




Application of Multigene Panels Testing for Hereditary Cancer Syndromes

 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Group
2.2. DNA Isolation
2.3. Library Preparation and Sequencing
2.4. Variant Classification and Bioinformatics Analysis
3. Results
3.1. Breast Cancer
3.2. Ovarian Cancer
3.3. Healthy Individuals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duggan, C.; Trapani, D.; Ilbawi, A.M.; Fidorova, E.; Laversanne, M.; Curigliano, G.; Bray, F.; Anderson, O.B. National health system characteristics, breast cancer stage at diagnosis, and breast cancer mortality: A population-based analysis. Lancet Oncol. 2021, 22, 1632–1642. [Google Scholar] [CrossRef]
- Lu, K.H.; Wood, M.E.; Daniels, M.; Burke, C.; Ford, J.; Kauff, N.D.; Kohlmann, W.; Lindor, N.M.; Mulvey, T.M.; Robinson, L.; et al. American Society of Clinical Oncology Expert Statement: Collection and use of a cancer family history for oncology providers. J. Clin. Oncol. 2014, 32, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D., 2nd; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In Proceedings of the 2019 IEEE 33rd International Parallel and Distributed Processing Symposium, IPDPS, Rio de Janeiro, Brazil, 20–24 May 2019; Institute of Electrical and Electronics Engineers Inc.: Piscataway, NJ, USA, 2019; pp. 314–324. [Google Scholar]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. Guideline Development Group, American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar]
- Robson, M.E.; Bradbury, A.R.; Arun, B.; Domchek, S.M.; Ford, J.M.; Hampel, H.L.; Lipkin, S.M.; Syngal, S.; Wollins, D.S.; Lindor, N.M. American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J. Clin. Oncol. 2015, 33, 3660–3667. [Google Scholar] [CrossRef]
- Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=2&id=1503 (accessed on 7 September 2022).
- Dos Santos, E.S.; Caputo, S.M.; Castera, L.; Gendrot, M.; Briaux, A.; Breault, M.; Krieger, S.; Rogan, P.K.; Mucaki, E.J.; Burke, L.J.; et al. Assessment of the functional impact of germline BRCA1/2 variants located in non-coding regions in families with breast and/or ovarian cancer predisposition. Breast Cancer Res. Treat. 2018, 168, 311–325. [Google Scholar] [CrossRef]
- Montalban, G.; Bonache, S.; Moles-Fernández, A.; Gisbert-Beamud, A.; Tenés, A.; Bach, V.; Carrasco, E.; López-Fernández, A.; Stjepanovic, N.; Balmaña, J.; et al. Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J. Med. Genet. 2019, 56, 63–74. [Google Scholar] [CrossRef]
- Dinicola, S.; Unfer, V.; Facchinetti, F.; Soulage, C.O.; Greene, N.D.; Bizzarri, M.; Laganà, A.S.; Chan, S.Y.; Bevilacqua, A.; Pkhaladze, L.; et al. Inositols: From Established Knowledge to Novel Approaches. Int. J. Mol. Sci. 2021, 22, 10575. [Google Scholar] [CrossRef]
- Brovkina, O.I.; Shigapova, L.; Chudakova, D.A.; Gordiev, M.G.; Enikeev, R.F.; Druzhkov, M.O.; Khodyrev, D.S.; Shagimardanova, E.I.; Nikitin, A.G.; Gusev, O.A. The Ethnic-Specific Spectrum of Germline Nucleotide Variants in DNA Damage Response and Repair Genes in Hereditary Breast and Ovarian Cancer Patients of Tatar Descent. Front. Oncol. 2018, 8, 421. [Google Scholar] [CrossRef] [PubMed]
- Broadinstitute/Picard: A Set of Command Line Tools (in Java) for Manipulating High-Throughput Sequencing (HTS) Data and Formats Such as SAM/BAM/CRAM and VCF. 2019. Available online: https://github.com/broadinstitute/picard (accessed on 10 May 2022).
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Kurian, A.W.; Li, Y.; Hamilton, A.S.; Ward, K.C.; Hawley, S.T.; Morrow, M.; McLeod, M.C.; Jagsi, R.; Katz, S.J. Gaps in Incorporating Germline Genetic Testing into Treatment Decision-Making for Early-Stage Breast Cancer. J. Clin. Oncol. 2017, 35, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Quarello, P.; Mascarin, M.; Banna, G.L.; Zecca, M.; Cinieri, S.; Peccatori, F.A.; Ferrari, A. Cancer Predisposition Genes in Adolescents and Young Adults (AYAs): A Review Paper from the Italian AYA Working Group. Curr. Oncol. Rep. 2022, 24, 843–860. [Google Scholar] [CrossRef] [PubMed]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Girard, E.; Eon-Marchais, S.; Olaso, R.; Renault, A.L.; Damiola, F.; Dondon, M.G.; Barjhoux, L.; Goidin, D.; Meyer, V.; Le Gal, D.; et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int. J. Cancer 2019, 144, 1962–1974. [Google Scholar] [CrossRef]
- Kwong, A.; Shin, V.Y.; Ho, J.C.; Kang, E.; Nakamura, S.; Teo, S.H.; Lee, A.S.; Sng, J.H.; Ginsburg, O.M.; Kurian, A.W.; et al. Comprehensive spectrum of BRCA1 and BRCA2 deleterious mutations in breast cancer in Asian countries. J. Med. Genet. 2016, 53, 15–23. [Google Scholar] [CrossRef]
- Wen, W.X.; Allen, J.; Lai, K.N.; Mariapun, S.; Hasan, S.N.; Ng, P.S.; Lee, D.S.; Lee, S.Y.; Yoon, S.Y.; Lim, J.; et al. Inherited mutations in BRCA1 and BRCA2 in an unselected multiethnic cohort of Asian patients with breast cancer and healthy controls from Malaysia. J. Med. Genet. 2018, 55, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.M.; Zheng, Y.B.; Gao, Y.; Ding, X.W.; Sun, Y.; Huang, Y.; Lou, C.J.; Pan, Z.W.; Peng, G.; Wang, X.J. Comprehensive mutation detection of BRCA1/2 genes reveals large genomic rearrangements contribute to hereditary breast and ovarian cancer in Chinese women. BMC Cancer 2019, 19, 551. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, G.; Li, X.; Ren, C.; Wang, Y.; Li, K.; Mok, H.; Cao, L.; Wen, L.; Jia, M.; et al. Comparison of BRCA versus non-BRCA germline mutations and associated somatic mutation profiles in patients with unselected breast cancer. Aging 2020, 12, 3140–3155. [Google Scholar] [CrossRef] [PubMed]
- Moles-Fernández, A.; Domènech-Vivó, J.; Tenés, A.; Balmaña, J.; Diez, O.; Gutiérrez-Enríquez, S. Role of Splicing Regulatory Elements and In Silico Tools Usage in the Identification of Deep Intronic Splicing Variants in Hereditary Breast/Ovarian Cancer Genes. Cancers 2021, 13, 3341. [Google Scholar] [CrossRef] [PubMed]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef]
- Boonen, R.A.C.M.; Vreeswijk, M.P.G.; van Attikum, H. CHEK2 variants: Linking functional impact to cancer risk. Trends Cancer Open Access 2022, 8, 759–770. [Google Scholar] [CrossRef]
- Starita, L.M.; Islam, M.M.; Banerjee, T.; Adamovich, A.I.; Gullingsrud, J.; Fields, S.; Shendure, J.; Parvin, J.D. A Multiplex Homology-Directed DNA Repair Assay Reveals the Impact of More Than 1000 BRCA1 Missense Substitution Variants on Protein Function. Am. J. Hum. Genet. 2018, 103, 498–508. [Google Scholar] [CrossRef]
- Sokolova, T.N.; Sokolenko, A.; Ni, V.; Preobrazhenskaya, E.; Iyevleva, A.; Aleksakhina, S.; Bessonov, A.; Gorodnova, T.; Anisimova, E.; Savonevich, E.; et al. Frequency and spectrum of founder and non-founder BRCA1/2 mutations in a large series of Russian breast cancer and ovarian cancer patients. Ann. Oncol. 2020, 31, S253. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, Y.; An, L.; Xi, C.; Wang, K.; Hong, D.; Shi, Y.; Zang, A.; Su, S.; Li, W. XRCC2 Arg188His polymorphism and colorectal cancer risk: A meta-analysis. J. Int. Med. 2021, 49, 10. [Google Scholar] [CrossRef]
- Kluźniak, W.; Wokołorczyk, D.; Rusak, B.; Huzarski, T.; Gronwald, J.; Stempa, K.; Rudnicka, H.; Kashyap, A.; Dębniak, T.; Jakubowska, A.; et al. Inherited variants in XRCC2 and the risk of breast cancer. Breast Cancer Res. Treat. 2019, 178, 657–663. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Giri, N.; Leathwood, L.; Risch, M.O.; Carr, A.G.; Alter, B.P. Risk of cancer in heterozygous relatives of patients with Fanconi anemia. Genet. Med. 2022, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Berwick, M.; Satagopan, J.M.; Ben-Porat, L.; Carlson, A.; Mah, K.; Henry, R.; Diotti, R.; Milton, K.; Pujara, K.; Landers, T.; et al. Genetic Heterogeneity among Fanconi Anemia Heterozygotes and Risk of Cancer. Cancer Res. 2007, 67, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Everman, D.B.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E. GeneReviews®; University of Washington: Seattle, WA, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1401/ (accessed on 14 February 2002).
- Demuth, I.; Wlodarski, M.; Tipping, A.J.; Morgan, N.V.; de Winter, J.P.; Thiel, M.; Gräsl, S.; Schindler, D.; D’Andrea, A.D.; Altay, C.; et al. Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur. J. Hum. Genet. 2000, 8, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Owens, K.M.; Bajpai, P.; Desouki, M.M.; Srinivasasainagendra, V.; Tiwari, H.K.; Singh, K.K. Mitochondrial DNA Polymerase POLG1 Disease Mutations and Germline Variants Promote Tumorigenic Properties. PLoS ONE 2015, 10, e0139846. [Google Scholar] [CrossRef]
- Wu, J.; Wei, Y.; Pan, J.; Jin, S.; Gu, W.; Gan, H.; Zhu, Y.; Ye, D.W. Prevalence of comprehensive DNA damage repair gene germline mutations in Chinese prostate cancer patients. Int. J. Cancer 2021, 148, 673–681. [Google Scholar] [CrossRef]
- Tung, N.; Domchek, S.M.; Stadler, Z.; Nathanson, K.L.; Couch, F.; Garber, J.E.; Offit, K.; Robson, M.E. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat. Rev. Clin. Oncol. 2016, 13, 581–588. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Inclusion Criteria: | ||
| № | Criteria: | Age (years) |
| 1. | Ovarian cancer | all |
| 2. | Breast cancer: | |
| - Female | ≤50 | |
| - Triple-negative molecular subtype | ≤60 | |
| - Male | all | |
| 3. | Colorectal cancer | ≤50 |
| 4. | Gastric cancer | ≤50 |
| 5. | Pancreatic cancer | all |
| 6. | Uterine cancer | ≤50 |
| 7. | Primary multiple tumors (≥2 cancers of one individual): | |
| Synchronous | all | |
| Metachronous | age of first diagnosis ≤ 50 | |
| 8. | Healthy individuals with ≥1 relatives (parents, children, brothers, sisters) with cancer | all |
| 9. | Healthy individuals with more than 10 colon polyps | all |
| Exclusion criteria | ||
| № | Criteria: | Age (years) |
| 1. | Refusal to participate in the study | all |
| 2. | Poor genomic DNA quantity (≤20 ng/µL) | all |
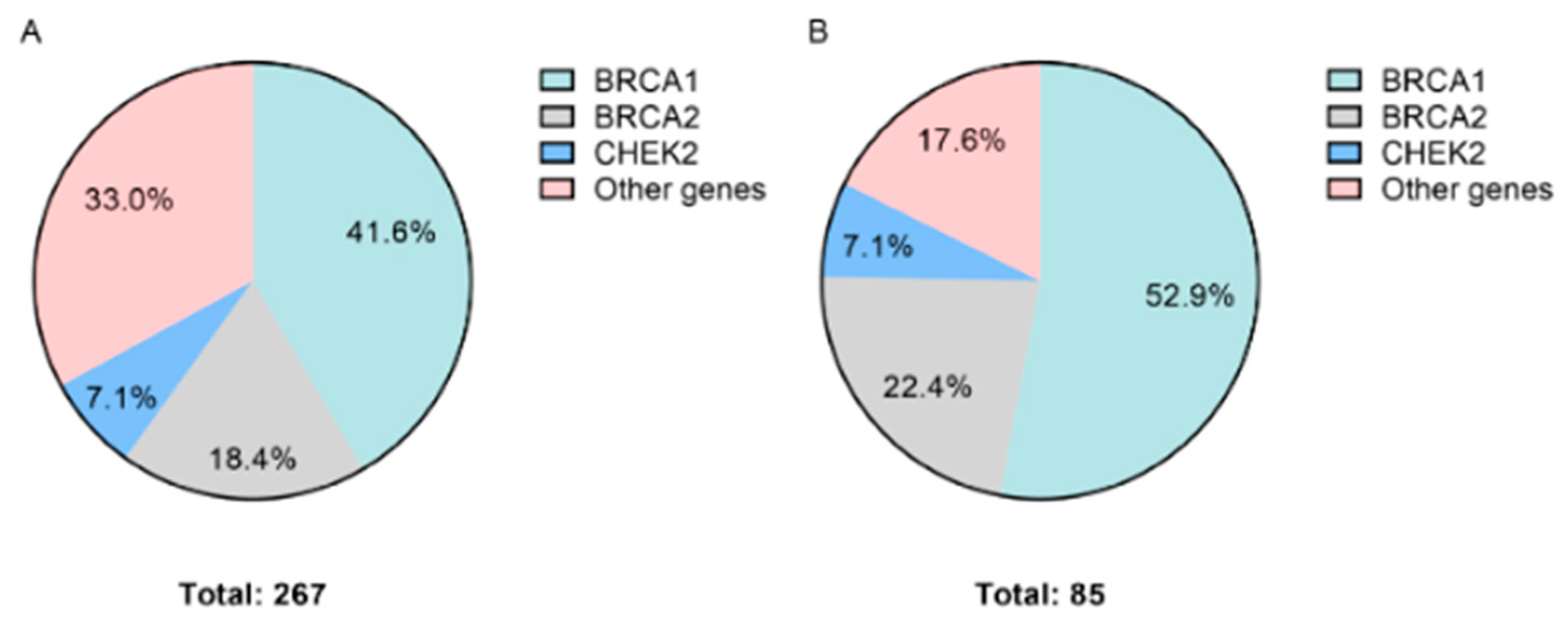
| Diagnosis | Total Cases | Cases with P/LP Variants (%) | Gene with P/LP Variants | Number of Variants (%) |
|---|---|---|---|---|
| Breast cancer | 800 | 267 (33.4) | ATM | 10 (3.7) |
| ATR | 2 (0.7) | |||
| BARD1 | 6 (2.2) | |||
| BLM | 3 (1.1) | |||
| BRCA1 | 111 (41.6) | |||
| BRCA2 | 49 (18.4) | |||
| BRIP1 | 1 (0.4) | |||
| CHEK2 | 19 (7.1) | |||
| CTNNA1 | 2 (0.7) | |||
| FANCA | 1 (0.4) | |||
| FANCC | 3 (1.1) | |||
| FANCD2 | 1 (0.4) | |||
| FANCI | 11 (4.1) | |||
| HOXB13 | 2 (0.7) | |||
| MLH1 | 1 (0.4) | |||
| MLH3 | 1 (0.4) | |||
| MSH6 | 3 (1.1) | |||
| MUTYH | 4 (1.5) | |||
| NBN | 2 (0.7) | |||
| NF1 | 1 (0.4) | |||
| NTHL1 | 2 (0.7) | |||
| PALB2 | 9 (3.4) | |||
| PMS1 | 2 (0.7) | |||
| POLE | 2 (0.7) | |||
| POLG | 10 (3.7) | |||
| RAD50 | 1 (0.4) | |||
| RAD51C | 2 (0.7) | |||
| SLX4 | 1 (0.4) | |||
| TP53 | 2 (0.7) | |||
| XRCC2 | 3 (1.1) | |||
| Gastric cancer | 13 | 2 (15.4) | CHEK2 | 1 (50.0) |
| XRCC2 | 1 (50.0) | |||
| Colon cancer | 27 | 6 (22.2) | BMPR1A | 1 (16.7) |
| MLH1 | 3 (50.0) | |||
| MSH2 | 2 (33.3) | |||
| Ovarian cancer | 209 | 85 (40.6) | ATM | 2 (2.4) |
| BABAM1 | 1 (1.2) | |||
| BLM | 1 (1.2) | |||
| BRCA1 | 45 (52.9) | |||
| BRCA2 | 19 (22.4) | |||
| BRIP1 | 1 (1.2) | |||
| CDH1 | 1 (1.2) | |||
| CHEK2 | 6 (7.1) | |||
| MSH6 | 1 (1.2) | |||
| MUTYH | 2 (2.4) | |||
| POLG | 2 (2.4) | |||
| RAD51C | 3 (3.5) | |||
| XRCC2 | 1 (1.2) | |||
| Pancreatic cancer | 10 | 4 (40.0) | ATM | 1 (25.0) |
| BLM | 2 (50.0) | |||
| MLH1 | 1 (25.0) | |||
| Healthy individuals (see Table 1) | 57 | 8 (14.0) | BLM | 2 (25.0) |
| BRCA1 | 1 (12.5) | |||
| CHEK2 | 1 (12.5) | |||
| MLH3 | 1 (12.5) | |||
| PMS1 | 1 (12.5) | |||
| POLD1 | 1 (12.5) | |||
| STK11 | 1 (12.5) | |||
| Multiple primary cancers | 8 | 6 (75.0) | BRCA2 | 1 (16.7) |
| CDH1 | 1 (16.7) | |||
| CHEK2 | 1 (16.7) | |||
| FANCG | 1 (16.7) | |||
| MSH2 | 1 (16.7) | |||
| TP53 | 1 (16.7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilyalov, A.; Nikolaev, S.; Shigapova, L.; Khatkov, I.; Danishevich, A.; Zhukova, L.; Smolin, S.; Titova, M.; Lisica, T.; Bodunova, N.; et al. Application of Multigene Panels Testing for Hereditary Cancer Syndromes. Biology 2022, 11, 1461. https://doi.org/10.3390/biology11101461
Bilyalov A, Nikolaev S, Shigapova L, Khatkov I, Danishevich A, Zhukova L, Smolin S, Titova M, Lisica T, Bodunova N, et al. Application of Multigene Panels Testing for Hereditary Cancer Syndromes. Biology. 2022; 11(10):1461. https://doi.org/10.3390/biology11101461
Chicago/Turabian StyleBilyalov, Airat, Sergey Nikolaev, Leila Shigapova, Igor Khatkov, Anastasia Danishevich, Ludmila Zhukova, Sergei Smolin, Marina Titova, Tatyana Lisica, Natalia Bodunova, and et al. 2022. "Application of Multigene Panels Testing for Hereditary Cancer Syndromes" Biology 11, no. 10: 1461. https://doi.org/10.3390/biology11101461
APA StyleBilyalov, A., Nikolaev, S., Shigapova, L., Khatkov, I., Danishevich, A., Zhukova, L., Smolin, S., Titova, M., Lisica, T., Bodunova, N., Shagimardanova, E., & Gusev, O. (2022). Application of Multigene Panels Testing for Hereditary Cancer Syndromes. Biology, 11(10), 1461. https://doi.org/10.3390/biology11101461