
Genome Analysis of Acinetobacter lwoffii Strains Isolated from Permafrost Soils Aged from 15 Thousand to 1.8 Million Years Revealed Their Close Relationships with Present-Day Environmental and Clinical Isolates

 , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains of A. lwoffii
2.2. Genome Sequencing and Assembly
2.3. Annotation and Analysis of the Genomes
2.4. Determination of the Ability of A. lwoffii Strains to Degrade Urea
3. Results and Discussion
3.1. General Genome Characteristics
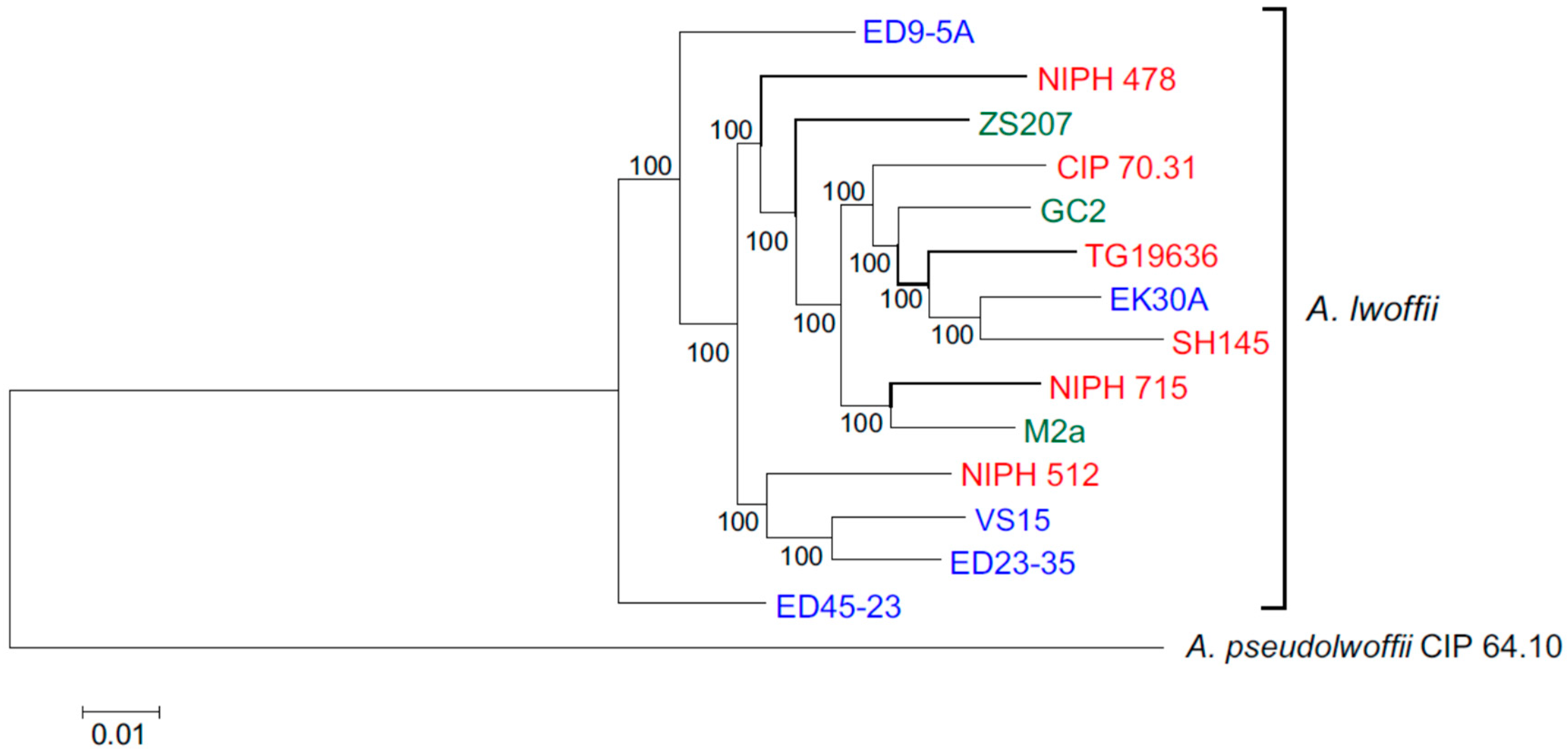
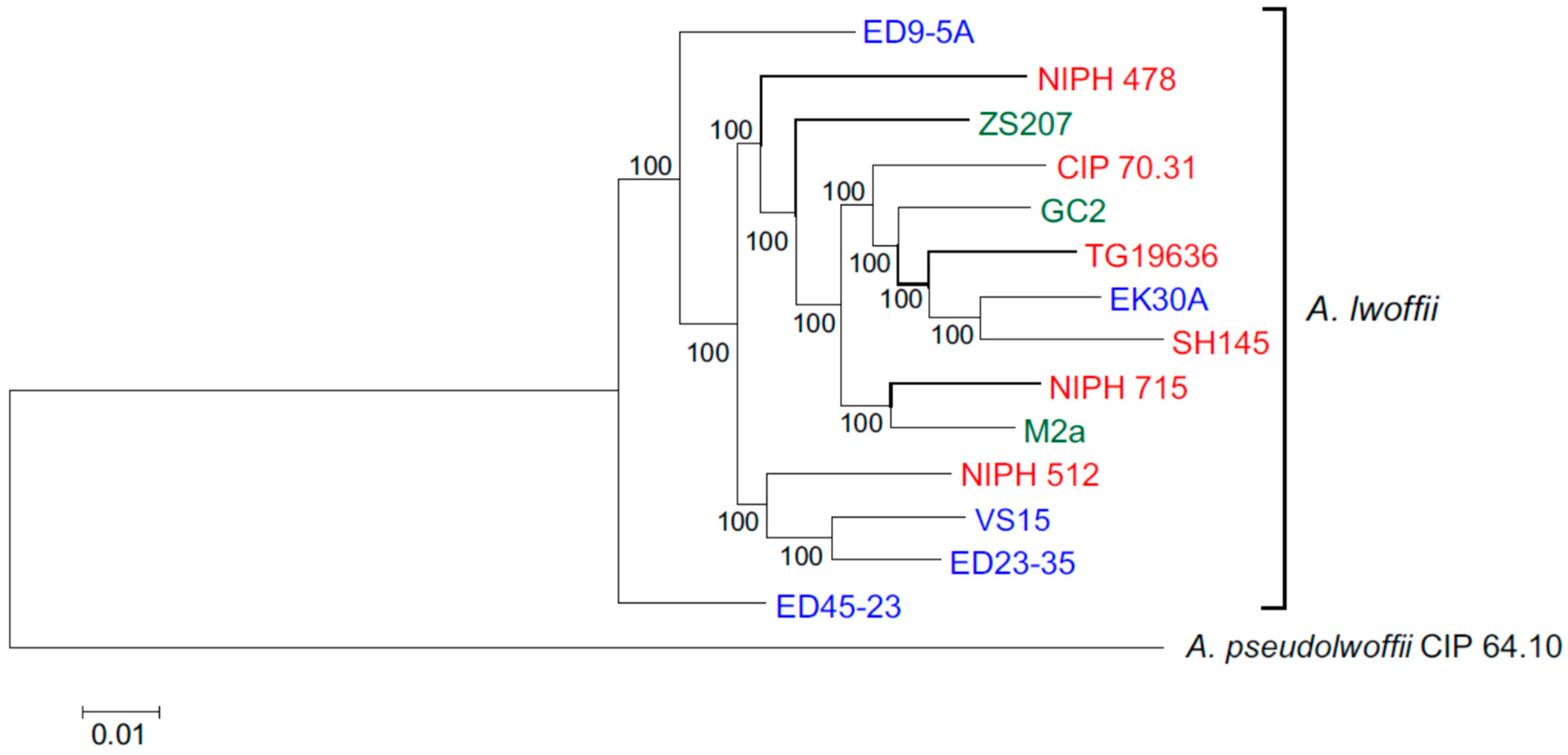
3.2. Core Genomes and Phylogenetic Relationships of Environmental and Clinical Strains of A. lwoffii
3.3. Antibiotics Resistance Genes
3.4. Mobile Elements
3.5. Plasmids
3.6. Integrated Phages
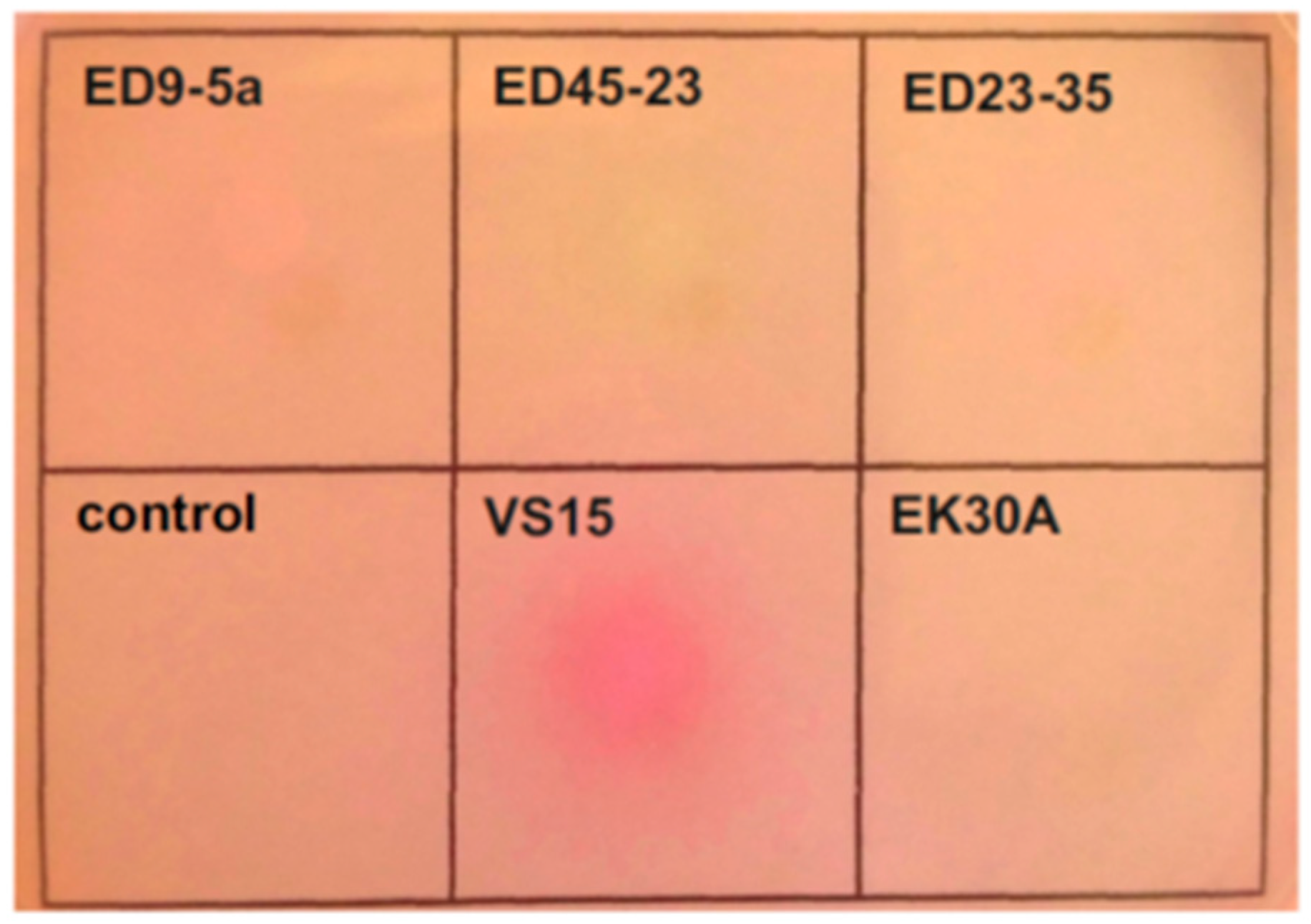
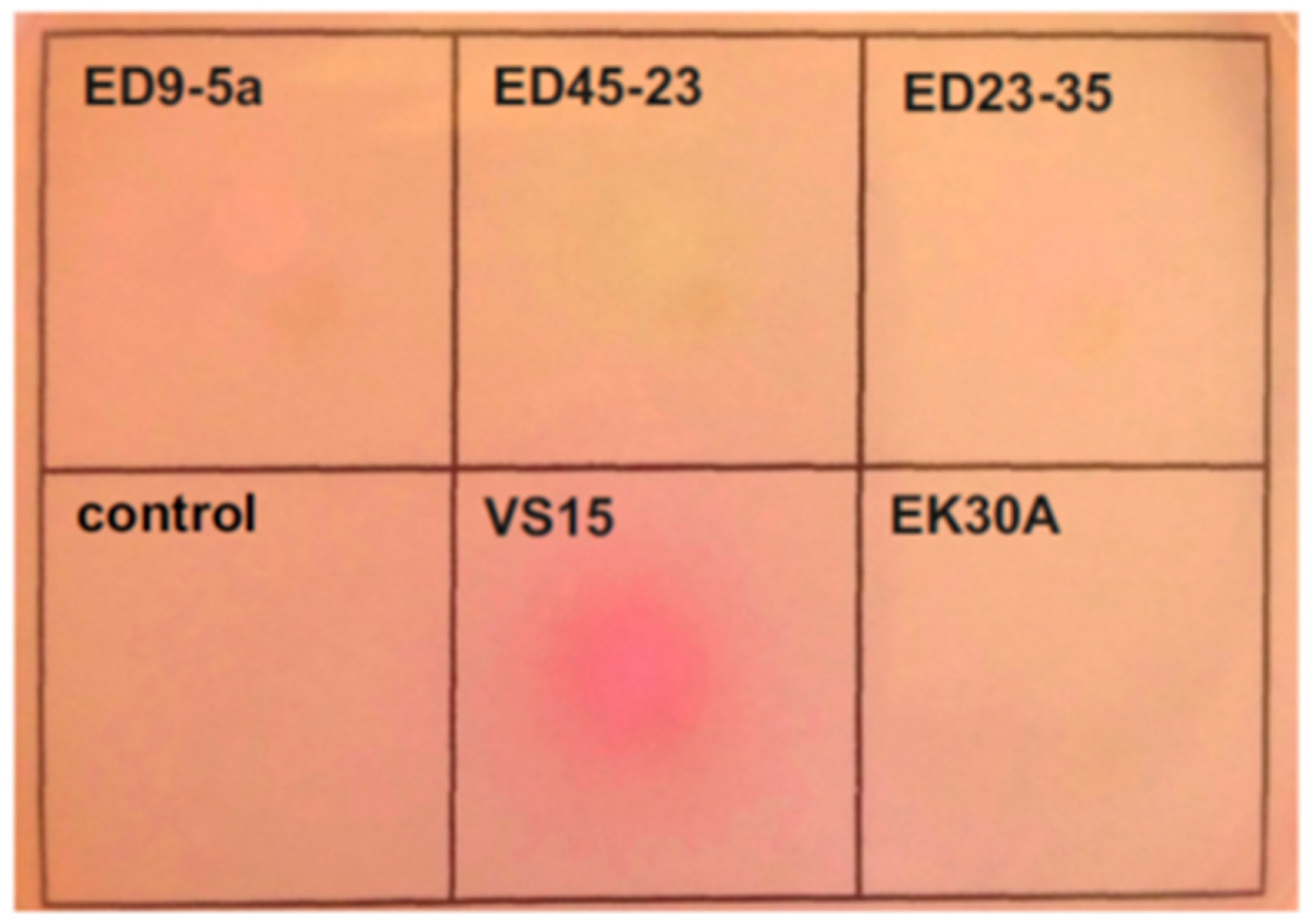
3.7. Urea Utilization Operon and its Functional Activity in Permafrost Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jansson, J.K.; Taş, N. The microbial ecology of permafrost. Nat. Rev. Microbiol. 2014, 12, 414–425. [Google Scholar] [CrossRef]
- Porfiriev, B.N.; Eliseev, D.O.; Streletskiy, D.A. Economic Assessment of Permafrost Degradation Effects on Road Infrastructure Sustainability under Climate Change in the Russian Arctic. Her. Russ. Acad. Sci. 2019, 89, 567–576. [Google Scholar] [CrossRef]
- Steven, B.; Léveillé, R.; Pollard, W.H.; Whyte, L.G. Microbial ecology and biodiversity in permafrost. Extremophiles 2006, 10, 259–267. [Google Scholar] [CrossRef]
- Zhang, D.C.; Brouchkov, A.; Griva, G.; Schinner, F.; Margesin, R. Isolation and characterization of bacteria from ancient siberian permafrost sediment. Biology 2013, 2, 85–106. [Google Scholar] [CrossRef]
- Steven, B.; Briggs, G.; McKay, C.P.; Pollard, W.H.; Greer, C.W.; Whyte, L.G. Characterization of the microbial diversity in a permafrost sample from the Canadian high Arctic using culture-dependent and culture-independent methods. FEMS Microbiol. Ecol. 2007, 59, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Vishnivetskaya, T.A.; Petrova, M.A.; Urbance, J.; Ponder, M.; Moyer, C.L.; Gilichinsky, D.A.; Tiedje, J.M. Bacterial community in ancient Siberian permafrost as characterized by culture and culture-independent methods. Astrobiology 2006, 6, 400–414. [Google Scholar] [CrossRef]
- Gilichinsky, D.A.; Vorobyova, E.A.; Erokhina, L.G.; Fyordorov-Davydov, D.G.; Chaikovskaya, N.R. Long-term preservation of microbial ecosystems in permafrost. Adv. Space Res. 1992, 12, 255–263. [Google Scholar] [CrossRef]
- Kashuba, E.; Dmitriev, A.A.; Kamal, S.M.; Melefors, O.; Griva, G.; Römling, U.; Ernberg, I.; Kashuba, V.; Brouchkov, A. Ancient permafrost staphylococci carry antibiotic resistance genes. Microb. Ecol. Health Dis. 2017, 28, 1345574. [Google Scholar] [CrossRef]
- Berendonk, T.U.; Manaia, C.M.; Fatta-kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.-N.; Kreuzinger, N.; et al. Tackling antibiotic resistance: The environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Perry, J.; Waglechner, N.; Wright, G. The prehistory of antibiotic resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025197. [Google Scholar] [CrossRef]
- Petrova, M.; Gorlenko, Z.; Mindlin, S. Molecular structure and translocation of a multiple antibiotic resistance region of a Psychrobacter psychrophilus permafrost strain. FEMS Microbiol. Lett. 2009, 296, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Petrova, M.; Gorlenko, Z.; Mindlin, S. Tn5045, a novel integron-containing antibiotic and chromate resistance transposon isolated from a permafrost bacterium. Res. Microbiol. 2011, 162, 337–345. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Kurakov, A.; Mindlin, S.; Beletsky, A.; Shcherbatova, N.; Rakitin, A.; Ermakova, A.; Mardanov, A.; Petrova, M. The ancient small mobilizable plasmid pALWED1.8 harboring a new variant of the non-cassette streptomycin/spectinomycin resistance gene aadA27. Plasmid 2016, 84–85, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Perron, G.; Whyte, L.; Turnbaugh, P.; Hanage, W.P.; Dantas, G.; Desai, M.M. Functional characterization of bacteria isolated from ancient arctic soil exposes diverse resistance mechanisms to modern antibiotics. PLoS ONE 2015, 10, e0069533. [Google Scholar]
- Afouda, P.; Dubourg, G.; Levasseur, A.; Fournier, P.E.; Delerce, J.; Mediannikov, O.; Diene, S.M.; Nahon, D.; Bourlès, D.; Rolain, J.M.; et al. Culturing Ancient Bacteria Carrying Resistance Genes from Permafrost and Comparative Genomics with Modern Isolates. Microorganisms 2020, 8, 1522. [Google Scholar] [CrossRef] [PubMed]
- Mindlin, S.; Minakhin, L.; Petrova, M.; Kholodii, G.; Minakhina, S.; Gorlenko, Z.; Nikiforov, V. Present-day mercury resistance transposons are common in bacteria preserved in permafrost grounds since the Upper Pleistocene. Res. Microbiol. 2005, 156, 994–1004. [Google Scholar] [CrossRef]
- Mindlin, S.; Petrenko, A.; Kurakov, A.; Beletsky, A.; Mardanov, A.; Petrova, M. Resistance of Permafrost and Modern Acinetobacter lwoffii Strains to Heavy Metals and Arsenic Revealed by Genome Analysis. BioMed. Res. Int. 2016, 2016, 3970831. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Park, W. Acinetobacter species as model microorganisms in environmental microbiology: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Hongsawat, P.; Vangnai, A.S. Biodegradation pathways of chloroanilines by Acinetobacter baylyi strain GFJ2. J. Hazard. Mater. 2011, 186, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Vaneechoutte, M.; Young, D.M.; Ornston, L.N.; De Baere, T.; Nemec, A.; Van Der Reijden, T.; Carr, E.; Tjernberg, I.; Dijkshoorn, L. Naturally transformable Acinetobacter sp. strain ADP1 belongs to the newly described species Acinetobacter baylyi. Appl. Environ. Microbiol. 2006, 72, 932–936. [Google Scholar] [CrossRef] [Green Version]
- Doughari, H.J.; Ndakidemi, P.A.; Human, I.S.; Benade, S. The Ecology, Biology and Pathogenesis of Acinetobacter spp.: An Overview. Microbes Environ. 2011, 26, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.C.; Hsueh, P.R.; Yang, P.C.; Luh, K.T. Clinical and microbiological characteristics of bacteremia caused by Acinetobacter lwoffii. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 501–505. [Google Scholar] [CrossRef]
- Regalado, N.; Martin, G.; Antony, S. Acinetobacter lwoffii: Bacteremia associated with acute gastroenteritis. Travel Med. Infect. Dis. 2009, 7, 316–317. [Google Scholar] [CrossRef]
- Turton, J.; Shah, J.; Ozongwu, C.; Pike, R. Incidence of Acinetobacter species other than A. baumannii among clinical isolates of Acinetobacter: Evidence for emerging species. J. Clin. Microbiol. 2010, 48, 1445–1449. [Google Scholar] [CrossRef] [Green Version]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Cerezales, M.; Xanthopoulou, K.; Wille, J.; Krut, O.; Seifert, H.; Gallego, L.; Higgins, P.G. Mobile Genetic Elements Harboring Antibiotic Resistance Determinants in Acinetobacter baumannii Isolates From Bolivia. Front. Microbiol. 2020, 11, 919. [Google Scholar] [CrossRef]
- López, M.; Rueda, A.; Florido, J.P.; Blasco, L.; Fernández-García, L.; Trastoy, R.; Fernández-Cuenca, F.; Martínez-Martínez, L.; Vila, J.; Pascual, A.; et al. Evolution of the Quorum network and the mobilome (plasmids and bacteriophages) in clinical strains of Acinetobacter baumannii during a decade. Sci. Rep. 2018, 8, 2523. [Google Scholar] [CrossRef] [Green Version]
- Walter, T.; Klim, J.; Jurkowski, M.; Gawor, J.; Köhling, I.; Słodownik, M.; Zielenkiewicz, U. Plasmidome of an environmental Acinetobacter lwoffii strain originating from a former gold and arsenic mine. Plasmid 2020, 110, 102505. [Google Scholar] [CrossRef]
- Veress, A.; Nagy, T.; Wilk, T.; Kömüves, J.; Olasz, F.; Kiss, J. Abundance of mobile genetic elements in an Acinetobacter lwoffii strain isolated from Transylvanian honey sample. Sci. Rep. 2020, 10, 2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acer, Ö.; Güven, K.; Poli, A.; Di Donato, P.; Leone, L.; Buono, L.; Güven, R.G.; Nicolaus, B.; Finore, I. Acinetobacter mesopotamicus sp. nov., petroleum-degrading bacterium, isolated from petroleum-contaminated soil in Diyarbakir, in the southeast of Turkey. Curr. Microbiol. 2020, 77, 3192–3200. [Google Scholar] [CrossRef]
- Nemec, A. Strain “Acinetobacter mesopotamicus” GC2 Does Not Represent a Novel Species, but Belongs to the Species Acinetobacter lwoffii as Revealed by Whole-Genome Sequence-Based Analysis. Curr. Microbiol. 2021, 78, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Mindlin, S.Z.; Petrova, M.A.; Gorlenko, Z.M.; Soina, V.S.; Khachikian, N.A.; Karaevskaya, E.A. Multidrug-resistant bacteria in permafrost: Isolation, biodiversity, phenotypic and genotypic analysis. In New Permafrost and Glacier Research, 1st ed.; Krugger, M.I., Stern, H.P., Eds.; Nova Science: Hauppauge, NY, USA, 2009; pp. 89–105. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterialgenome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Korf, I.; Gish, W. MPBLAST: Improved BLAST performance with multiplexed queries. Bioinformatics 2000, 16, 1052–1053. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar]
- Rodriguez, R.L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Christensen, W.B. Urea decomposition as a means of differentiating Proteus and paracolon cultures from each other and from Salmonella and Shigella types. J. Bacteriol. 1946, 52, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez, R.L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinitschke, S.; Denger, K.; Cook, A.M.; Smits, T.H.M. The DUF81 protein TauE in Cupriavidus necator H16, a sulfite exporter in the metabolism of C2 sulfonates. Microbiology 2007, 153, 3055–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.M.; Denger, K. Dissimilation of the C2 sulfonates. Arch. Microbiol. 2002, 179, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.; Dubnau, D. DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2004, 2, 241–249. [Google Scholar] [CrossRef]
- Domingues, S.; Rosário, N.; Cândido, Â.; Neto, D.; Nielsen, K.M.; Da Silva, G.J. Competence for Natural Transformation Is Common among Clinical Strains of Resistant Acinetobacter spp. Microorganisms 2019, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.L.; Neu, H.M.; Alamneh, Y.A.; Reddinger, R.M.; Jacobs, A.C.; Singh, S.; Abu-Taleb, R.; Michel, S.L.J.; Zurawski, D.V.; Merrell, D.S. Characterization of Acinetobacter baumannii copper resistance reveals a role in virulence. Front. Microbiol. 2020, 11, 16. [Google Scholar] [CrossRef]
- Figueiredo, S.; Poirel, L.; Seifert, H.; Mugnier, P.; Benhamou, D.; Nordmann, P. OXA-134, a naturally occurring carbapenem-hydrolyzing class D beta-lactamase from Acinetobacter lwoffii. Antimicrob. Agents Chemother. 2010, 54, 5372–5375. [Google Scholar] [CrossRef] [Green Version]
- Ermakova, A.Y.; Beletsky, A.V.; Mardanov, A.V.; Petrova, M.A.; Ravin, N.V.; Rakitin, A.L. A Novel Plasmid pALWVS1.4 from Acinetobacter lwoffii Strain VS15, Carrying the Chloramphenicol Resistance Gene. Microbiology 2020, 89, 637–640. [Google Scholar] [CrossRef]
- Siguier, P.; Gourbeyre, E.; Varani, A.; Ton-Hoang, B.; Chandler, M. Everyman’s Guide to Bacterial Insertion Sequences. Microbiol. Spectr. 2015, 3, MDNA3-0030-2014. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A. Cooperation is fleeting in the world of transposable elements. PLoS Comput. Biol. 2006, 2, e162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, H.; Dijkshoorn, L.; Gerner-Smidt, P.; Pelzer, N.; Tjernberg, I.; Vaneechoutte, M. Distribution of Acinetobacter species on human skin: Comparison of phenotypic and genotypic identification methods. J. Clin. Microbiol. 1997, 35, 2819–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.; Ackermann, H.W.; Kropinski, A.M.; Lavigne, R.; Sutton, J.M.; Reynolds, D.M. Comparative Analysis of 37 Acinetobacter Bacteriophages. Viruses 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Mobley, H.L.; Island, M.D.; Hausinger, R.P. Molecular biology of microbial ureases. Microbiol. Rev. 1995, 59, 451–480. [Google Scholar] [CrossRef] [PubMed]
- Eitinger, T.; Suhr, J.; Moore, L.; Smith, J.A. Secondary transporters for nickel and cobalt ions: Theme and variations. Biometals 2005, 18, 399–405. [Google Scholar] [CrossRef]
- Beckers, G.; Bendt, A.K.; Krämer, R.; Burkovski, A. Molecular identification of the urea uptake system and transcriptional analysis of urea transporter-and urease-encoding genes in Corynebacterium glutamicum. J. Bacteriol. 2004, 186, 7645–7652. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Strain | Isolation Depth (m) | Age of Permafrost (Thousand Years) | Resistance to Antibiotics | Resistance to Heavy Metals |
|---|---|---|---|---|
| ED23-35 | 4.5 | 20–40 | Sm, Sp | Hg, Cr, Co, Cd, Zn, Ni |
| ED45-23 | 2.9 | 20–40 | - | Hg, As, Cu |
| ED9-5A | 6.5 | 15–30 | - | Hg, As, Cr, Cd, Zn, Cu |
| VS15 | 34.0 | 20–40 | Amp, Cm, Sm, Sp | Co, Cd, Zn, Cu |
| EK30A | 47.9 | 1600–1800 | Amp, Sm, Sp | Cr, Co, Cd, Cu |
| Strain | Chromosome | Plasmids | ||
|---|---|---|---|---|
| Size (bp) | Name | Size (bp) | Resistance Genes 1 | |
| ED23-35 | 3,160,760 | pALWED 1.1 | 287,861 | tet(H), mer, chr, czc, nreB |
| pALWED 1.2 | 48,955 | |||
| pALWED 1.3 | 16,067 | chr | ||
| pALWED 1.4 | 14,116 | |||
| pALWED 1.5 | 6715 | |||
| pALWED 1.7 | 4861 | |||
| pALWED 1.8 | 4135 | aadA27 | ||
| ED45-23 | 3,260,192 | pALWED 2.1 | 196,557 | mer, ars, cop |
| pALWED 2.2 | 43,270 | |||
| pALWED 2.3 | 22,769 | |||
| pALWED 2.4 | 11,092 | |||
| pALWED 2.5 | 10,584 | |||
| pALWED 2.6 | 9201 | |||
| pALWED 2.7 | 8816 | |||
| pALWED 2.8 | 8120 | |||
| pALWED 2.9 | 6308 | |||
| ED9-5A | 3,231,133 | pALWED 3.6 | 185,756 | mer, ars, cop, czc |
| pALWED 3.1 | 138,030 | |||
| pALWED 3.5 | 16,567 | chr | ||
| pALWED 3.2 | 15,656 | |||
| pALWED 3.7 | 9958 | |||
| pALWED 3.3 | 8055 | |||
| VS15 | 3,260,140 | pALWVS 1.1 | 134,096 | cop, czc |
| pALWVS 1.2 | 15,780 | |||
| pALWVS 1.4 | 11,964 | |||
| pALWVS 1.3 | 10,985 | |||
| pALWVS 1.5 | 4677 | |||
| pALWED 1.8 | 4135 | aadA27 | ||
| EK30A | 3,183,510 | pALWEK 1.1 | 209,982 | |
| pALWEK 1.2 | 12,172 | |||
| pALWEK 1.12 | 11,382 | |||
| pALWEK 1.3 | 10,347 | |||
| pALWEK 1.10 | 9202 | |||
| pALWEK 1.13 | 8910 | |||
| pALWEK 1.4 | 8635 | cflA | ||
| pALWEK 1.5 | 8227 | chr | ||
| pALWEK 1.7 | 6691 | |||
| pALWEK 1.8 | 5324 | |||
| pALWEK 1.14 | 4760 | |||
| pALWEK 1.15 | 4677 | |||
| pALWED 1.8 | 4135 | aadA27 | ||
| pALWEK 1.17 | 4130 | |||
| pALWEK 1.16 | 2621 | |||
| Parameter | Strain | ||||
|---|---|---|---|---|---|
| ED23-35 | ED45-23 | ED9-5A | VS15 | EK30A | |
| Predicted genes | 3560 | 3644 | 3653 | 3496 | 3495 |
| Protein-coding genes | 3453 | 3537 | 3547 | 3392 | 3387 |
| Protein-coding genes with predicted function | 2362 (68,4%) | 2463 (69.6%) | 2543 (71.7%) | 2404 (70.9%) | 2405 (71.0%) |
| tRNA genes | 86 | 86 | 85 | 83 | 87 |
| G + C content (chromosome) | 43.22 | 43.20 | 43.21 | 43.26 | 43.01 |
| Strain | Source | Protein-Coding Genes | Genes in the Core Genome |
|---|---|---|---|
| Environmental group | 2224 | ||
| ED23-35 | Permafrost | 3453 | |
| ED45-23 | Permafrost | 3537 | |
| ED9-5A | Permafrost | 3547 | |
| VS15 | Permafrost | 3392 | |
| EK30A | Permafrost | 3387 | |
| ZS207 | Gold mine | 3230 | |
| M2a | Honey | 3533 | |
| GC2 | Petroleum-contaminated soil | 3267 | |
| Clinical group | 2266 | ||
| SH145 | Skin | 3134 | |
| NIPH 715 | Pus | 3314 | |
| CIP 70.31 | Gangrenous lesion | 3503 | |
| NIPH 478 | Ear swab | 3088 | |
| NIPH 512 | Unknown | 3237 | |
| TG19636 | Urine | 3412 | |
| Gene | Protein | Strain Group | |
|---|---|---|---|
| Clinical (6) | Environmental (8) | ||
| cat | Chloramphenicol acetyltransferase | 6 | 8 |
| blaOXA-134 | OXA-134 family class D β-lactamase | 6 | 8 |
| macAB, tolC | Drug efflux ABC-type transporter | 6 | 8 |
| sul2 | Dihydropteroate synthase | 3 1 | 0 |
| aph(3″)-Ib | Aminoglycoside 3′-phosphotransferase | 1 2 | 0 |
| aph(6)-I | Aminoglycoside-6-phosphotransferase | 1 2 | 0 |
| aadA27 | Streptomycin- spectinomycin 3″-adenylyltransferase | 0 | 3 |
| tet(H) | Tetracycline efflux MSF transporter | 0 | 1 |
| cflA | Drug efflux MSF transporter Bcr/CflA family | 0 | 1 |
| Strain | ED23-35 | ED45-23 | ED9-5A | VS15 | EK30A | GC2 | ZS207 1 | M2a 1 |
|---|---|---|---|---|---|---|---|---|
| Total number of copies of IS elements | 78 | 70 | 107 | 98 | 133 | 25 | 86 | 205 |
| Number of IS elements | 25 | 29 | 34 | 33 | 39 | 23 | 40 | 49 |
| Number of families of IS elements | 9 | 9 | 11 | 11 | 11 | 10 | 15 | 13 |
| Composite transposons | 2 | 4 | 0 | 3 | 4 | 0 | 0 | 2 |
| Assembly quality (number of scaffolds) 2 | C/P | C/P | C/P | C/P | C/P | 285 | C/P | 277 |
| Strain | SH145 | NIPH 715 | CIP 70.31 | NIPH 478 | NIPH 512 | TG19636 |
|---|---|---|---|---|---|---|
| Total number of copies of IS elements | 7 | 90 | 115 | 19 | 57 | 19 |
| Number of IS elements | 7 | 30 | 31 | 8 | 18 | 19 |
| Number of families of IS elements | 4 | 19 | 11 | 3 | 9 | 10 |
| Composite transposons | 0 | 0 | 0 | 0 | 0 | 0 |
| Assembly quality (number of scaffolds) | 76 | 26 | 12 | 9 | 12 | 245 |
| Strain | Phage | Length, (kb) | Position | Location | Similar Prophages (Coverage 1/Identity, %) |
|---|---|---|---|---|---|
| ED23-35 | ED23-35-3 | 34.1 | 1709101-1743234 | Chromosome | - |
| ED23-35-6 | 20 | 2260258-2280259 | Chromosome | EK30A-7 (30/93.4) | |
| ED45-23 | ED45-23-1 | 33.6 | 1543115-1576799 | Chromosome | - |
| ED45-23-4 | 43.7 | 2081979-2125746 | Chromosome | - | |
| ED45-23-7 | 50.4 | 3208722-3259176 | Chromosome | VS15-6 (45/91.4) EK30A-8 (30/94.3) | |
| ED9-5A | ED9-5A-1 | 37.8 | 994113-1031993 | Chromosome | - |
| ED9-5A-3 | 39 | 2063965-2103060 | Chromosome | - | |
| ED9-5A-6 | 18.9 | 972-19932 | pALWED 3.6 | ED23-35-6 (36/93.8) | |
| VS15 | VS15-6 | 49.6 | 2753607-2803257 | Chromosome | ED45-23-7 (46/91.4) |
| EK30A | EK30A-7 | 36.6 | 1504782-1541418 | Chromosome | - |
| EK30A-8 | 24.5 | 2890359-2914858 | Chromosome | ED45-23-7 (55/94.3) VS15-6 (38/95.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakitin, A.L.; Ermakova, A.Y.; Beletsky, A.V.; Petrova, M.; Mardanov, A.V.; Ravin, N.V. Genome Analysis of Acinetobacter lwoffii Strains Isolated from Permafrost Soils Aged from 15 Thousand to 1.8 Million Years Revealed Their Close Relationships with Present-Day Environmental and Clinical Isolates. Biology 2021, 10, 871. https://doi.org/10.3390/biology10090871
Rakitin AL, Ermakova AY, Beletsky AV, Petrova M, Mardanov AV, Ravin NV. Genome Analysis of Acinetobacter lwoffii Strains Isolated from Permafrost Soils Aged from 15 Thousand to 1.8 Million Years Revealed Their Close Relationships with Present-Day Environmental and Clinical Isolates. Biology. 2021; 10(9):871. https://doi.org/10.3390/biology10090871
Chicago/Turabian StyleRakitin, Andrey L., Alexandra Y. Ermakova, Alexey V. Beletsky, Mayya Petrova, Andrey V. Mardanov, and Nikolai V. Ravin. 2021. "Genome Analysis of Acinetobacter lwoffii Strains Isolated from Permafrost Soils Aged from 15 Thousand to 1.8 Million Years Revealed Their Close Relationships with Present-Day Environmental and Clinical Isolates" Biology 10, no. 9: 871. https://doi.org/10.3390/biology10090871
APA StyleRakitin, A. L., Ermakova, A. Y., Beletsky, A. V., Petrova, M., Mardanov, A. V., & Ravin, N. V. (2021). Genome Analysis of Acinetobacter lwoffii Strains Isolated from Permafrost Soils Aged from 15 Thousand to 1.8 Million Years Revealed Their Close Relationships with Present-Day Environmental and Clinical Isolates. Biology, 10(9), 871. https://doi.org/10.3390/biology10090871