
The Impact of Host Genotype, Intestinal Sites and Probiotics Supplementation on the Gut Microbiota Composition and Diversity in Sheep

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sampling
- Samples from 3 different sheep breeds, with each breed belonging to one group. Colon contents and back-fat samples were collected from SHS (n = 12), BHBS (n = 14), and SHBS (n = 12), which were raised concurrently in the same manage condition, with the same forage in the Breeding Sheep Farm of state-owned Bayan Farm (Inner Mongolia Autonomous Region, China) from 4-month to 8-month age. The final body weights (FBW) ± SD of SHS, BHBS, and SHBS were 40.38 ± 6.00 kg, 38.90 ± 5.37 kg, and 37.47 ± 5.77 kg, respectively.
- Samples from different intestinal sites. TS (female, 4-month-old, n = 6) were purchased from Ningxia livestock farm (Ningxia Hui Autonomous Region, China) and fed with commercial diets and raised in the same condition. After feeding 3 months, the intestinal content samples were collected from jejunum, cecum, colon, and feces from TS with 33.80 ± 4.20 kg (FBW ± SD), (Figure S1a).
- Samples from sheep with probiotics administration, along with feeding. TS (female, n = 7) were purchased from Ningxia livestock farm (Ningxia Hui Autonomous Region, China) and fed with freeze-dried bifidobacteria (1010 CFU/g, 0.03% of basal diets), which was mixed into commercial diets. Sampling feces at T1 (4-month-old, 24.80 ± 1.22 kg, nonfeeding bifidobacteria), T2 (5-month-old, 30.90 ± 1.42 kg, feeding bifidobacteria 30 days), T3 (6-month-old, 36.68 ± 1.99 kg, feeding bifidobacteria 60 days), and T4 (7-month-old, 40.11 ± 2.14 kg, after stop feeding bifidobacteria 30 days). Due to the unexpected death of a sheep, there were 6 samples of T4 group (Figure S1b).
2.2. DNA Isolation and 16S rRNA Sequencing
2.3. RNA Extraction and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.4. Processing of 16S rRNA Sequencing Data
2.5. Statistical Analysis
3. Results
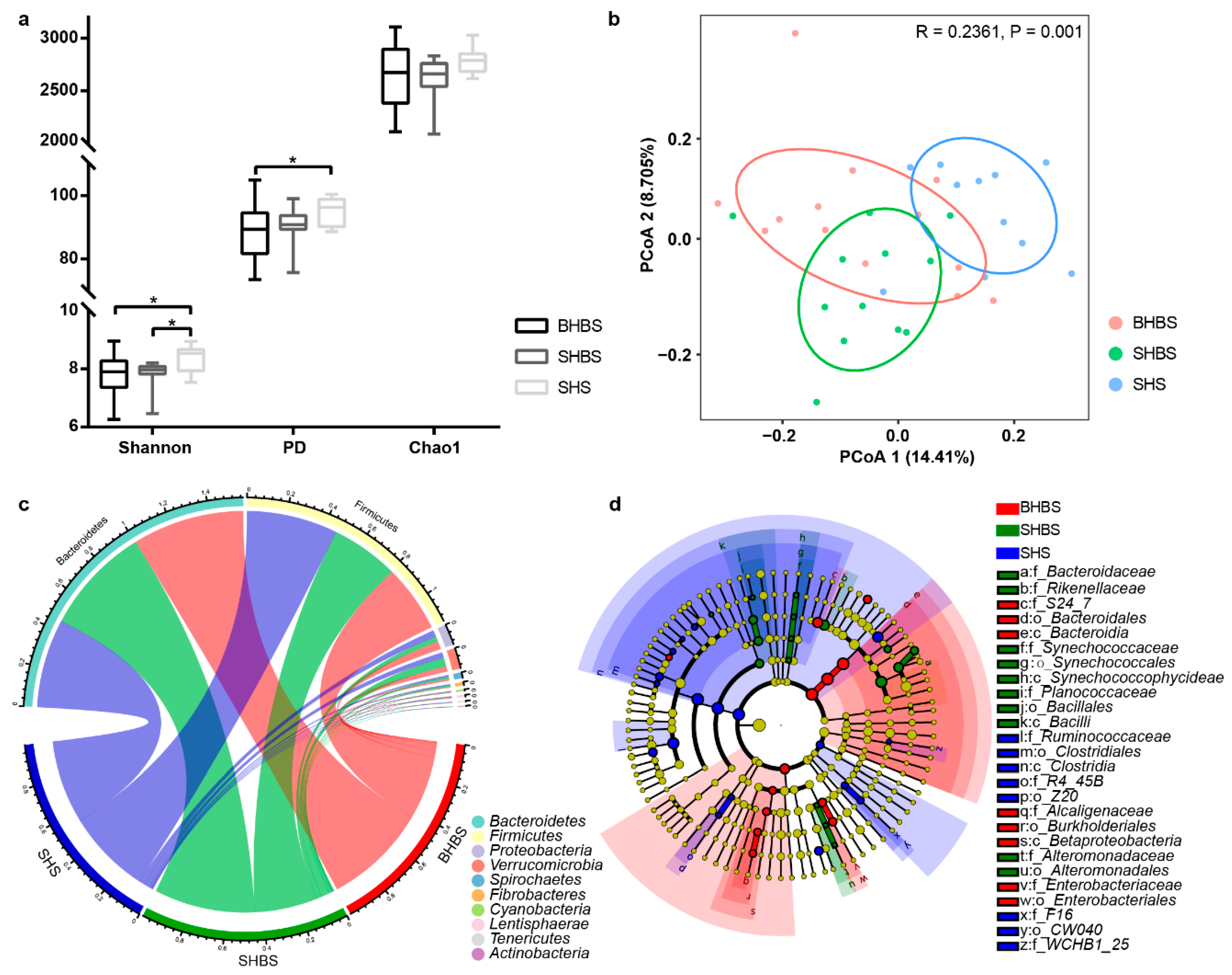
3.1. Bacterial Community Variation of BHBS, SHBS, and SHS in the Same Habitat
3.1.1. Diversity and Taxonomic Analysis
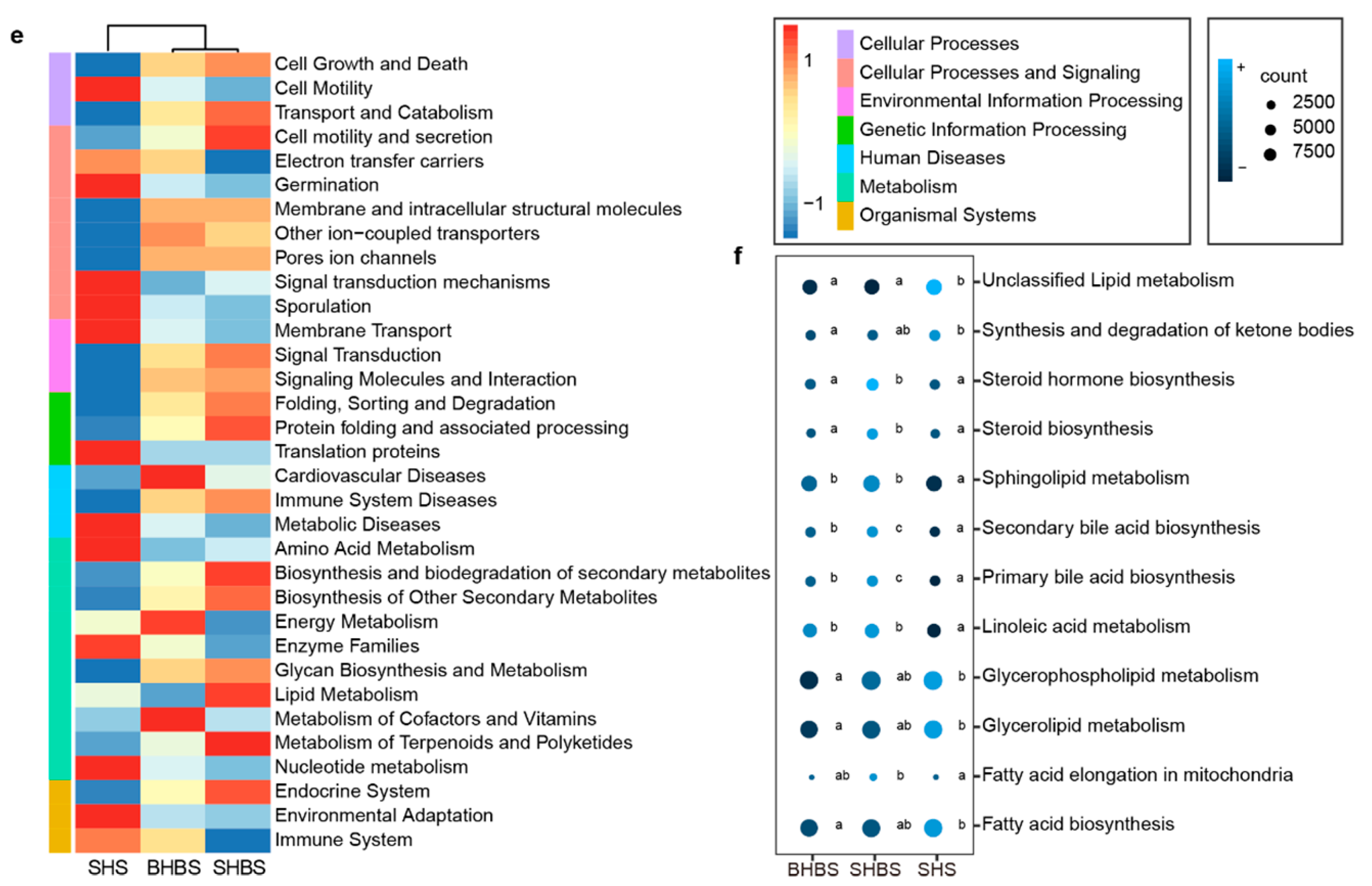
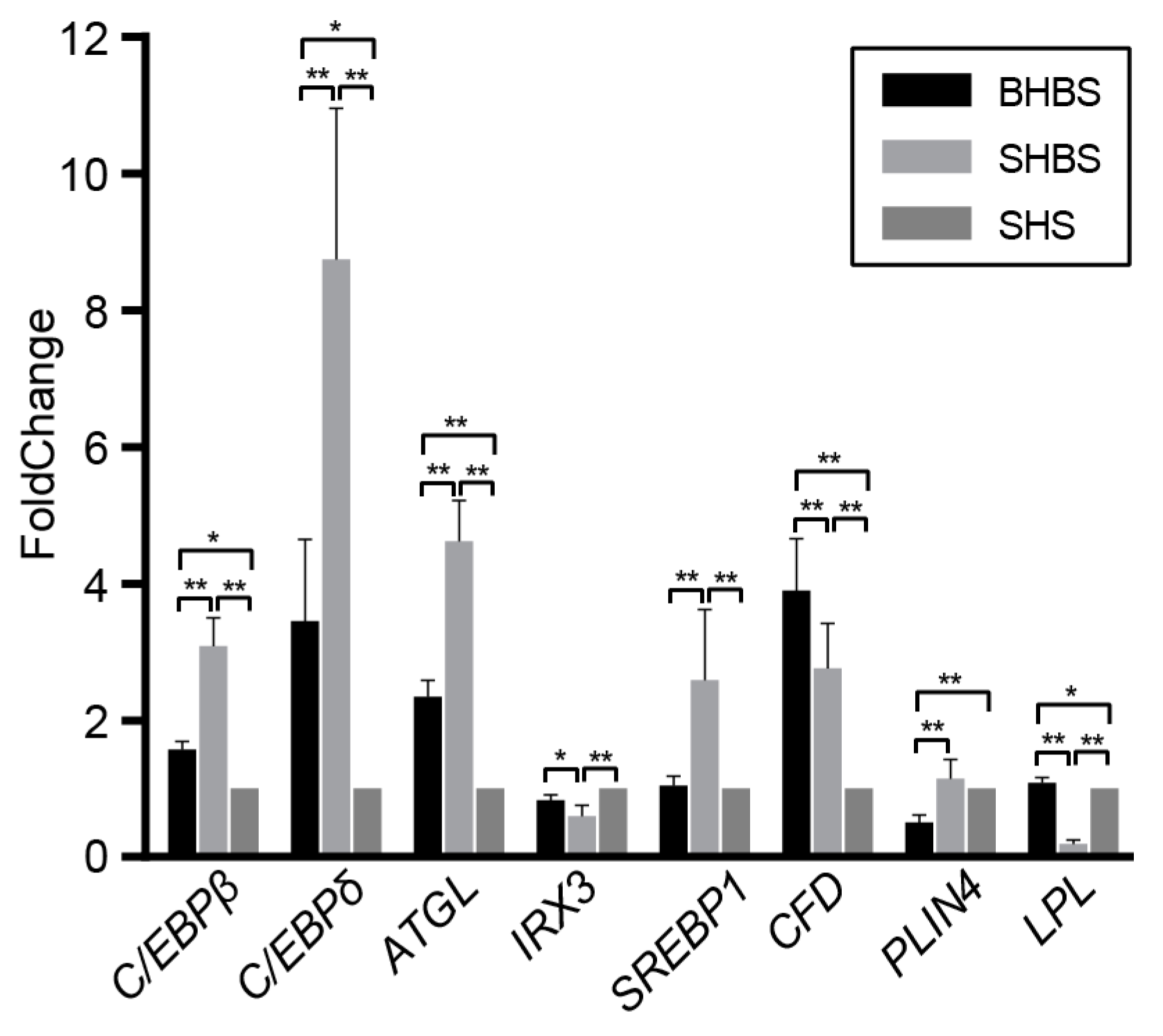
3.1.2. Functional Prediction and Lipid-Relevant Genes Detection
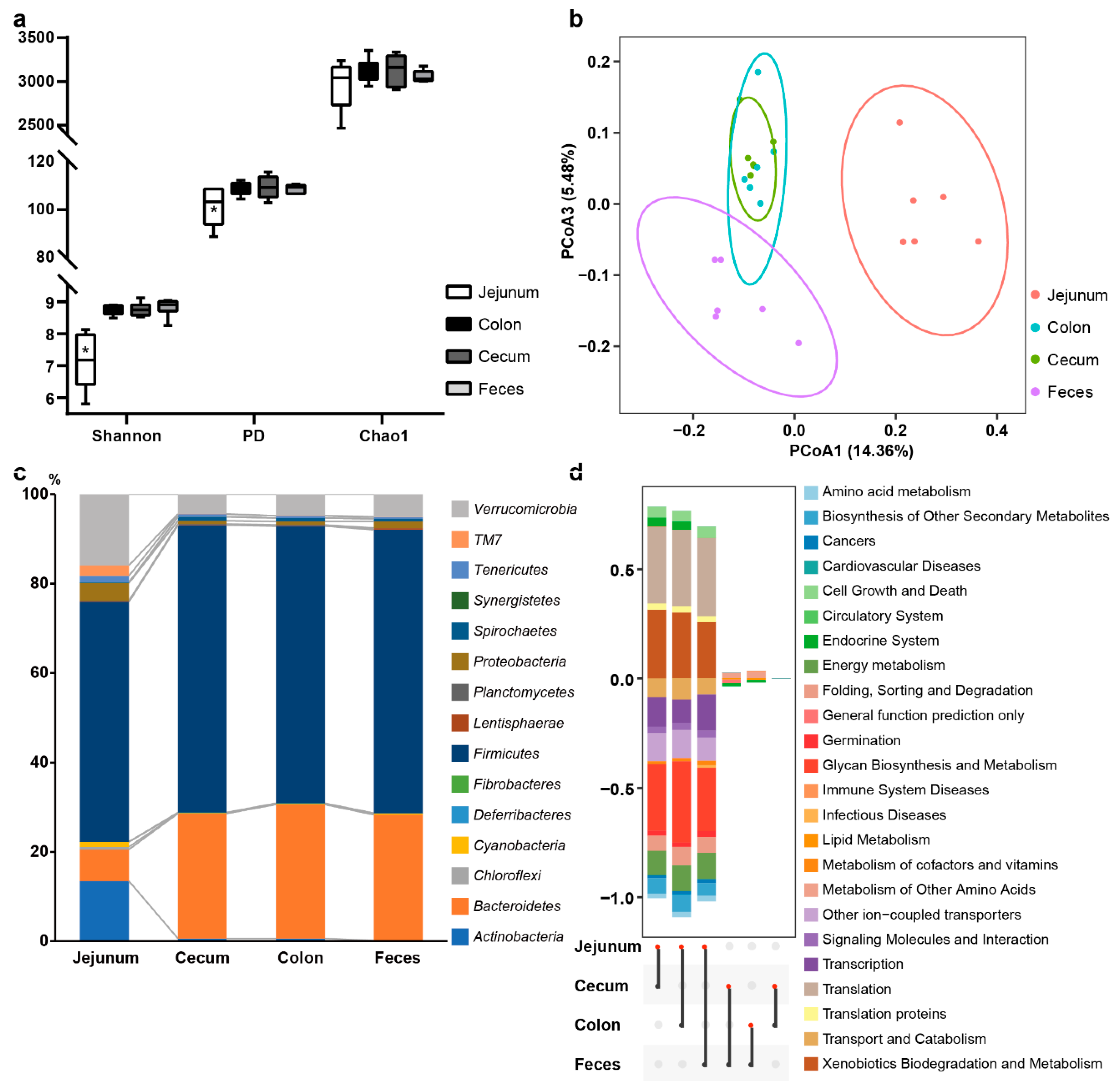
3.2. Bacterial Community Variation in Different Intestinal Sites
3.3. Gut Bacterial Community Variation along with Feeding Time of Probiotics
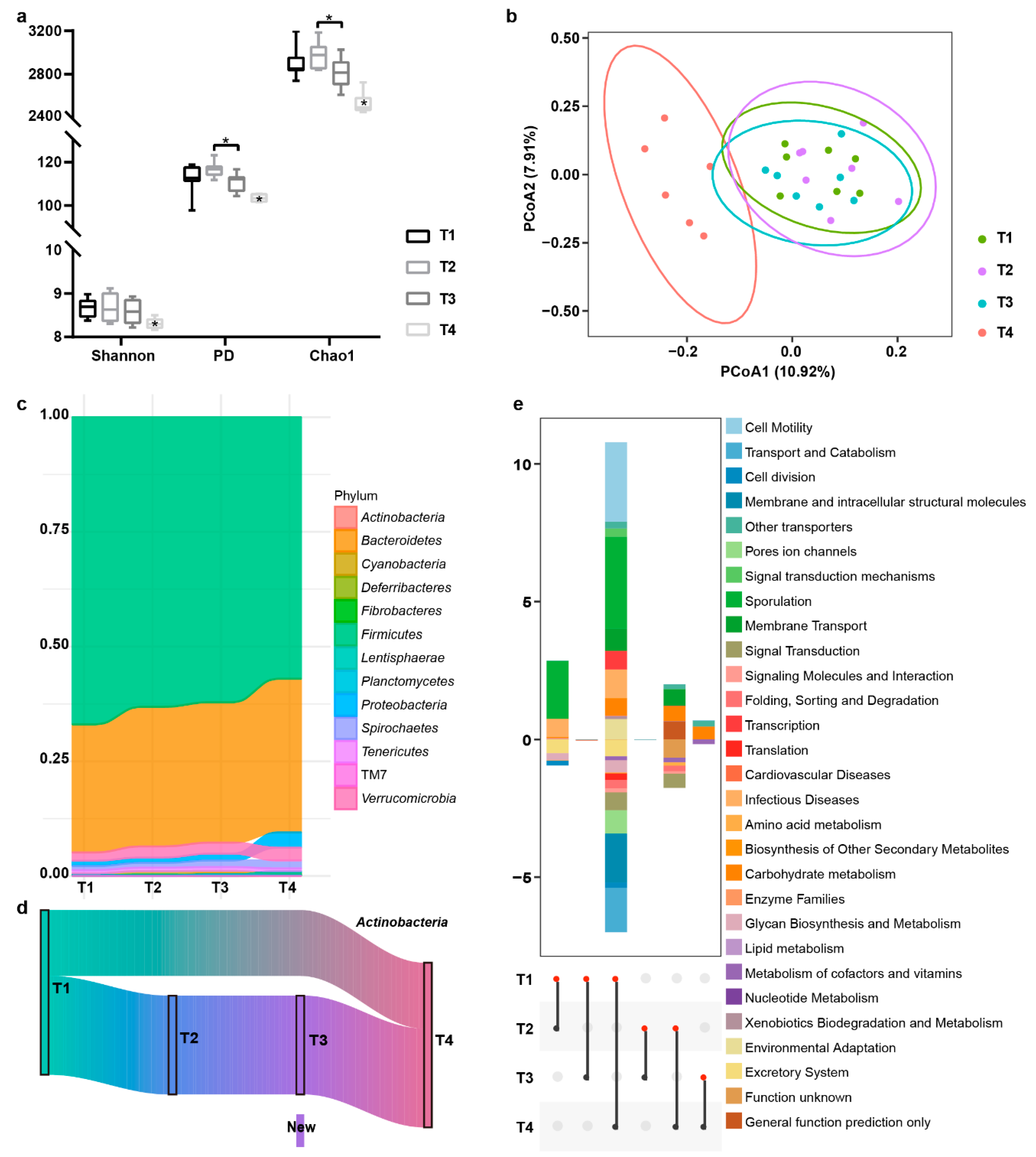
3.3.1. Analysis of the Fluctuation in Diversity and Bacterial Taxa
3.3.2. Predictive Function Analysis
4. Discussion
4.1. Host Genotype Affected Intestinal Microbial Compositions in Sheep with Shared Geography
4.2. The Relevance of Microbial Communities between Intestinal Sites
4.3. Dynamic Intestinal Microbial Communities Relevant to Probiotics Administration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kataoka, K. The intestinal microbiota and its role in human health and disease. J. Med. Investig. 2016, 63, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [Green Version]
- Clauss, M.; Hume, I.D.; Hummel, J. Evolutionary adaptations of ruminants and their potential relevance for modern production systems. Animal 2010, 4, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrand, S. Animal production in a sustainable agriculture. Environ. Dev. Sustain. 2012, 15, 999–1036. [Google Scholar] [CrossRef]
- Sanchez-Morate, E.; Gimeno-Mallench, L.; Stromsnes, K.; Sanz-Ros, J.; Román-Domínguez, A.; Parejo-Pedrajas, S.; Inglés, M.; Olaso, G.; Gambini, J.; Mas-Bargues, C. Relationship between Diet, Microbiota, and Healthy Aging. Biomedicines 2020, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Yao, X.; Zuo, C.; Qi, Y.; Chen, D.; Ma, W. The gut bacterial diversity of sheep associated with different breeds in Qinghai province. BMC Vet. Res. 2020, 16, 254. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, M.; Tiezzi, F.; Howard, J.; Huang, Y.J.; Gray, K.A.; Schillebeeckx, C.; McNulty, N.P.; Maltecca, C. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Zhi, D.; Da, L.; Liu, M.; Cheng, C.; Zhang, Y.; Wang, X.; Li, X.; Tian, Z.; Yang, Y.; He, T.; et al. Whole Genome Sequencing of Hulunbuir Short-Tailed Sheep for Identifying Candidate Genes Related to the Short-Tail Phenotype. G3 Genes Genomes Genet. 2018, 8, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P. Livestock Breeds of China; FAO: Rome, Italy, 1984; p. 46. [Google Scholar]
- Zhao, Y. Sheep Production, 2nd ed.; China Agricultural Press: Beijing, China, 2011. [Google Scholar]
- Zhong, T.; Han, J.L.; Guo, J.; Zhao, Q.J.; Fu, B.L.; He, X.H.; Jeon, J.T.; Guan, W.J.; Ma, Y.H. Genetic diversity of Chinese indigenous sheep breeds inferred from microsatellite markers. Small Rumin. Res. 2010, 90, 88–94. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australas. J. Anim. Sci. 2017, 30, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Ingala, M.R.; Simmons, N.B.; Wultsch, C.; Krampis, K.; Speer, K.A.; Perkins, S.L. Comparing Microbiome Sampling Methods in a Wild Mammal: Fecal and Intestinal Samples Record Different Signals of Host Ecology, Evolution. Front. Microbiol. 2018, 9, 803. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Sun, C.; Zheng, J.; Wen, C.; Ji, C.; Zhang, D.; Chen, Y.; Hou, Z.; Yang, N. Efficacy of Fecal Sampling as a Gut Proxy in the Study of Chicken Gut Microbiota. Front. Microbiol. 2019, 10, 2126. [Google Scholar] [CrossRef] [Green Version]
- Byndloss, M.X.; Pernitzsch, S.R.; Bäumler, A.J. Healthy hosts rule within: Ecological forces shaping the gut microbiota. Mucosal Immunol. 2018, 11, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Bhalodi, A.A.; van Engelen, T.S.R.; Virk, H.S.; Wiersinga, W.J. Impact of antimicrobial therapy on the gut microbiome. J. Antimicrob. Chemother. 2019, 74, i6–i15. [Google Scholar] [CrossRef] [Green Version]
- Abd El-Hack, M.E.; El-Saadony, M.T.; Shafi, M.E.; Qattan, S.Y.A.; Batiha, G.E.; Khafaga, A.F.; Abdel-Moneim, A.-M.E.; Alagawany, M. Probiotics in poultry feed: A comprehensive review. J. Anim. Physiol. Anim. Nutr. 2020, 104, 1835–1850. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; He, Y.; Gao, S.; Liao, Z.; Lai, T.; Zhou, H.; Chen, Q.; Li, L.; Gao, H.; Lu, W. The effect of a diet based on rice straw co-fermented with probiotics and enzymes versus a fresh corn Stover-based diet on the rumen bacterial community and metabolites of beef cattle. Sci. Rep. 2020, 10, 10721. [Google Scholar] [CrossRef]
- Mani, S.; Aiyegoro, O.A.; Adeleke, M.A. Characterization of Rumen Microbiota of Two Sheep Breeds Supplemented With Direct-Fed Lactic Acid Bacteria. Front. Vet. Sci. 2020, 7, 570074. [Google Scholar] [CrossRef]
- Gibson, G.R.; Roberfroid, M.B. Dietary Modulation of the Human Colonic Microbiota-Introducing the Concept of Prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [CrossRef]
- Chi, Y.; Wang, Y.; Zhao, Y.; Zhu, J.; Lin, Y. Study on Cloning and Expression Patterns of Goat C/EBPα, C/EBPβ and C/EBPδ Genes. Acta Agric. Boreali-Sin. 2020, 35, 231–238. (In Chinese) [Google Scholar] [CrossRef]
- Zhang, Y.; Li, L.; Wang, Q.; Zhan, S.; Wang, L.; Zhong, T.; Guo, J.; Zhang, H. Fibroblast growth factor 21 induces lipolysis more efficiently than it suppresses lipogenesis in goat adipocytes. Cytotechnology 2018, 70, 1423–1433. [Google Scholar] [CrossRef]
- Fan, H. Transcriptomic Difference Analysis for Tail Adipose Tissue of Hulun Buir Sheep. Ph.D. Dissertation, Gansu Agricultural University, Lanzhou, China, 2016. (In Chinese). [Google Scholar]
- Liu, L.; Trent, C.M.; Fang, X.; Son, N.H.; Jiang, H.; Blaner, W.S.; Hu, Y.; Yin, Y.X.; Farese, R.V., Jr.; Homma, S.; et al. Cardiomyocyte-specific Loss of Diacylglycerol Acyltransferase 1 (DGAT1) Reproduces the Abnormalities in Lipids Found in Severe Heart Failure. J. Biol. Chem. 2014, 289, 29881–29891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Wang, R.; Luo, J.; Ren, F.; Gu, Z.; Zhao, Y.; Zhao, L. The Core and Distinction of the Gut Microbiota in Chinese Populations across Geography and Ethnicity. Microorganisms 2020, 8, 1579. [Google Scholar] [CrossRef]
- Mamun, M.A.A.; Sandeman, M.; Rayment, P.; Brook-Carter, P.; Scholes, E.; Kasinadhuni, N.; Piedrafita, D.; Greenhill, A.R. The composition and stability of the faecal microbiota of Merino sheep. J. Appl. Microbiol. 2020, 128, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Abbas, W.; Howard, J.T.; Paz, H.A.; Hales, K.E.; Wells, J.E.; Kuehn, L.A.; Erickson, G.E.; Spangler, M.L.; Fernando, S.C. Influence of host genetics in shaping the rumen bacterial community in beef cattle. Sci. Rep. 2020, 10, 15101. [Google Scholar] [CrossRef]
- Sun, W. Differential Analysis of Adipocyte Morphology and Fat Metabolism-Related Genes Expression in Two Strains of Hulun Buir Sheep. MA Dissertation, Sichuan Agricultural University, Ya’an, China, 2017. (In Chinese). [Google Scholar]
- Bestion, E.; Jacob, S.; Zinger, L.; Di Gesu, L.; Richard, M.; White, J.; Cote, J. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat. Ecol. Evol. 2017, 1, 161. [Google Scholar] [CrossRef]
- Fan, H.; Hou, Y.; Sahana, G.; Gao, H.; Zhu, C.; Du, L.; Zhao, F.; Wang, L. A Transcriptomic Study of the Tail Fat Deposition in Two Types of Hulun Buir Sheep According to Tail Size and Sex. Animals 2019, 9, 655. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schaefer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Huang, F.; Zhao, A.; Lei, S.; Zhang, Y.; Xie, G.; Chen, T.; Qu, C.; Rajani, C.; Dong, B.; et al. Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice. BMC Biol. 2017, 15, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Zhao, H.; Ma, Z.; Zhang, S.; Li, M.; Zheng, Z.; Ren, X.; Ho, C.-T.; Bai, N. Anti-Obesity and Gut Microbiota Modulation Effect of Secoiridoid-Enriched Extract from Fraxinus mandshurica Seeds on High-Fat Diet-Fed Mice. Molecules 2020, 25, 4001. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, T.; Nava, G.M.; Olvera-Ramírez, A.M.; Ronquillo, D.; Camacho, M.; Zavala, G.A.; Caamaño, M.C.; Acevedo-Whitehouse, K.; Rosado, J.L.; García, O.P. Gut Bacterial Families Are Associated with Body Composition and Metabolic Risk Markers in School-Aged Children in Rural Mexico. Child. Obes. 2020, 16, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Z.; He, Y.; Li, P.; Zhou, H.; Zeng, N. Regional distribution of Christensenellaceae and its associations with metabolic syndrome based on a population-level analysis. PeerJ 2020, 8, e9591. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.Y.; Zeng, F.; Guo, W.L.; Li, T.T.; Jia, R.B.; Huang, Z.R.; Lv, X.C.; Zhang, J.; Liu, B. Effect of Grifola frondosa 95% ethanol extract on lipid metabolism and gut microbiota composition in high-fat diet-fed rats. Food Funct. 2018, 9, 6268–6278. [Google Scholar] [CrossRef]
- Huang, S.; Ji, S.; Yan, H.; Hao, Y.; Zhang, J.; Wang, Y.; Cao, Z.; Li, S. The day-to-day stability of the ruminal and fecal microbiota in lactating dairy cows. Microbiologyopen 2020, 9, e990. [Google Scholar] [CrossRef]
- Mao, S.; Zhang, M.; Liu, J.; Zhu, W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: Membership and potential function. Sci. Rep. 2015, 5, 16116. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Hong, S.W.; Park, B.-Y.; Yoo, J.G.; Oh, M.-H. Characterisation of the bacterial community in the gastrointestinal tracts of elk (Cervus canadensis). Antonie Leeuwenhoek 2019, 112, 225–235. [Google Scholar] [CrossRef]
- Yang, H.; Huang, X.; Fang, S.; Xin, W.; Huang, L.; Chen, C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci. Rep. 2016, 6, 27427. [Google Scholar] [CrossRef] [Green Version]
- Karasov, W.H.; Douglas, A.E. Comparative Digestive Physiology. Compr. Physiol. 2013, 3, 741–783. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shao, M.; Huang, H.; Wang, S.; Ma, L.; Wang, H.; Hu, L.; Wei, K.; Zhu, R. The Dynamic Distribution of Small-Tail Han Sheep Microbiota across Different Intestinal Segments. Front. Microbiol. 2018, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Behr, C.; Sperber, S.; Jiang, X.; Strauss, V.; Kamp, H.; Walk, T.; Herold, M.; Beekmann, K.; Rietjens, I.M.C.M.; van Ravenzwaay, B. Microbiome-related metabolite changes in gut tissue, cecum content and feces of rats treated with antibiotics. Toxicol. Appl. Pharmacol. 2018, 355, 198–210. [Google Scholar] [CrossRef]
- Kozik, A.J.; Nakatsu, C.H.; Chun, H.; Jones-Hall, Y.L. Comparison of the fecal, cecal, and mucus microbiome in male and female mice after TNBS-induced colitis. PLoS ONE 2019, 14, e0225079. [Google Scholar] [CrossRef]
- Grimm, P.; Combes, S.; Pascal, G.; Cauquil, L.; Julliand, V. Dietary composition and yeast/microalgae combination supplementation modulate the microbial ecosystem in the caecum, colon and faeces of horses. Br. J. Nutr. 2020, 123, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yang, H. Transcriptome Analysis of the Clostridioides difficile Response to Different Doses of Bifidobacterium breve. Front. Microbiol. 2020, 11, 1863. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; Edwards, A.N.; Sarker, M.R.; Paredes-Sabja, D. Sporulation and Germination in Clostridial Pathogens. Microbiol. Spectr. 2019, 7, 7. [Google Scholar] [CrossRef]
- Laghi, L.; Mastromarino, P.; Elisei, W.; Capobianco, D.; Zhu, C.L.; Picchio, M.; Giorgetti, G.; Brandimarte, G.; Tursi, A. Impact of treatments on fecal microbiota and fecal metabolome in symptomatic uncomplicated diverticular disease of the colon: A pilot study. J. Biol. Regul. Homeost. Agents 2018, 32, 1421–1432. [Google Scholar] [PubMed]
- Ali, N.; Gong, H.; Giwa, A.S.; Yuan, Q.; Wang, K. Metagenomic analysis and characterization of acidogenic microbiome and effect of pH on organic acid production. Arch. Microbiol. 2019, 201, 1163–1171. [Google Scholar] [CrossRef]
- Abildgaard, A.; Kern, T.; Pedersen, O.; Hansen, T.; Lund, S.; Wegener, G. A diet-induced gut microbiota component and related plasma metabolites are associated with depressive-like behaviour in rats. Eur. Neuropsychopharmacol. 2020, 43, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Terada-Ikeda, C.; Kitabatake, M.; Hiraku, A.; Kato, K.; Yasui, S.; Imakita, N.; Ouji-Sageshima, N.; Iwabuchi, N.; Hamada, K.; Ito, T. Maternal supplementation with Bifidobacterium breve M-16V prevents their offspring from allergic airway inflammation accelerated by the prenatal exposure to an air pollutant aerosol. PLoS ONE 2020, 15, e0238923. [Google Scholar] [CrossRef]
- Hossain, M.; Park, D.S.; Rahman, M.S.; Ki, S.J.; Lee, Y.R.; Imran, K.M.; Yoon, D.; Heo, J.; Lee, T.J.; Kim, Y.S. Bifidobacterium longum DS0956 and Lactobacillus rhamnosus DS0508 culture-supernatant ameliorate obesity by inducing thermogenesis in obese-mice. Benef. Microbes 2020, 11, 361–373. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Z.; Wang, X.; Bao, Q.; Wang, R.; Duan, Z. The Impact of Host Genotype, Intestinal Sites and Probiotics Supplementation on the Gut Microbiota Composition and Diversity in Sheep. Biology 2021, 10, 769. https://doi.org/10.3390/biology10080769
Wang X, Zhang Z, Wang X, Bao Q, Wang R, Duan Z. The Impact of Host Genotype, Intestinal Sites and Probiotics Supplementation on the Gut Microbiota Composition and Diversity in Sheep. Biology. 2021; 10(8):769. https://doi.org/10.3390/biology10080769
Chicago/Turabian StyleWang, Xiaoqi, Zhichao Zhang, Xiaoping Wang, Qi Bao, Rujing Wang, and Ziyuan Duan. 2021. "The Impact of Host Genotype, Intestinal Sites and Probiotics Supplementation on the Gut Microbiota Composition and Diversity in Sheep" Biology 10, no. 8: 769. https://doi.org/10.3390/biology10080769
APA StyleWang, X., Zhang, Z., Wang, X., Bao, Q., Wang, R., & Duan, Z. (2021). The Impact of Host Genotype, Intestinal Sites and Probiotics Supplementation on the Gut Microbiota Composition and Diversity in Sheep. Biology, 10(8), 769. https://doi.org/10.3390/biology10080769