmeier, W.; Yamato, M.; Okano, T. Afadin requirement for cytokine expressions in keratinocytes during chemically induced inflammation in mice. Genes Cells 2014, 19, 842–852. [Google Scholar] [CrossRef]
- Brancati, F.; Fortugno, P.; Bottillo, I.; Lopez, M.; Josselin, E.; Boudghene-Stambouli, O.; Agolini, E.; Bernardini, L.; Bellacchio, E.; Iannicelli, M.; et al. Mutations in PVRL4, encoding cell adhesion molecule nectin-4, cause ectodermal dysplasia-syndactyly syndrome. Am. J. Hum. Genet. 2010, 87, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Saadi, I.; Alkuraya, F.S.; Gisselbrecht, S.S.; Goessling, W.; Cavallesco, R.; Turbe-Doan, A.; Petrin, A.L.; Harris, J.; Siddiqui, U.; Grix, A.W., Jr.; et al. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am. J. Hum. Genet. 2011, 89, 44–55. [Google Scholar] [CrossRef]
- Hall, E.G.; Wenger, L.W.; Wilson, N.R.; Undurty-Akella, S.S.; Standley, J.; Augustine-Akpan, E.A.; Kousa, Y.A.; Acevedo, D.S.; Goering, J.P.; Pitstick, L.; et al. SPECC1L regulates palate development downstream of IRF6. Hum. Mol. Genet. 2020, 29, 845–858. [Google Scholar] [CrossRef]
- Goering, J.P.; Wenger, L.W.; Stetsiv, M.; Moedritzer, M.; Hall, E.G.; Isai, D.G.; Jack, B.M.; Umar, Z.; Rickabaugh, M.K.; Czirok, A.; et al. In-frame deletion of SPECC1L microtubule association domain results in gain-of-function phenotypes affecting embryonic tissue movement and fusion events. Hum. Mol. Genet. 2021, 31, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.; Kumari, P.; Sepulveda Rincon, L.; Gu, R.; Ji, Y.; Kumar, S.; Zhou, C.J. Wnt signaling in orofacial clefts: Crosstalk, pathogenesis and models. Dis. Model. Mech. 2019, 12, dmm037051. [Google Scholar] [CrossRef]
- Nopoulos, P.; Richman, L.; Andreasen, N.C.; Murray, J.C.; Schutte, B. Abnormal brain structure in adults with Van der Woude syndrome. Clin. Genet. 2007, 71, 511–517. [Google Scholar] [CrossRef]
- Kousa, Y.A.; Zhu, H.; Fakhouri, W.D.; Lei, Y.; Kinoshita, A.; Roushangar, R.R.; Patel, N.K.; Agopian, A.J.; Yang, W.; Leslie, E.J.; et al. The TFAP2A-IRF6-GRHL3 genetic pathway is conserved in neurulation. Hum. Mol. Genet. 2019, 28, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar]
- Tessier, P. Anatomical classification facial, cranio-facial and latero-facial clefts. J. Maxillofac. Surg. 1976, 4, 69–92. [Google Scholar] [CrossRef]
- Shah, J.; Guerrera, D.; Vasileva, E.; Sluysmans, S.; Bertels, E.; Citi, S. PLEKHA7: Cytoskeletal adaptor protein at center stage in junctional organization and signaling. Int. J. Biochem. Cell. Biol. 2016, 75, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Schutte, B.C.; Richardson, R.J.; Bjork, B.C.; Knight, A.S.; Watanabe, Y.; Howard, E.; de Lima, R.L.; Daack-Hirsch, S.; Sander, A.; et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002, 32, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Zucchero, T.M.; Cooper, M.E.; Maher, B.S.; Daack-Hirsch, S.; Nepomuceno, B.; Ribeiro, L.; Caprau, D.; Christensen, K.; Suzuki, Y.; Machida, J.; et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N. Engl. J. Med. 2004, 351, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, C.R.; Kinoshita, A.; Kondo, S.; Yang, B.; Sajan, S.; Trout, K.J.; Malik, M.I.; Dunnwald, M.; Goudy, S.L.; Lovett, M.; et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 2006, 38, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Antiguas, A.; DeMali, K.A.; Dunnwald, M. IRF6 Regulates the Delivery of E-Cadherin to the Plasma Membrane. J. Investig. Dermatol. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Biggs, L.C.; Naridze, R.; DeMali, K.A.; Lusche, D.F.; Kuhl, S.; Soll, D.R.; Schutte, B.C.; Dunnwald, M. Interferon Regulatory Factor 6 regulates keratinocyte migration. J. Cell Sci. 2014, 127, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Kaibuchi, K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2001, 2, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Tagashira, T.; Fukuda, T.; Miyata, M.; Nakamura, K.; Fujita, H.; Takai, Y.; Hirata, K.I.; Rikitake, Y. Afadin Facilitates Vascular Endothelial Growth Factor-Induced Network Formation and Migration of Vascular Endothelial Cells by Inactivating Rho-Associated Kinase Through ArhGAP29. Arter. Thromb. Vasc. Biol. 2018, 38, 1159–1169. [Google Scholar] [CrossRef] [PubMed]






| Pharyngeal Arch | Ectoderm | Endoderm | Mesoderm | Neuroectoderm | ||||
| Pharyngeal cleft | Pharyngeal pouch | Skeletal | Visceral | Arterial | Muscular | Motor nervous | Sensory nervous | |
| First (mandibular) | External acoustic meatus, helical crus, tragus (anterior 3 hillocks of His) | Auditory tube, tympanic membrane * | Mandible (Meckel’s cartilage), maxilla ƒ, palatine bone ƒ, malleus, incus, teeth | Body of tongue (anterior 2/3) | External carotid, maxillary | Muscles of mastication, tensor tympani, tensor veli palatini ^, mylohyoid, anterior belly of digastric | CN V, maxillary division V3 | CN V, lingual nerve |
| Second (hyoid) | Helix, antihelix, antitragus, lobule (posterior 3 hillocks of His) | Tonsillar fossa | Stapes, styloid process, superior hyoid body | Midtongue, thyroid, tonsil | Stapedial | Muscles of facial expression, stapedius, hyoid, posterior belly of digastric | CN VII | CN VII, chorda tympani (taste) |
| Third | ---- | Inferior parathyroid, thymus | Inferior hyoid body, great cornu hyoid | Root of tongue (posterior 1/3), epiglottis, thymus, carotid body | Internal carotid | Stylopharyngeus | CN IX | CN IX |
| Fourth | ---- | Superior parathyroid | Thyroid and laryngeal cartilages | Epiglottis, superior parathyroid | Aorta (left), subclavius (right) | Pharyngeal constrictors, levator veli palatini ^, palatoglossus ^, palatopharyngeus ^ | CN X, superior laryngeal | Auricular nerve to external acoustic meatus |
| Fifth | ---- | ---- | ---- | ---- | ---- | ---- | ---- | ---- |
| Sixth | ---- | Telopharyngeal body, parafollicular (“C”) cells | Cricoid, aretynoid, corniculate cartilages | Larynx | Pulmonary arteries, ductus arteriosus | Cricothyroid, laryngeal muscles, pharyngeal constrictors | CN X, inferior laryngeal | CN X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antiguas, A.; Paul, B.J.; Dunnwald, M. To Stick or Not to Stick: Adhesions in Orofacial Clefts. Biology 2022, 11, 153. https://doi.org/10.3390/biology11020153
Antiguas A, Paul BJ, Dunnwald M. To Stick or Not to Stick: Adhesions in Orofacial Clefts. Biology. 2022; 11(2):153. https://doi.org/10.3390/biology11020153
Chicago/Turabian StyleAntiguas, Angelo, Brian J. Paul, and Martine Dunnwald. 2022. "To Stick or Not to Stick: Adhesions in Orofacial Clefts" Biology 11, no. 2: 153. https://doi.org/10.3390/biology11020153
APA StyleAntiguas, A., Paul, B. J., & Dunnwald, M. (2022). To Stick or Not to Stick: Adhesions in Orofacial Clefts. Biology, 11(2), 153. https://doi.org/10.3390/biology11020153