
Epigenetics for Crop Improvement in Times of Global Change


 , ,
, , 
 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
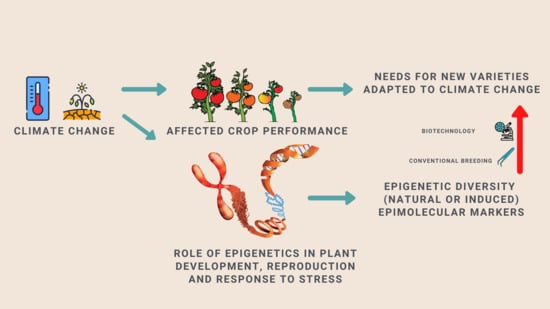
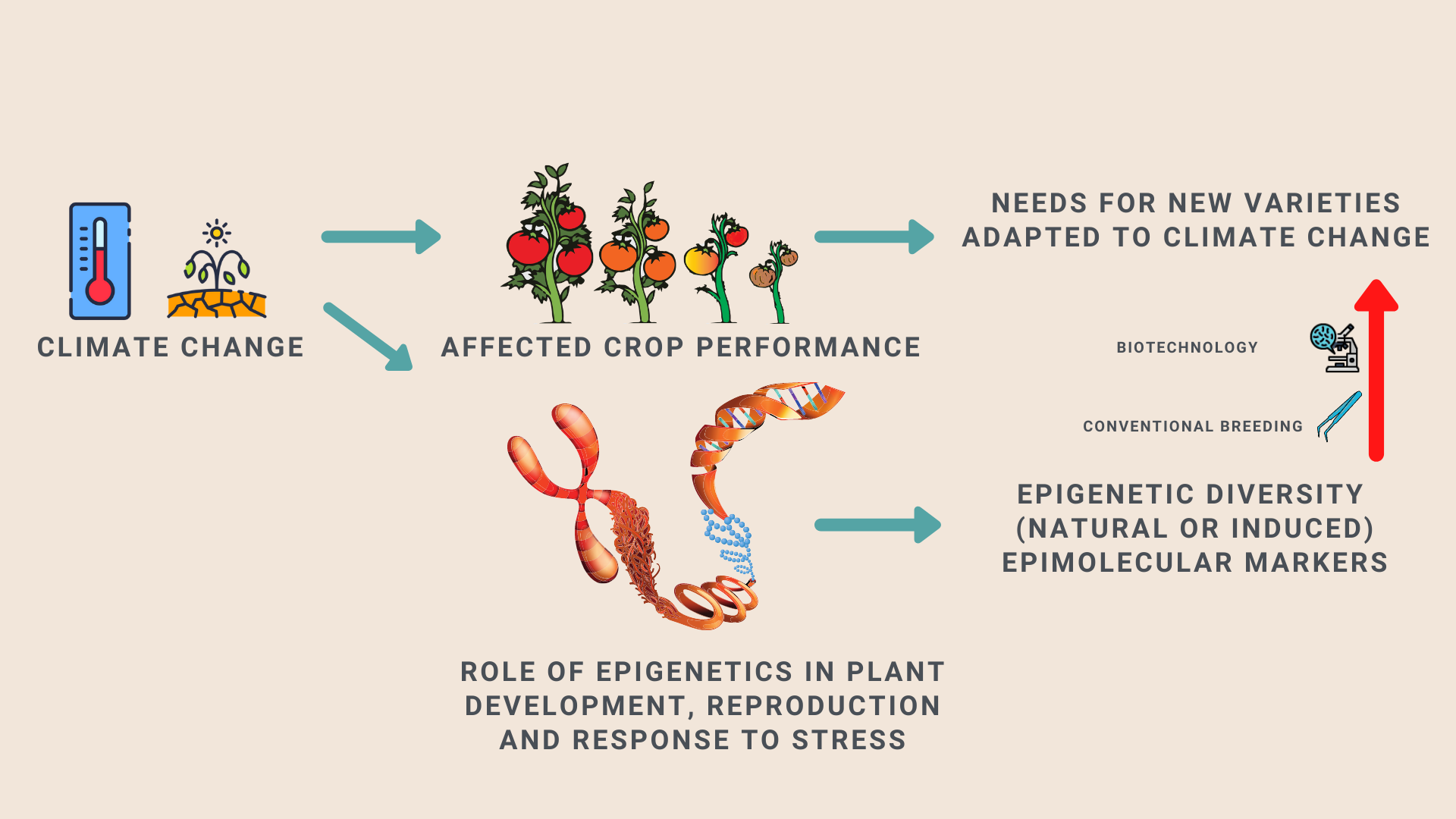
1. Introduction
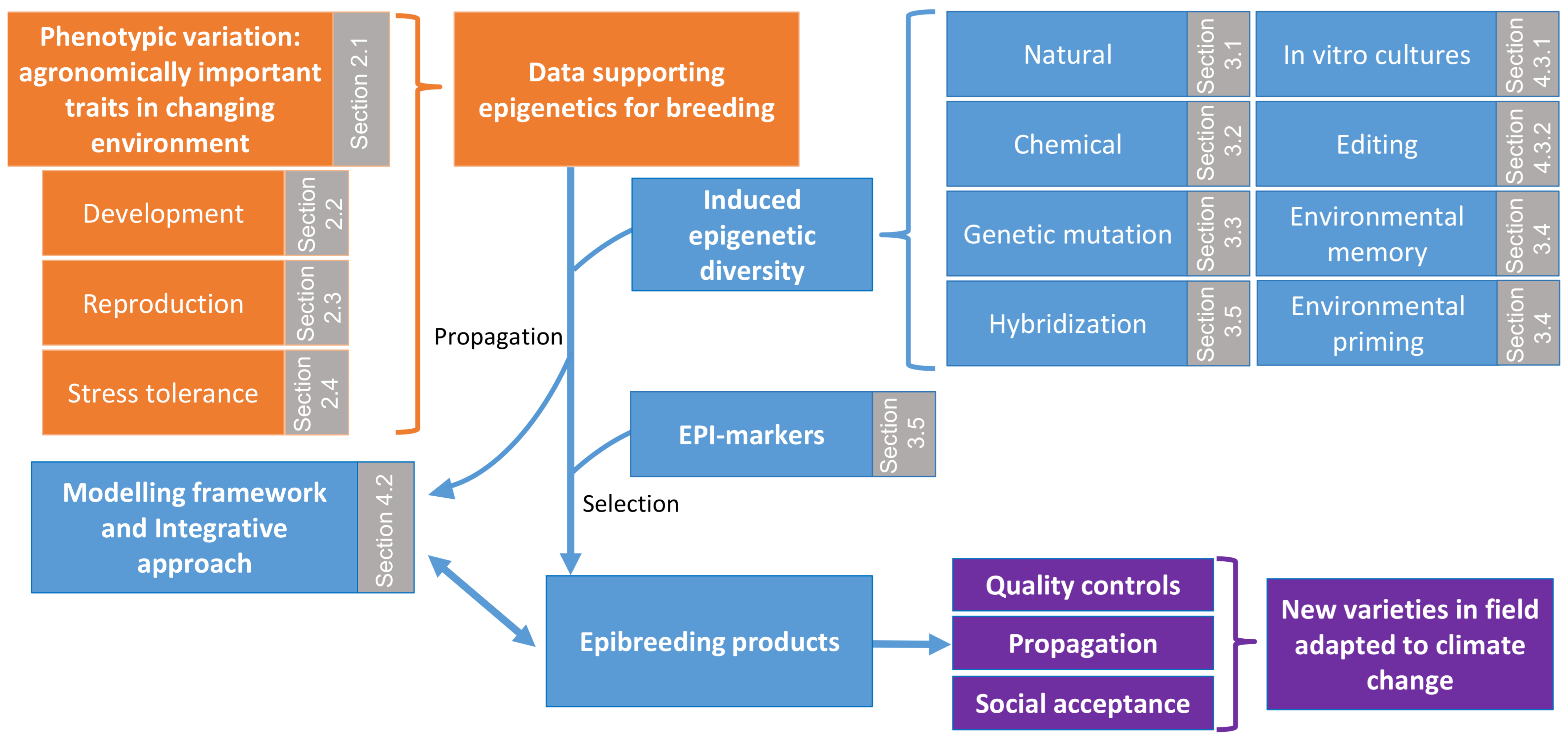
2. From Epigenetics to Crop Improvement: Lessons from Arabidopsis and other Model Plant Species
2.1. Epimutations Contribute to Phenotypic Variation in Model Plants
2.2. Epigenetic Control of Plant Development
2.3. Epigenetic Control of Plant Reproduction and Meiosis
2.4. Epigenetic Control of Plant Response to Stress
3. Epigenetic Advances in Crop Improvement: Exploiting Epigenetic Diversity
3.1. Natural Epi-Alleles
3.2. Chemically Induced Epigenetic Diversity
3.3. Inducing Epigenetic Diversity through Genetic Mutation
3.4. Environmentally Induced Epigenetic Diversity
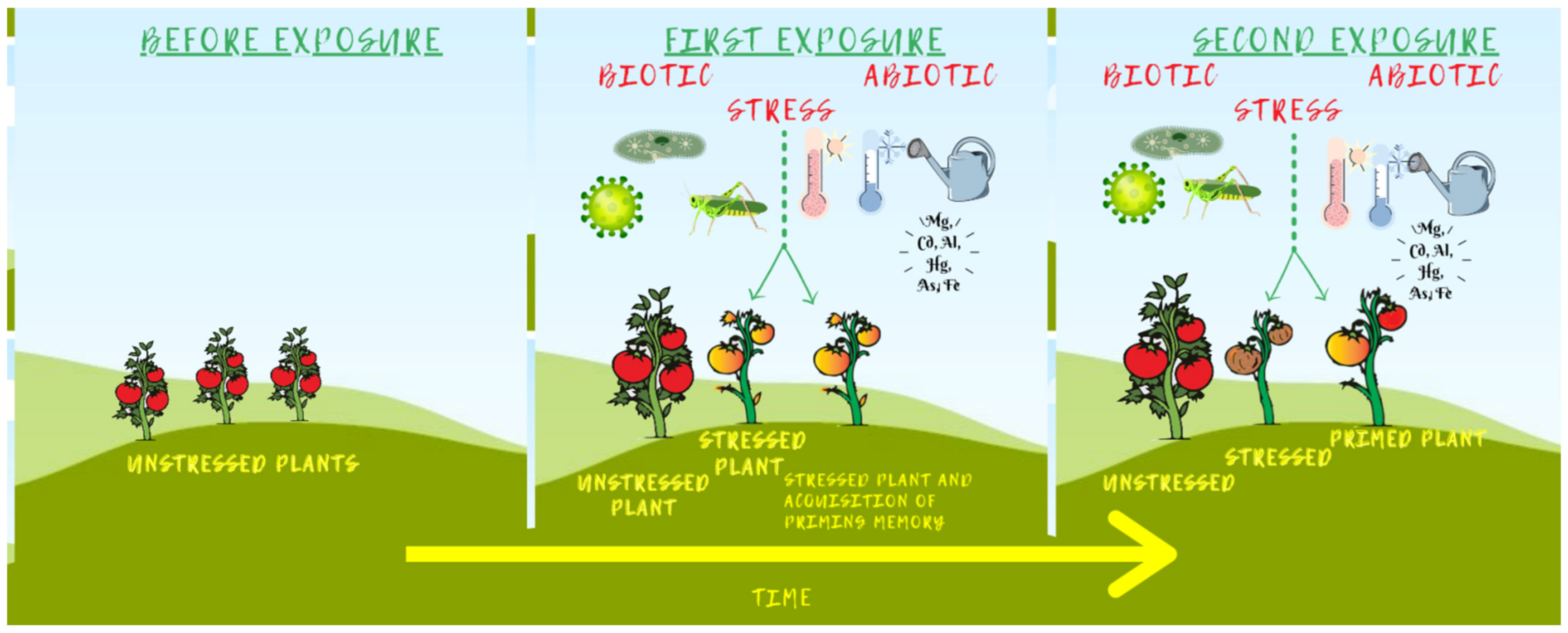
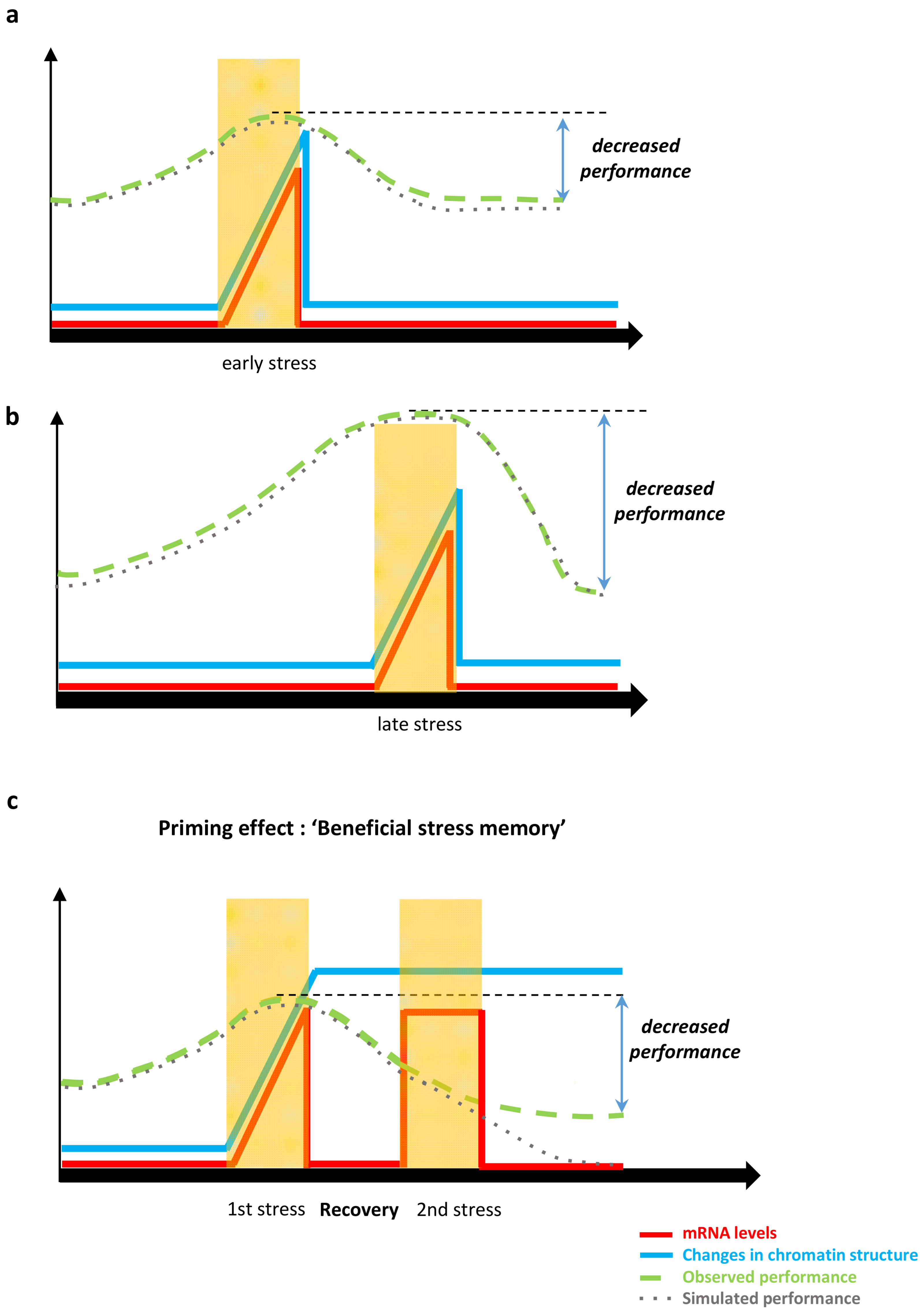
3.4.1. Epigenetic Stress Memory and Priming
3.4.2. Clonal Propagation and Uses
3.5. Hybridisation and Epigenetic as a Predictive Marker of Hybrid Performance
4. Gaps in Knowledge and Future Challenges
4.1. Improving Strategies for Studying the Role of Epigenetics in Crops under Changing Environment
4.2. Modelling Epigenetically Regulated Complex Traits in Crops
4.2.1. The Need to Link Epigenetic Marks to Phenotypes into Modelling Frames
4.2.2. Modelling Epigenetic Regulation Induced by Environmental Stress
4.3. Biotechnologies and Epi/Genome Editing
4.3.1. Targeting Epigenetics for In Vitro Regeneration to Improve and Accelerate Crop Breeding
4.3.2. Epigenetic Editing
5. Conclusions
- Identification of new epigenetically regulated traits
- Facilitate the selection of elite genotypes for the development of new cultivars/varieties
- Understand how epigenetic mechanisms trigger resistance/tolerance to multiple stresses and evaluate their stability
- Improve integrative approaches, statistics, and modelling for crops using epigenetics
- Reduce loss of genetic variability
- Use epigenetics and priming for enhanced management of a/biotic stresses in crops
- Reduce efforts on molecular breeding
- Clarify epigenetic mechanisms for public acceptance
- Requirement for further research in plant epigenetics and synergy between academic and private or public partners.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Topic | Epigenetic Modification | Main Conclusions towards Crop Improvement | Reference |
|---|---|---|---|---|
| Arabidopsis, rice, maize, and other plants | Identification of a sexual-lineage-specific DNA methylation signatures occurred by RNA-directed DNA methylation (RdDM) during plant gametogenesis. | DNA methylation | The clarification of genes and oligonucleotides involved in the modulation of RNA-directed DNA methylation and their mapping in sequence genomes will be of extreme interest to develop new molecular markers associated with fertility, male sterility, and self-incompatibility. | [327] |
| Arabidopsis, tomato | Rootstock epigenetic variation in a comparative analysis in Arabidopsis and tomato. | Small RNA | They showed how the enhanced plant vigour phenotypes of the MSH1 system is reproducible in tomato field size experiments and therefore demonstrated how epigenetic perturbation strategies can be used in crops. | [20] |
| Arabidopsis, white clover | Diminishing the differences between memorised and wild-type plants by DNA demethylating chemical. | DNA methylation | Studies focused on description of DNA methylation in stress memory phenomenon. | [141] |
| Basket willow, spinach, Arabidopsis | More frequent flowering after treatment by DNA demethylating chemical. | DNA methylation | Artificial induction of flowering. | [131] |
| Canola | In an isogenic canola population, the authors showed how energy use efficiency can be selected artificially through an epigenetic feature to increase yield in hybrids. | DNA methylation and histone modifications | The shaping of the epigenome has the potential to artificially increase yield in crops. | [231] |
| Cork oak | Interplay between epigenetic markers related to the acclimation of cork oak plants to high temperatures. | DNA methylation and histone modifications | Increased DNA methylation under high temperature. Dynamics of methylation/demethylation patterns over stress. DNA methylation and histone H3 acetylation have opposite effects and a particular dynamic. | [328] |
| Cotton | Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. | DNA methylation | DNA methylation is suggested to affect photoperiodic flowering time and seed dormancy. | [303] |
| Grapevine | Epigenetic memory induced by stress. | DNA methylation | Conservation of DNA methylation changes in response to medium-high temperatures in regenerated plants. | [195] |
| Grapevine, poplar | Locally established unique epigenetic marks used for authentication/declaration of origin. | DNA methylation | Authentication of plant origin; use of locally adapted clones. | [203] |
| Grapevine, fruit, woody- crop, and forest trees | Woody plants grafting and epigenetic changes. | All chromatin interactions | Woody species grafting is a promising agriculture technology for generating improved woody plants that can face environmental challenges without major compromise in yield and quality and with low input requirements. | [14] |
| Madagascar periwinkle | Production of medicinal secondary metabolites. | DNA methylation | An epigenetic regulation of specialised metabolisms (alkaloids) was unravelled in C roseus, notably targeting transcription factors, which in turn may control the expression of enzyme-encoding genes. This could be exploited to improve the production of secondary metabolites for pharmaceutical applications using plant biotechnologies. | [71] |
| Maize | Defence priming to herbivores. | DNA methylation | Possibility to increase plant defence by application of volatiles related to this mechanism. | [329] |
| Maize | The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. | DNA methylation | The methylation map will provide an invaluable resource for epigenetic studies in maize and how methylation patterns can be used to predict key phenotypes. | [240] |
| Maize | DNA methylation variation (and specific DMRs) as a powerful phenotypic predictor, independent of genetic polymorphism data. | DNA methylation | A first effort to perform genome-wide association analysis using epigenetic data in a crop species. | [78] |
| Maize | Analysis of DNA methylation in different growth zones of maize leaves and transcriptional analysis of genes involved in chromatin remodeling, cell cycle progression, and growth regulation. | DNA methylation | DNA methylation controls cell division in maize leaves and correlates with the mitotic exit and entering cell expansion. | [28] |
| Maize | Investigation of the diversity of DNA methylation states and their association to genotype and gene expression in maize inbred lines. | DNA methylation | Many genes located near the identified DMRs have tissue-specific expression. The expression patterns of over 300 of these genes strongly correlate with the methylation state and are often stably inherited. | [70] |
| Maize | H3K4me3 and H3K27me3 changes involved in the memory of drought stress. Floral patterning is affected in response to stress as a possible consequence of epigenetic changes. | Histone modifications | Coordinated transcriptomic and epigenomic reprogramming of maize plants in response to a main abiotic stress with an impact on plant development and recovery to the stress. Identification of different types of memory genes that may be used as future targets to enhance plant resilience to stress. Identification of putative stress marks which are not associated to direct transcriptional changes. | [192] |
| Maize | Parental divergence in sRNA are strong predictors for grain yield in the hybrids. | Small RNA | Epigenetic measurements may be used as complementary biomarkers in crops. | [220] |
| Maize, rice | Epigenetic mechanisms involved in meiotic events during pollen development. | Non-coding mechanisms | Mapping of genes encoding 21-nt phasiRNAs will allow the development of epi-molecular markers usable for the selection of genotypes with different rates of occurrence of meiotic events. | [84] |
| Maize, wheat, barley, rice, chickpea, pea, tomato | Possible applications of epigenetics in climate-smart crop breeding. | DNA methylation and chromatin modifications | Gaining insight into epigenetic mechanisms will allow improvement of crop adaptation and resilience to environmental stresses, producing a next generation of stable climate-smart crops. | [182] |
| Oil palm | Epiallele responsible for poor fruit production in oil palm. | DNA methylation and small RNA | The ability to predict and cull mantling at the plantlet stage will facilitate the introduction of higher-performing clones and optimise environmentally sensitive land resources. | [108] |
| Poplar | Memory of drought stress in cultivated trees. | DNA methylation | Epigenetic memory in the meristem of stressful environmental conditions occurred during the preceding summer period. This memory may facilitate tree acclimation through priming for cuttings. Clonal propagation of primed trees. | [65] |
| Poplar | Drought tolerance in trees. | DNA methylation | RNAi-ddm1 lines are more tolerant to drought stress. DNA methylation controls hormonal pathway genes (salicylic acid, cytokinins, ethylene) and some transcription factors, but also the activation of TEs that induce mutations potentially near or in genes. This, taking place in the shoot apical meristem, may be transmitted mitotically to primed organs and to the next generation. Confirmation is needed. | [155] |
| Rapeseed, white oak | DNA hypomethylation characterises somatic embryogenesis initiation in quercus trees. | DNA methylation | DNA hypomethylation characterises somatic embryogenesis initiation in clonal propagation techniques of forest trees | [280,285] |
| Rapeseed | DNA methylation changes during pollen development and cell reprogramming in somatic embryogenesis. | DNA methylation | DNA hypomethylation is required for plant cell reprogramming to initiate microspore embryogenesis and doubled haploid production for crop breeding. | [96] |
| Rapeseed, barley | Epigenetic modulators that reduce DNA methylation promote cell reprogramming and microspore embryogenesis for double haploid production. | DNA methylation | DNA de-methylating agents promote cell reprogramming in microspore embryogenesis and doubled-haploid production, favouring acceleration of crop breeding programs. | [128] |
| Rapeseed, barley | Small molecules that produce H3K9 de-methylation to promote cell reprogramming and somatic embryogenesis in crop species. | Histone modifications | Novel small molecules that decrease histone H3K9 methylation levels promote cell reprogramming in microspore embryogenesis and doubled haploid production, favouring acceleration of crop breeding programs. | [284] |
| Rice | Long-term semantic memory to salinity stress. | DNA methylation | Rice is considered a salt-sensitive crop; molecular processes involved in memory to stress may help to breed more resistant plants. | [172] |
| Rice | Phasing analysis of the transcriptome and epigenome in a rice hybrid. | DNA methylation | Developed a phasing pipeline that provides insights into alternative splicing, interaction networks, trans-acting regulation, and the inheritance of DNA methylation in rice. | [40] |
| Rice | Exploring the role of DNA methylation variations in rice adaptation to drought stress. | DNA methylation | Multi-generational drought improves drought adaptability of offspring, which could be linked to non-random appearance of drought-induced transgenerational epimutations. Some of the genes related to these epimutations are directly involved in stress-responsive pathways. | [45] |
| Rice | Identification of DNA methylation transgenerational inherited changes in heavy-metal-responsive genes. | DNA methylation | How plants can cope better with heavy metal stress through heritable changes in DNA methylation. | [193] |
| Rice | A large-scale whole-genome sequencing analysis to assess the specificity of genome editing by Cas9 and Cpf1 nucleases in rice. | Whole-genome sequencing | Cas9 and Cpf1 nucleases are very specific in generating targeted DNA modifications, and off-targeting can be avoided by designing guide RNAs with high specificity. | [51] |
| Rice | DNA methylation and H3K9me2 was shown to repress plant crossover hotspots. | DNA methylation and chromatin modifications | Important implications in the creation of genetic variability produced by breeding activities, because it allows better selection of parental genotypes usable for artificial crossings. | [89] |
| Rice, pea, tomato | Controlled recombination through counting on crossovers can facilitate plant breeding. | Epigenetic modifications and crossovers | Use of genome editing reagents that induce double-stranded breaks (DSBs) or modify the epigenome at desired sites of recombination, and manipulation of cofactors, are increasingly applicable approaches for achieving this goal. These strategies for ‘controlled recombination’ have potential to reduce the time and expense associated with traditional breeding, reveal currently inaccessible genetic diversity, and increase control over the inheritance of preferred haplotypes. | [83] |
| Rubber trees | Chilling-induced DNA demethylation is associated with the cold tolerance of Hevea brasiliensis | DNA methylation | Chilling treatments induced methylation changes and transcriptional activity of methylation and cold-stress-related genes. | [51] |
| Soybean | DNA methylation reprogramming during soybean seed development. | DNA methylation | DNA methylation dynamically changes during soybean seed maturation, affecting the expression of multiple genes. Majority of the DMR genes in the CHH context are downregulated, and closely linked to DNA replication and cell division. This seems to be a protective mechanism that keeps transposons silent to prevent inactivation of genes essential for seed development. | [31] |
| Soybean | DNA methylation patterns in soybean root hairs. | DNA methylation | DMRs in each methylation context have distinct methylation patterns between root hairs and stripped roots, and under heat stress. At normal temperature, root hairs are more hypermethylated than stripped roots. Upon heat stress, both cell types are hypomethylated in each context, especially in the CHH context. | [59] |
| Soybean | DNA methylation and histone modifications of salt-responsive transcription factor genes. | DNA methylation and histone modifications | Salinity stress was shown to affect the methylation status of several transcription factors (one MYB, one b-ZIP, and two AP2/DREB family members). For some of them, DNA methylated transcription factors were correlated with an increased level of histone H3K4 trimethylation and H3K9 acetylation, and/or a reduced level of H3K9 demethylation in various parts of the promoter or coding regions. | [330] |
| Sugar beet | Tolerance to bolting. | DNA methylation | Tolerance to bolting is an agronomic trait for biennial cultivated sugar beet. Bolting is associated with the use of sucrose root stock and should be avoided in the field. Here, tolerance to bolting was correlated to epigenomic polymorphism in DNA methylation, notably in genes involved in cold acclimation, hormonal pathway genes, and flowering genes. | [63,242] |
| Tobacco | Abiotic stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. | DNA methylation | Aluminum stress, salt, and low temperature treatments induced demethylation patterns. These results suggested a close correlation between methylation and expression of NtGPDL upon abiotic stresses with a cause–effect relationship. | [331] |
| Tobacco, potato | Reactivation of silenced transgenes by DNA demethylating chemicals. | DNA methylation | More efficient genetic transformation of plants. | [137] |
| Tobacco, rapeseed onion, barley, cork oat | Method to evaluate global DNA methylation changes and nuclear pattern distribution in a variety of crop and forest species. | DNA methylation | Method to estimate differences in global DNA methylation levels among different cell types and organs during development, which can help to evaluate epigenetic reprogramming events associated with plant growth and adaptation. | [281,282] |
| Tomato | Epigenetic marks in an adaptive water stress-responsive gene in tomato roots under normal and drought conditions. | DNA methylation | Drought induces the removal of methyl marks in the regulatory region (at 77 of the 142 CNN sites) DNA methylation involved in drought acclimation. | [332] |
| Tomato | A DEMETER-like DNA demethylase governs tomato fruit ripening. | DNA methylation | Active DNA demethylation is central to the control of ripening in tomato. RNAi SlDML2 knockdown results in ripening inhibition via hypermethylation and repression of the expression of genes encoding ripening transcription factors and rate-limiting enzymes of key biochemical processes such as carotenoid synthesis. | [75] |
| Tomato | Chilling-induced tomato flavor loss is associated with altered volatile synthesis and transient changes in DNA methylation. | DNA methylation | Changes in DNA methylation are associated with reduced levels of specific volatiles and reductions in transcripts encoding key volatile synthesis enzymes during fruit ripening. RNAs encoding transcription factors essential for ripening, including RIPENING INHIBITOR (RIN), NONRIPENING, and COLORLESS NONRIPENING, are reduced in response to chilling and may be responsible for reduced transcript levels in many downstream genes during chilling. Those reductions are accompanied by major changes in the methylation status of promoters. | [333] |
| Tomato | Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. | DNA methylation | DNA methylation changes through fruit ripening: the epigenome is not static during development and may have been selected to ensure the fidelity of developmental processes, such as ripening. | [334] |
| Tomato | Relationships between genome methylation, levels of non-coding RNAs, mRNAs, and metabolites in ripening tomato fruit. | DNA methylation | Multiple changes in gene methylation were linked to the ethylene pathway and ripening processes. | [335] |
| Tomato | Naturally occurring epialleles determine vitamin E accumulation in tomato fruits. | DNA methylation | Vitamin E content is controlled by mQTL9-2-6—an expression QTL associated with differential methylation of a SINE retrotransposon located in the promoter region of VTE3—that catalyses one of the final steps in the biosynthesis of vitamin E. These findings indicate, therefore, that naturally occurring epialleles are responsible for regulation of a nutritionally important metabolic QTL. | [115] |
| Wheat | The contribution of epigenetic modifications to the expression divergence of three TaEXPA1 homoeologs in hexaploid heat. | DNA methylation and chromatin modifications | Epigenetic modifications contribute to the expression divergence of three TaEXPA1 homoeologs during wheat development. | [30] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, P.; Davis, S.J.; Creutzig, F.; Fuss, S.; Minx, J.C.; Gabrielle, B.; Kato, E.; Jackson, R.B.; Cowie, A.; Kriegler, E.; et al. Biophysical and economic limits to negative CO2 emissions. Nat. Clim. Chang. 2015, 6, 42–50. [Google Scholar] [CrossRef]
- Wollenberg, E.; Richards, M.; Smith, P.; Havlík, P.; Obersteiner, M.; Tubiello, F.N.; Herold, M.; Gerber, P.; Carter, S.; Reisinger, A.; et al. Reducing emissions from agriculture to meet the 2 °C target. Glob. Chang. Biol. 2016, 22, 3859–3864. [Google Scholar] [CrossRef]
- Frank, S.; Havlik, P.; Soussana, J.-F.; Levesque, A.; Valin, H.; Wollenberg, E.; Kleinwechter, U.; Fricko, O.; Gusti, M.; Herrero, M.; et al. Reducing greenhouse gas emissions in agriculture without compromising food security? Environ. Res. Lett. 2017, 12, 105004. [Google Scholar] [CrossRef]
- Ruane, A.C.; Antle, J.; Elliott, J.; Folberth, C.; Hoogenboom, G.; Mason-D’Croz, D.; Müller, C.; Porter, C.; Phillips, M.M.; Raymundo, R.M.; et al. Biophysical and economic implications for agriculture of +1.5° and +2.0 °C global warming using AgMIP Coordinated Global and Regional Assessments. Clim. Res. 2018, 76, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Van Meijl, H.; Havlik, P.; Lotze-Campen, H.; Stehfest, E.; Witzke, P.; Domínguez, I.P.; Bodirsky, B.L.; Van Dijk, M.; Doelman, J.; Fellmann, T.; et al. Comparing impacts of climate change and mitigation on global agriculture by 2050. Environ. Res. Lett. 2018, 13, 064021. [Google Scholar] [CrossRef]
- Lobell, D.; Field, C.B. Global scale climate–crop yield relationships and the impacts of recent warming. Environ. Res. Lett. 2007, 2. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.; Huang, Y.; Huang, M.; Yao, Y.; Bassu, S.; Ciais, P.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326–9331. [Google Scholar] [CrossRef] [PubMed]
- Hartman, G.L.; West, E.D.; Herman, T.K. Crops that feed the World 2. Soybean—worldwide production, use, and constraints caused by pathogens and pests. Food Secur. 2011, 3, 5–17. [Google Scholar] [CrossRef]
- Oerke, E.-C. Crop losses to pests. J. Agric. Sci. 2005, 144, 31–43. [Google Scholar] [CrossRef]
- Oerke, E.-C.; Dehne, H.-W. Safeguarding production—Losses in major crops and the role of crop protection. Crop. Prot. 2004, 23, 275–285. [Google Scholar] [CrossRef]
- Ojolo, S.P.; Cao, S.; Priyadarshani, S.V.G.N.; Li, W.; Yan, M.; Aslam, M.; Zhao, H.; Qin, Y. Regulation of Plant Growth and Development: A Review From a Chromatin Remodeling Perspective. Front. Plant. Sci. 2018, 9, 1232. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, G.; Kudapa, H.; Ramalingam, A.; Choudhary, D.; Sinha, P.; Garg, V.; Singh, V.K.; Patil, G.B.; Pandey, M.K.; Nguyen, H.T.; et al. Epigenetics and epigenomics: Underlying mechanisms, relevance, and implications in crop improvement. Funct. Integr. Genom. 2020, 20, 739–761. [Google Scholar] [CrossRef] [PubMed]
- Mercé, C.; Bayer, P.E.; Fernandez, C.T.; Batley, J.; Edwards, D. Induced Methylation in Plants as a Crop Improvement Tool: Progress and Perspectives. Agronomy 2020, 10, 1484. [Google Scholar] [CrossRef]
- Kapazoglou, A.; Tani, E.; Avramidou, E.V.; Abraham, E.M.; Gerakari, M.; Megariti, S.; Doupis, G.; Doulis, A.G. Epigenetic Changes and Transcriptional Reprogramming Upon Woody Plant Grafting for Crop Sustainability in a Changing Environment. Front. Plant. Sci. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Bodadilla, R. Histone Methylation—A Cornerstone for Plant Responses to Environmental Stresses? In Abiotic and Biotic Stress in Plants—Recent Advances and Future Perspectives; IntechOpen: London, UK, 2016. [Google Scholar]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Singh, J.; Mishra, V.; Wang, F.; Huang, H.-Y.; Pikaard, C.S. Reaction Mechanisms of Pol IV, RDR2, and DCL3 Drive RNA Channeling in the siRNA-Directed DNA Methylation Pathway. Mol. Cell 2019, 75, 576–589.e5. [Google Scholar] [CrossRef]
- Yang, X.; Kundariya, H.; Xu, Y.-Z.; Sandhu, A.; Yu, J.; Hutton, S.F.; Zhang, M.; MacKenzie, S.A. MutS HOMOLOG1-Derived Epigenetic Breeding Potential in Tomato. Plant. Physiol. 2015, 168, 222–232. [Google Scholar] [CrossRef]
- Kundariya, H.; Yang, X.; Morton, K.; Sanchez, R.; Axtell, M.J.; Hutton, S.F.; Fromm, M.; Mackenzie, S.A. MSH1-induced heritable enhanced growth vigor through grafting is associated with the RdDM pathway in plants. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Raju, S.K.K.; Shao, M.; Sanchez, R.; Xu, Y.; Sandhu, A.; Graef, G.; Mackenzie, S. An epigenetic breeding system in soybean for increased yield and stability. Plant. Biotechnol. J. 2018, 16, 1836–1847. [Google Scholar] [CrossRef]
- Pecinka, A.; Chevalier, C.; Colas, I.; Kalantidis, K.; Varotto, S.; Krugman, T.; Michailidis, C.; Vallés, M.-P.; Muñoz, A.; Pradillo, M. Chromatin dynamics during interphase and cell division: Similarities and differences between model and crop plants. J. Exp. Bot. 2019, 71, 5205–5222. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Henry, H.A.; Tian, L. Brachypodium histone deacetylase BdHD1 positively regulates ABA and drought stress responses. Plant. Sci. 2019, 283, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Tsikou, D.; Yan, Z.; Holt, D.B.; Abel, N.B.; Reid, D.E.; Madsen, L.H.; Bhasin, H.; Sexauer, M.; Stougaard, J.; Markmann, K. Systemic control of legume susceptibility to rhizobial infection by a mobile microRNA. Science 2018, 362, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Boycheva, I.; Vassileva, V.; Revalska, M.; Zehirov, G.; Iantcheva, A. Different functions of the histone acetyltransferase HAC1 gene traced in the model species Medicago truncatula, Lotus japonicus and Arabidopsis thaliana. Protoplasma 2016, 254, 697–711. [Google Scholar] [CrossRef]
- Rose, R.J. Somatic Embryogenesis in the Medicago truncatula Model: Cellular and Molecular Mechanisms. Front. Plant. Sci. 2019, 10, 267. [Google Scholar] [CrossRef]
- Deng, X.; Song, X.; Wei, L.; Liu, C.; Cao, X. Epigenetic regulation and epigenomic landscape in rice. Natl. Sci. Rev. 2016, 3, 309–327. [Google Scholar] [CrossRef]
- Candaele, J.; Demuynck, K.; Mosoti, D.; Beemster, G.; Inzé, D.; Nelissen, H. Differential Methylation during Maize Leaf Growth Targets Developmentally Regulated Genes. Plant. Physiol. 2014, 164, 1350–1364. [Google Scholar] [CrossRef]
- Huang, J.; Lynn, J.S.; Schulte, L.; Vendramin, S.; McGinnis, K. Chapter Two—Epigenetic Control of Gene Expression in Maize. In International Review of Cell and Molecular Biology; Galluzzi, L., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 328, pp. 25–48. [Google Scholar]
- Hu, Z.; Han, Z.; Song, N.; Chai, L.; Yao, Y.; Peng, H.; Ni, Z.; Sun, Q. Epigenetic modification contributes to the expression divergence of three TaEXPA1 homoeologs in hexaploid wheat (Triticum aestivum). N. Phytol. 2013, 197, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- An, Y.-Q.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Yao, N.; Schmitz, R.J.; Johannes, F. Epimutations define a fast-ticking molecular clock in plants. Trends Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D.; Colot, V. Epialleles in plant evolution. Genome Biol. 2012, 13, 1–6. [Google Scholar] [CrossRef]
- Johannes, F.; Schmitz, R.J. Spontaneous epimutations in plants. N. Phytol. 2018, 221, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis thaliana. Science 2009, 327, 92–94. [Google Scholar] [CrossRef]
- Denkena, J.; Johannes, F.; Colomé-Tatché, M. Region-level epimutation rates in Arabidopsis thaliana. Heredity 2021, 1–13. [Google Scholar] [CrossRef]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Exposito-Alonso, M.; Weng, M.-L.; Rutter, M.T.; Fenster, C.B.; Weigel, D. Mutation Bias Shapes Gene Evolution in Arabidopsis Thaliana. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hofmeister, B.T.; Denkena, J.; Colomé-Tatché, M.; Shahryary, Y.; Hazarika, R.; Grimwood, J.; Mamidi, S.; Jenkins, J.; Grabowski, P.P.; Sreedasyam, A.; et al. A genome assembly and the somatic genetic and epigenetic mutation rate in a wild long-lived perennial Populus trichocarpa. Genome Biol. 2020, 21, 1–27. [Google Scholar] [CrossRef]
- Hofmeister, B.; Lee, K.; Rohr, N.A.; Hall, D.; Schmitz, R.J. Stable inheritance of DNA methylation allows creation of epigenotype maps and the study of epiallele inheritance patterns in the absence of genetic variation. Genome Biol. 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-W.; Lu, Y.; Shao, L.; Zhang, J.; Li, H.; Chen, L.-L. Phasing analysis of the transcriptome and epigenome in a rice hybrid reveals the inheritance and difference in DNA methylation and allelic transcription regulation. Plant. Commun. 2021, 100185. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed]
- Baduel, P.; Colot, V. The epiallelic potential of transposable elements and its evolutionary significance in plants. Philos. Trans. Soc. Biol. Sci. 2021, 376, 20200123. [Google Scholar] [CrossRef]
- Wendte, J.M.; Zhang, Y.; Dizaji, Y.S.; Shi, X.; Hazarika, R.R.; Shahryary, Y.; Johannes, F.; Schmitz, R.J. Epimutations are associated with CHROMOMETHYLASE 3-induced de novo DNA methylation. eLife 2019, 8, e47891. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA Methylome Is Stable under Transgenerational Drought Stress. Plant. Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Becker, C.; Marconi, G.; Durr, J.; Price, J.; Hagmann, J.; Papareddy, R.; Putra, H.; Kageyama, J.; Becker, J.; et al. Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. eLife 2016, 5, e13546. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.J.; Donoghue, M.T.A.; Barros, P.M.; Saibo, N.J.; Santos, A.P.; Oliveira, M.M. Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance. Epigenomes 2019, 3, 4. [Google Scholar] [CrossRef]
- Lang-Mladek, C.; Popova, O.; Kiok, K.; Berlinger, M.; Rakic, B.; Aufsatz, W.; Jonak, C.; Hauser, M.-T.; Luschnig, C. Transgenerational Inheritance and Resetting of Stress-Induced Loss of Epigenetic Gene Silencing in Arabidopsis. Mol. Plant. 2010, 3, 594–602. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Kovalchuk, I. Transgenerational changes in plant physiology and in transposon expression in response to UV-C stress in Arabidopsis thaliana. Plant. Signal. Behav. 2014, 9, e976490. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Huang, S.-S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef]
- Tang, X.; Liu, G.; Zhou, J.; Ren, Q.; You, Q.; Tian, L.; Xin, X.; Zhong, Z.; Liu, B.; Zheng, X.; et al. A large-scale whole-genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 (Cas12a) nucleases in rice. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Schmitz, R.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nat. Cell Biol. 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Omalley, R.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Widman, N.; Feng, S.; Jacobsen, S.E.; Pellegrini, M. Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 2013, 9, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt Stress Induced Variation in DNA Methylation Pattern and Its Influence on Gene Expression in Contrasting Rice Genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef] [PubMed]
- Lafos, M.; Kroll, P.; Hohenstatt, M.L.; Thorpe, F.L.; Clarenz, O.; Schubert, D. Dynamic Regulation of H3K27 Trimethylation during Arabidopsis Differentiation. PLoS Genet. 2011, 7, e1002040. [Google Scholar] [CrossRef]
- Baubec, T.; Finke, A.; Scheid, O.M.; Pecinka, A. Meristem-specific expression of epigenetic regulators safeguards transposon silencing in Arabidopsis. EMBO Rep. 2014, 15, 446–452. [Google Scholar] [CrossRef]
- Hossain, S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J.; et al. Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. N. Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef]
- Placette, C.L.; Faivre-Rampant, P.; Delaunay, A.; Street, N.; Brignolas, F.; Maury, S. Methylome of DN ase I sensitive chromatin in P opulus trichocarpa shoot apical meristematic cells: A simplified approach revealing characteristics of gene-body DNA methylation in open chromatin state. N. Phytol. 2012, 197, 416–430. [Google Scholar] [CrossRef]
- Hsieh, P.-H.; He, S.; Buttress, T.; Gao, H.; Couchman, M.; Fischer, R.L.; Zilberman, D.; Feng, X. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 15132–15137. [Google Scholar] [CrossRef]
- Kordyum, E.L.; Mosyakin, S.L. Endosperm of Angiosperms and Genomic Imprinting. Life 2020, 10, 104. [Google Scholar] [CrossRef]
- Hébrard, C.; Peterson, D.; Willems, G.; Delaunay, A.; Jesson, B.; Lefèbvre, M.; Barnes, S.; Maury, S. Epigenomics and bolting tolerance in sugar beet genotypes. J. Exp. Bot. 2015, 67, 207–225. [Google Scholar] [CrossRef]
- Conde, D.; Le Gac, A.; Perales, M.; Dervinis, C.; Kirst, M.; Maury, S.; González-Melendi, P.; Allona, I. Chilling-responsive DEMETER-LIKE DNA demethylase mediates in poplar bud break. Plant. Cell Environ. 2017, 40, 2236–2249. [Google Scholar] [CrossRef] [PubMed]
- Le Gac, A.-L.; Placette, C.L.; Chauveau, D.; Segura, V.; Delaunay, A.; Fichot, R.; Marron, N.; Le Jan, I.; Berthelot, A.; Bodineau, G.; et al. Winter-dormant shoot apical meristem in poplar trees shows environmental epigenetic memory. J. Exp. Bot. 2018, 69, 4821–4837. [Google Scholar] [CrossRef] [PubMed]
- Sow, M.D.; Allona, I.; Ambroise, C.; Conde, D.; Fichot, R.; Gribkova, S.; Jorge, V.; Le-Provost, G.; Pâques, L.; Plomion, C.; et al. Chapter Twelve—Epigenetics in Forest Trees: State of the Art and Potential Implications for Breeding and Management in a Context of Climate Change. In Advances in Botanical Research; Mirouze, M., Bucher, E., Gallusci, P., Eds.; Plant Epigenetics Coming of Age for Breeding Applications; Academic Press: London, UK, 2018; Volume 88, pp. 387–453. [Google Scholar]
- Le Gac, A.-L.; Lafon-Placette, C.; Delaunay, A.; Maury, S. Developmental, genetic and environmental variations of global DNA methylation in the first leaves emerging from the shoot apical meristem in poplar trees. Plant. Signal. Behav. 2019, 14, 1596717. [Google Scholar] [CrossRef]
- Whittaker, C.; Dean, C. The FLC Locus: A Platform for Discoveries in Epigenetics and Adaptation. Annu. Rev. Cell Dev. Biol. 2017, 33, 555–575. [Google Scholar] [CrossRef]
- Maury, S.; Sow, M.D.; Le Gac, A.-L.; Genitoni, J.; Placette, C.L.; Mozgova, I. Phytohormone and Chromatin Crosstalk: The Missing Link for Developmental Plasticity? Front. Plant. Sci. 2019, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and Genetic Influences on DNA Methylation Variation in Maize Populations. Plant. Cell 2013, 25, 2783–2797. [Google Scholar] [CrossRef]
- De Bernonville, T.D.; Maury, S.; Delaunay, A.; Daviaud, C.; Chaparro, C.; Tost, J.; O’Connor, S.E.; Courdavault, V. Developmental Methylome of the Medicinal Plant Catharanthus roseus Unravels the Tissue-Specific Control of the Monoterpene Indole Alkaloid Pathway by DNA Methylation. Int. J. Mol. Sci. 2020, 21, 6028. [Google Scholar] [CrossRef]
- Schmid, M.W.; Giraldo-Fonseca, A.; Rövekamp, M.; Smetanin, D.; Bowman, J.L.; Grossniklaus, U. Extensive epigenetic reprogramming during the life cycle of Marchantia polymorpha. Genome Biol. 2018, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Van Dooren, T.J.M.; Silveira, A.B.; Gilbault, E.; Jiménez-Gómez, J.M.; Martin, A.; Bach, L.; Tisné, S.; Quadrana, L.; Loudet, O.; Colot, V. Mild drought in the vegetative stage induces phenotypic, gene expression, and DNA methylation plasticity in Arabidopsis but no transgenerational effects. J. Exp. Bot. 2020, 71, 3588–3602. [Google Scholar] [CrossRef]
- Bhatia, H.; Khemka, N.; Jain, M.; Garg, R. Genome-wide bisulphite-sequencing reveals organ-specific methylation patterns in chickpea. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; How-Kit, A.; Stammitti, L.; Teyssier, E.; Rolin, D.; Mortain-Bertrand, A.; Halle, S.; Liu, M.; Kong, J.; Wu, C.; et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. USA 2015, 112, 10804–10809. [Google Scholar] [CrossRef]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.-K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, G.; Hermanson, P.J.; Xu, Q.; Sun, C.; Chen, W.; Kan, Q.; Li, M.; Crisp, P.; Yan, J.; et al. Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 2019, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- McKey, D.; Elias, M.; Pujol, B.; Duputié, A. The evolutionary ecology of clonally propagated domesticated plants. N. Phytol. 2010, 186, 318–332. [Google Scholar] [CrossRef]
- Latutrie, M.; Gourcilleau, D.; Pujol, B. Epigenetic variation for agronomic improvement: An opportunity for vegetatively propagated crops. Am. J. Bot. 2019, 106, 1281–1284. [Google Scholar] [CrossRef]
- Nybom, H.; Lācis, G. Recent Large-Scale Genotyping and Phenotyping of Plant Genetic Resources of Vegetatively Propagated Crops. Plants 2021, 10, 415. [Google Scholar] [CrossRef]
- Wibowo, A.; Becker, C.; Durr, J.; Price, J.; Spaepen, S.; Hilton, S.; Putra, H.; Papareddy, R.; Saintain, Q.; Harvey, S.; et al. Partial maintenance of organ-specific epigenetic marks during plant asexual reproduction leads to heritable phenotypic variation. Proc. Natl. Acad. Sci. USA 2018, 115, E9145–E9152. [Google Scholar] [CrossRef]
- Taagen, E.; Bogdanove, A.J.; Sorrells, M.E. Counting on Crossovers: Controlled Recombination for Plant Breeding. Trends Plant. Sci. 2020, 25, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Yang, L.; Liang, W.; Zhang, D.; Meyers, B.C. Dynamic changes of small RNAs in rice spikelet development reveal specialized reproductive phasiRNA pathways. J. Exp. Bot. 2016, 67, 6037–6049. [Google Scholar] [CrossRef]
- Zhai, J.; Zhang, H.; Arikit, S.; Huang, K.; Nan, G.-L.; Walbot, V.; Meyers, B.C. Spatiotemporally dynamic, cell-type–dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc. Natl. Acad. Sci. USA 2015, 112, 3146–3151. [Google Scholar] [CrossRef]
- Howell, M.D.; Fahlgren, N.; Chapman, E.J.; Cumbie, J.S.; Sullivan, C.M.; Givan, S.; Kasschau, K.D.; Carrington, J.C. Genome-Wide Analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 Pathway in Arabidopsis Reveals Dependency on miRNA- and tasiRNA-Directed Targeting. Plant. Cell 2007, 19, 926–942. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xia, R.; Zhao, B.; An, Y.-Q.; Dardick, C.D.; Callahan, A.M.; Liu, Z. Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant. Biol. 2012, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Shilo, S.; Melamed-Bessudo, C.; Dorone, Y.; Barkai, N.; Levy, A.A. DNA Crossover Motifs Associated with Epigenetic Modifications Delineate Open Chromatin Regions in Arabidopsis. Plant. Cell 2015, 27, 2427–2436. [Google Scholar] [CrossRef]
- Yelina, N.E.; Lambing, C.; Hardcastle, T.J.; Zhao, X.; Santos, B.; Henderson, I. DNA methylation epigenetically silences crossover hot spots and controls chromosomal domains of meiotic recombination in Arabidopsis. Genes Dev. 2015, 29, 2183–2202. [Google Scholar] [CrossRef] [PubMed]
- Mirouze, M.; Lieberman-Lazarovich, M.; Aversano, R.; Bucher, E.; Nicolet, J.; Reinders, J.; Paszkowski, J. Loss of DNA methylation affects the recombination landscape in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 5880–5885. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Zhao, X.; Tock, A.J.; Lambing, C.; Underwood, C.; Hardcastle, T.J.; Serra, H.; Kim, J.; Cho, H.S.; Kim, J.; et al. Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions. Genome Res. 2018, 28, 532–546. [Google Scholar] [CrossRef]
- Higo, H.; Tahir, M.; Takashima, K.; Miura, A.; Watanabe, K.; Tagiri, A.; Ugaki, M.; Ishikawa, R.; Eiguchi, M.; Kurata, N.; et al. DDM1 (Decrease in DNA Methylation) genes in rice (Oryza sativa). Mol. Genet. Genom. 2012, 287, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Nonomura, K.-I. Histone H3 modifications are widely reprogrammed during male meiosis I in rice dependently on MEL1 Argonaute protein. J. Cell Sci. 2016, 129, 3553–3561. [Google Scholar] [CrossRef]
- Dukowic-Schulze, S.; Liu, C.; Chen, C. Not just gene expression: 3D implications of chromatin modifications during sexual plant reproduction. Plant. Cell Rep. 2017, 37, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, K.-I. Small RNA pathways responsible for non-cell-autonomous regulation of plant reproduction. Plant. Reprod. 2018, 31, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Solís, M.-T.; Rodríguez-Serrano, M.; Meijón, M.; Canal-Villanueva, M.J.F.; Cifuentes, A.; Risueño, M.C.; Testillano, P.S. DNA methylation dynamics and MET1a-like gene expression changes during stress-induced pollen reprogramming to embryogenesis. J. Exp. Bot. 2012, 63, 6431–6444. [Google Scholar] [CrossRef] [PubMed]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijó, J.; Becker, J.; et al. Reprogramming of DNA Methylation in Pollen Guides Epigenetic Inheritance via Small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Olmedo-Monfil, V.; Duran-Figueroa, N.; Arteaga-Vazquez, M.A.; Demesa-Arevalo, E.; Autran, D.; Grimanelli, D.; Slotkin, R.K.; Martienssen, R.A.; Vielle-Calzada, J.-P. Control of female gamete formation by a small RNA pathway in Arabidopsis. Nat. Cell Biol. 2010, 464, 628–632. [Google Scholar] [CrossRef]
- Chang, Y.; Zhu, C.; Jiang, J.; Zhang, H.; Zhu, J.; Duan, C. Epigenetic regulation in plant abiotic stress responses. J. Integr. Plant. Biol. 2019, 62, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, J.; Han, J. Chromatin remodeling factors regulate environmental stress responses in plants. J. Integr. Plant. Biol. 2021, 63, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cruz, D.; Troyee, A.N.; Becker, C. Epigenetics in plant organismic interactions. Curr. Opin. Plant. Biol. 2021, 61, 102060. [Google Scholar] [CrossRef]
- McCue, A.D.; Panda, K.; Nuthikattu, S.; Choudury, S.G.; Thomas, E.N.; Slotkin, R.K. ARGONAUTE 6 bridges transposable element m RNA -derived si RNA s to the establishment of DNA methylation. EMBO J. 2014, 34, 20–35. [Google Scholar] [CrossRef]
- Bokszczanin, K.L.; Fragkostefanakis, S.; Bostan, H.; Bovy, A.; Chaturvedi, P.; Chiusano, M.L.; Firon, N.; Iannacone, R.; Jegadeesan, S.; Klaczynskid, K.; et al. Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Front. Plant. Sci. 2013, 4, 315. [Google Scholar] [CrossRef] [PubMed]
- Bokszczanin, K.L.; Krezdorn, N.; Fragkostefanakis, S.; Müller, S.; Rycak, L.; Chen, Y.; Hoffmeier, K.; Kreutz, J.; Paupière, M.J.; Chaturvedi, P.; et al. Identification of novel small ncRNAs in pollen of tomato. BMC Genom. 2015, 16, 714. [Google Scholar] [CrossRef] [PubMed]
- Pecinka, A.; Scheid, O.M. Stress-Induced Chromatin Changes: A Critical View on Their Heritability. Plant. Cell Physiol. 2012, 53, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nat. Cell Biol. 1999, 401, 157–161. [Google Scholar] [CrossRef]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.-E.; Kok, S.-Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nat. Cell Biol. 2015, 525, 533–537. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Z.; Qin, R.; Qiu, Y.; Wang, J.-L.; Cui, X.; Gu, L.; Zhang, X.; Guo, X.; Wang, D.; et al. Identification and Characterization of an Epi-Allele of FIE1 Reveals a Regulatory Linkage between Two Epigenetic Marks in Rice. Plant. Cell 2012, 24, 4407–4421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, J.; Cao, X.; Song, X. Epigenetic Mutation of RAV6 Affects Leaf Angle and Seed Size in Rice. Plant. Physiol. 2015, 169, 2118–2128. [Google Scholar] [CrossRef]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A. A transposon-induced epigenetic change leads to sex determination in melon. Nat. Cell Biol. 2009, 461, 1135–1138. [Google Scholar] [CrossRef]
- Miura, K.; Agetsuma, M.; Kitano, H.; Yoshimura, A.; Matsuoka, M.; Jacobsen, S.E.; Ashikari, M. A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. USA 2009, 106, 11218–11223. [Google Scholar] [CrossRef]
- Manning, K.; Tor, M.; Poole, M.; Hong, Y.; Thompson, A.; King, G.; Giovannoni, J.J.; Seymour, G. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Bender, J.; Fink, G.R. Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 1995, 83, 725–734. [Google Scholar] [CrossRef]
- Quadrana, L.; Almeida, J.; Asís, R.; Duffy, T.; Dominguez, P.G.; Bermudez, L.F.; Conti, G.; Da Silva, J.V.C.; Peralta, I.E.; Colot, V.; et al. Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 2014, 5, 4027. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.B.; Trontin, C.; Cortijo, S.; Barau, J.; Del-Bem, L.-E.; Loudet, O.; Colot, V.; Vincentz, M. Extensive Natural Epigenetic Variation at a De Novo Originated Gene. PLoS Genet. 2013, 9, e1003437. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; et al. A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M. Epigenetics and crop improvement. Trends Genet. 2013, 29, 241–247. [Google Scholar] [CrossRef]
- Springer, N.M.; Schmitz, R. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 2017, 18, 563–575. [Google Scholar] [CrossRef]
- Mondal, P.; Natesh, J.; Penta, D.; Meeran, S.M. Progress and promises of epigenetic drugs and epigenetic diets in cancer prevention and therapy: A clinical update. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Cheng, J.; Jones, P. Zebularine: A new drug for epigenetic therapy. Biochem. Soc. Trans. 2004, 32, 910–912. [Google Scholar] [CrossRef] [PubMed]
- Baránek, M.; Otmar, M.; Krečmerová, M.; Eichmeier, A.; Moudrá, J.; Mynarzová, Z. Effect of Different DNA Demethylating Agents on in vitro Cultures of Peach Rootstock GF 677. Not. Bot. Horti Agrobot. Cluj-Napoca 2019, 47. [Google Scholar] [CrossRef]
- Chen, R.; Chen, X.; Huo, W.; Zheng, S.; Lin, Y.; Lai, Z. Transcriptome analysis of azacitidine (5-AzaC)-treatment affecting the development of early somatic embryogenesis in longan. J. Hortic. Sci. Biotechnol. 2020, 96, 311–323. [Google Scholar] [CrossRef]
- Quinga, L.A.P.; Fraga, H.; Vieira, L.D.N.; Guerra, M.P. Epigenetics of long-term somatic embryogenesis in Theobroma cacao L.: DNA methylation and recovery of embryogenic potential. Plant. Cell Tissue Organ. Cult. 2017, 131, 295–305. [Google Scholar] [CrossRef]
- Fraga, H.; Vieira, L.D.N.; Caprestano, C.A.; Steinmacher, D.A.; Micke, G.A.; Spudeit, D.A.; Pescador, R.; Guerra, M.P. 5-Azacytidine combined with 2,4-D improves somatic embryogenesis of Acca sellowiana (O. Berg) Burret by means of changes in global DNA methylation levels. Plant. Cell Rep. 2012, 31, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, A.; Juzoń, K.; Krzewska, M.; Dziurka, M.; Dubas, E.; Kopeć, P.; Zieliński, K.; Żur, I. Chemically-induced DNA de-methylation alters the effectiveness of microspore embryogenesis in triticale. Plant. Sci. 2019, 287, 110189. [Google Scholar] [CrossRef]
- Solís, M.-T.; El-Tantawy, A.A.; Cano, V.; Risueño, M.C.; Testillano, P.S. 5-azacytidine promotes microspore embryogenesis initiation by decreasing global DNA methylation, but prevents subsequent embryo development in rapeseed and barley. Front. Plant. Sci. 2015, 6, 472. [Google Scholar] [CrossRef]
- Zhao, Q.; Du, Y.; Wang, H.; Rogers, H.J.; Yu, C.; Liu, W.; Zhao, M.; Xie, F. 5-Azacytidine promotes shoot regeneration during Agrobacterium-mediated soybean transformation. Plant. Physiol. Biochem. 2019, 141, 40–50. [Google Scholar] [CrossRef]
- Dennis, E.; Peacock, W. Epigenetic regulation of flowering. Curr. Opin. Plant. Biol. 2007, 10, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Peng, X.-Y.; Yu, Y.-C.; Sun, Z.-Y.; Han, L. The Effects of DNA Methylation Inhibition on Flower Development in the Dioecious Plant Salix Viminalis. Forests 2019, 10, 173. [Google Scholar] [CrossRef]
- Li, S.F.; Zhang, G.J.; Yuan, J.H.; Deng, C.L.; Lu, L.D.; Gao, W.J. Effect of 5-azaC on the growth, flowering time and sexual phenotype of spinach. Russ. J. Plant. Physiol. 2015, 62, 670–675. [Google Scholar] [CrossRef]
- Ogneva, Z.V.; Suprun, A.; Dubrovina, A.S.; Kiselev, K.V. Effect of 5-azacytidine induced DNA demethylation on abiotic stress tolerance in Arabidopsis thaliana. Plant. Prot. Sci. 2019, 55, 73–80. [Google Scholar] [CrossRef]
- Nishimura, H.; Himi, E.; Eun, C.-H.; Takahashi, H.; Qian, Q.; Tsugane, K.; Maekawa, M. Transgenerational activation of an autonomous DNA transposon, Dart1-24, by 5-azaC treatment in rice. Theor. Appl. Genet. 2019, 132, 3347–3355. [Google Scholar] [CrossRef]
- Konečná, K.; Sováková, P.; Anteková, K.; Fajkus, J.; Fojtová, M. Distinct Responses of Arabidopsis Telomeres and Transposable Elements to Zebularine Exposure. Int. J. Mol. Sci. 2021, 22, 468. [Google Scholar] [CrossRef] [PubMed]
- Boonjing, P.; Masuta, Y.; Nozawa, K.; Kato, A.; Ito, H. The effect of Zebularine on the heat-activated retrotransposon ONSEN in Arabidopsis thaliana and Vigna angularis. Genes Genet. Syst. 2020, 95, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, K.; Kikuta, Y. Nucleoside derivatives of 5-methylcytosine suppress 5-azacytidine-induced reactivation of a silent transgene in suspension-cultured tobacco cells. Plant. Biotechnol. 2021, 38, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Tyč, D.; Nocarová, E.; Sikorová, L.; Fischer, L. 5-Azacytidine mediated reactivation of silenced transgenes in potato (Solanum tuberosum) at the whole plant level. Plant. Cell Rep. 2017, 36, 1311–1322. [Google Scholar] [CrossRef]
- Verhoeven, K.J.; van Gurp, T. Transgenerational Effects of Stress Exposure on Offspring Phenotypes in Apomictic Dandelion. PLoS ONE 2012, 7, e38605. [Google Scholar] [CrossRef]
- González, A.P.R.; Preite, V.; Verhoeven, K.J.F.; Latzel, V. Transgenerational Effects and Epigenetic Memory in the Clonal Plant Trifolium repens. Front. Plant. Sci. 2018, 9, 1677. [Google Scholar] [CrossRef]
- Yang, X.; Sanchez, R.; Kundariya, H.; Maher, T.; Dopp, I.; Schwegel, R.; Virdi, K.; Axtell, M.J.; MacKenzie, S.A. Segregation of an MSH1 RNAi transgene produces heritable non-genetic memory in association with methylome reprogramming. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Castillo, A.M.; Valero-Rubira, I.; Burrell, M.; Allué, S.; Costar, M.A.; Vallés, M.P. Trichostatin A Affects Developmental Reprogramming of Bread Wheat Microspores towards an Embryogenic Route. Plants 2020, 9, 1442. [Google Scholar] [CrossRef]
- Wójcikowska, B.; Botor, M.; Morończyk, J.; Wójcik, A.; Nodzynski, T.; Karcz, J.; Gaj, M.D. Trichostatin A Triggers an Embryogenic Transition in Arabidopsis Explants via an Auxin-Related Pathway. Front. Plant. Sci. 2018, 9, 1353. [Google Scholar] [CrossRef]
- Bie, X.M.; Dong, L.; Li, X.H.; Wang, H.; Gao, X.-Q.; Li, X.G. Trichostatin A and sodium butyrate promotes plant regeneration in common wheat. Plant. Signal. Behav. 2020, 15, 1820681. [Google Scholar] [CrossRef]
- Becker, C.; Weigel, D. Epigenetic variation: Origin and transgenerational inheritance. Curr. Opin. Plant. Biol. 2012, 15, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Meyerowitz, E.M. Hypermethylated SUPERMAN Epigenetic Alleles in Arabidopsis. Science 1997, 277, 1100–1103. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Sakai, H.; Finnegan, E.; Cao, X.; Meyerowitz, E.M. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 2000, 10, 179–186. [Google Scholar] [CrossRef]
- Soppe, W.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J. The Late Flowering Phenotype of fwa Mutants Is Caused by Gain-of-Function Epigenetic Alleles of a Homeodomain Gene. Mol. Cell 2000, 6, 791–802. [Google Scholar] [CrossRef]
- Stokes, T.L.; Kunkel, B.N.; Richards, E.J. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 2002, 16, 171–182. [Google Scholar] [CrossRef]
- Saze, H.; Kakutani, T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J. 2007, 26, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Kakutani, T.; Jeddeloh, J.A.; Flowers, S.K.; Munakata, K.; Richards, E.J. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc. Natl. Acad. Sci. USA 1996, 93, 12406–12411. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Ton, J. The epigenetic machinery controlling transgenerational systemic acquired resistance. Plant. Signal. Behav. 2012, 7, 615–618. [Google Scholar] [CrossRef]
- Furci, L.; Jain, R.; Stassen, J.; Berkowitz, O.; Whelan, J.; Roquis, D.; Baillet, V.; Colot, V.; Johannes, F.; Ton, J. Identification and characterisation of hypomethylated DNA loci controlling quantitative resistance in Arabidopsis. eLife 2019, 8, e40655. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Sow, M.D.; Le Gac, A.; Fichot, R.; Lanciano, S.; Delaunay, A.; Le Jan, I.; Lesage-Descauses, M.; Citerne, S.; Caius, J.; Brunaud, V.; et al. RNAi suppression of DNA methylation affects drought stress response and genome integrity in transgenic poplar. N. Phytol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the Impact of Transgenerational Epigenetic Variation on Complex Traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Reinders, J.; Wulff, B.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef]
- Johannes, F.; Tatche, M.C. Quantitative Epigenetics Through Epigenomic Perturbation of Isogenic Lines. Genetics 2011, 188, 215–227. [Google Scholar] [CrossRef][Green Version]
- Cortijo, S.; Wardenaar, R.; Colomé-Tatché, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.-M.; Wincker, P.; et al. Mapping the Epigenetic Basis of Complex Traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Roux, F.; Tatche, M.C.; Edelist, C.; Wardenaar, R.; Guerche, P.; Hospital, F.; Colot, V.; Jansen, R.C.; Johannes, F. Genome-Wide Epigenetic Perturbation Jump-Starts Patterns of Heritable Variation Found in Nature. Genetics 2011, 188, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Kooke, R.; Johannes, F.; Wardenaar, R.; Becker, F.; Etcheverry, M.; Colot, V.; Vreugdenhil, D.; Keurentjes, J.J. Epigenetic Basis of Morphological Variation and Phenotypic Plasticity in Arabidopsis thaliana. Plant. Cell 2015, 27, 337–348. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Latzel, V.; Fischer, M.; Bossdorf, O. Understanding the evolutionary potential of epigenetic variation: A comparison of heritable phenotypic variation in epiRILs, RILs, and natural ecotypes of Arabidopsis thaliana. Heredity 2018, 121, 257–265. [Google Scholar] [CrossRef]
- Kooke, R.; Morgado, L.; Becker, F.F.M.; Van Eekelen, H.; Hazarika, R.R.; Zheng, Q.; De Vos, R.C.; Johannes, F.; Keurentjes, J.J. Epigenetic mapping of the Arabidopsis metabolome reveals mediators of the epigenotype-phenotype map. Genome Res. 2018, 29, 96–106. [Google Scholar] [CrossRef]
- Lauss, K.; Wardenaar, R.; Oka, R.; van Hulten, M.H.A.; Guryev, V.; Keurentjes, J.J.B.; Stam, M.; Johannes, F. Parental DNA Methylation States Are Associated with Heterosis in Epigenetic Hybrids. Plant. Physiol. 2017, 176, 1627–1645. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.; Reinders, J.; Bédiée, A.; Balsera, C.; Bucher, E.; Theiler, G.; Granier, C.; Paszkowski, J. Heterosis and inbreeding depression of epigenetic Arabidopsis hybrids. Nat. Plants 2015, 1, 15092. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Yuliang, A.; Wu, Y.; Chen, Y.-R.; et al. Mutation of a major CG methylase in rice causes genome-wide hypomethylation, dysregulated genome expression, and seedling lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Zaunbrecher, V.M.; Song, J.; Wendt, J.; Rosenbaum, H.; Madzima, T.F.; Sloan, A.E.; Huang, J.; et al. Genetic Perturbation of the Maize Methylome. Plant. Cell 2014, 26, 4602–4616. [Google Scholar] [CrossRef]
- Yamauchi, T.; Johzuka-Hisatomi, Y.; Terada, R.; Nakamura, I.; Iida, S. The MET1b gene encoding a maintenance DNA methyltransferase is indispensable for normal development in rice. Plant. Mol. Biol. 2014, 85, 219–232. [Google Scholar] [CrossRef]
- Xu, Y.-Z.; Arrieta-Montiel, M.P.; Virdi, K.; de Paula, W.B.; Widhalm, J.; Basset, G.J.; Davila, J.I.; Elthon, T.E.; Elowsky, C.G.; Sato, S.J.; et al. MutS HOMOLOG1 Is a Nucleoid Protein That Alters Mitochondrial and Plastid Properties and Plant Response to High Light. Plant. Cell 2011, 23, 3428–3441. [Google Scholar] [CrossRef]
- Abdelnoor, R.V.; Yule, R.; Elo, A.; Christensen, A.C.; Meyer-Gauen, G.; Mackenzie, S.A. Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc. Natl. Acad. Sci. USA 2003, 100, 5968–5973. [Google Scholar] [CrossRef]
- Virdi, K.; Laurie, J.D.; Xu, Y.-Z.; Yu, J.; Shao, M.-R.; Sanchez, R.; Kundariya, H.; Wang, D.; Riethoven, J.-J.; Wamboldt, Y.; et al. ArabidopsisMSH1 mutation alters the epigenome and produces heritable changes in plant growth. Nat. Commun. 2015, 6, 6386. [Google Scholar] [CrossRef]
- Amaral, M.N.D.; Auler, P.A.; Rossatto, T.; Barros, P.M.; Oliveira, M.M.; Braga, E.J.B. Long-term somatic memory of salinity unveiled from physiological, biochemical and epigenetic responses in two contrasting rice genotypes. Physiol. Plant. 2020, 170. [Google Scholar] [CrossRef] [PubMed]
- Srikant, T.; Drost, H.-G. How Stress Facilitates Phenotypic Innovation through Epigenetic Diversity. Front. Plant. Sci. 2021, 11. [Google Scholar] [CrossRef]
- Sampaio, B.L.; Edrada-Ebel, R.; Da Costa, F. Effect of the environment on the secondary metabolic profile of Tithonia diversifolia: A model for environmental metabolomics of plants. Sci. Rep. 2016, 6, 29265. [Google Scholar] [CrossRef]
- Borges, C.V.; Minatel, I.O.; Gomez-Gomez, H.A.; Lima, G.P.P. Medicinal Plants: Influence of Environmental Factors on the Content of Secondary Metabolites. In Medicinal Plants and Environmental Challenges; Ghorbanpour, M., Varma, A., Eds.; Springer: Cham, Switzerland, 2017; pp. 259–277. [Google Scholar] [CrossRef]
- Luo, X.; He, Y. Experiencing winter for spring flowering: A molecular epigenetic perspective on vernalization. J. Integr. Plant. Biol. 2019, 62, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Guo, W.; Huang, Y. Genetic and epigenetic regulation of phenotypic variation in invasive plants—Linking research trends towards a unified framework. NeoBiota 2019, 49, 77–103. [Google Scholar] [CrossRef]
- Marin, P.; Genitoni, J.; Barloy, D.; Maury, S.; Gibert, P.; Ghalambor, C.K.; Vieira, C. Biological invasion: The influence of the hidden side of the (epi)genome. Funct. Ecol. 2019, 34, 385–400. [Google Scholar] [CrossRef]
- Mozgova, I.; Mikulski, P.; Pecinka, A.; Farrona, S. Epigenetic Mechanisms of Abiotic Stress Response and Memory in Plants. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications: Transcriptional Regulation and Chromatin Remodelling in Plants; Alvarez-Venegas, R., De-la-Peña, C., Casas-Mollano, J.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–64. ISBN 9783030147600. [Google Scholar]
- Roberts, M.R.; López Sánchez, A. Plant Epigenetic Mechanisms in Response to Biotic Stress. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications: Transcriptional Regulation and Chromatin Remodelling in Plants; Alvarez-Venegas, R., De-la-Peña, C., Casas-Mollano, J.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 65–113. ISBN 9783030147600. [Google Scholar]
- Iwasaki, M. Chromatin resetting mechanisms preventing transgenerational inheritance of epigenetic states. Front. Plant. Sci. 2015, 6, 380. [Google Scholar] [CrossRef] [PubMed]
- Varotto, S.; Tani, E.; Abraham, E.; Krugman, T.; Kapazoglou, A.; Melzer, R.; Radanović, A.; Miladinović, D. Epigenetics: Possible applications in climate-smart crop breeding. J. Exp. Bot. 2020, 71, 5223–5236. [Google Scholar] [CrossRef]
- Molinier, J.; Ries, G.; Zipfel, C.; Hohn, B. Transgeneration memory of stress in plants. Nat. Cell Biol. 2006, 442, 1046–1049. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Genetic and Epigenetic Effects of Plant–Pathogen Interactions: An Evolutionary Perspective. Mol. Plant. 2011, 4, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Paszkowski, J.; Grossniklaus, U. Selected aspects of transgenerational epigenetic inheritance and resetting in plants. Curr. Opin. Plant. Biol. 2011, 14, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Arora, R. Priming memory invokes seed stress-tolerance. Environ. Exp. Bot. 2013, 94, 33–45. [Google Scholar] [CrossRef]
- Oberkofler, V.; Pratx, L.; Bäurle, I. Epigenetic regulation of abiotic stress memory: Maintaining the good things while they last. Curr. Opin. Plant. Biol. 2021, 61, 102007. [Google Scholar] [CrossRef]
- Bäurle, I. Can’t remember to forget you: Chromatin-based priming of somatic stress responses. Semin. Cell Dev. Biol. 2018, 83, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollunder, J.; Meins, F.M., Jr.; Kovalchuk, I. Transgenerational Adaptation of Arabidopsis to Stress Requires DNA Methylation and the Function of Dicer-Like Proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Sánchez, A.L.; Pascual-Pardo, D.; Furci, L.; Roberts, M.R.; Ton, J. Costs and Benefits of Transgenerational Induced Resistance in Arabidopsis. Front. Plant. Sci. 2021, 12, 248. [Google Scholar] [CrossRef]
- Forestan, C.; Farinati, S.; Zambelli, F.; Pavesi, G.; Rossi, V.; Varotto, S. Epigenetic signatures of stress adaptation and flowering regulation in response to extended drought and recovery in Zea mays. Plant. Cell Environ. 2019, 43, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.; Miao, Y.; Xu, L.; Zhang, Y.; Yuan, C.; Wang, J.; Zhuang, T.; Lin, X.; Jiang, L.; Wang, N.; et al. Transgenerational memory of gene expression changes induced by heavy metal stress in rice (Oryza sativa L.). BMC Plant. Biol. 2019, 19, 282. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.; Ribeyre, Z.; Vigneaud, J.; Sow, M.D.; Fichot, R.; Messier, C.; Pinto, G.; Nolet, P.; Maury, S. Advances and Promises of Epigenetics for Forest Trees. Forests 2020, 11, 976. [Google Scholar] [CrossRef]
- Baránek, M.; Čechová, J.; Raddová, J.; Holleinová, V.; Ondrušíková, E.; Pidra, M. Dynamics and Reversibility of the DNA Methylation Landscape of Grapevine Plants (Vitis vinifera) Stressed by In Vitro Cultivation and Thermotherapy. PLoS ONE 2015, 10, e0126638. [Google Scholar] [CrossRef]
- Crisp, P.A.; Ganguly, D.; Eichten, S.R.; Borevitz, J.; Pogson, B.J. Reconsidering plant memory: Intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2016, 2, e1501340. [Google Scholar] [CrossRef]
- Torti, S.; Schlesier, R.; Thümmler, A.; Bartels, D.; Römer, P.; Koch, B.; Werner, S.; Panwar, V.; Kanyuka, K.; von Wirén, N.; et al. Transient reprogramming of crop plants for agronomic performance. Nat. Plants 2021, 7, 159–171. [Google Scholar] [CrossRef]
- Papikian, A.; Liu, W.; Gallego-Bartolome, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guarino, F.; Cicatelli, A.; Brundu, G.; Heinze, B.; Castiglione, S. Epigenetic Diversity of Clonal White Poplar (Populus alba L.) Populations: Could Methylation Support the Success of Vegetative Reproduction Strategy? PLoS ONE 2015, 10, e0131480. [Google Scholar] [CrossRef] [PubMed]
- Schönberger, B.; Chen, X.; Mager, S.; Ludewig, U. Site-Dependent Differences in DNA Methylation and Their Impact on Plant Establishment and Phosphorus Nutrition in Populus trichocarpa. PLoS ONE 2016, 11, e0168623. [Google Scholar] [CrossRef]
- Broeck, A.V.; Cox, K.; Brys, R.; Castiglione, S.; Cicatelli, A.; Guarino, F.; Heinze, B.; Steenackers, M.; Mijnsbrugge, K.V. Variability in DNA Methylation and Generational Plasticity in the Lombardy Poplar, a Single Genotype Worldwide Distributed Since the Eighteenth Century. Front. Plant. Sci. 2018, 9, 1635. [Google Scholar] [CrossRef]
- Pereira, W.J.; Pappas, M.D.C.R.; Campoe, O.C.; Stape, J.L.; Grattapaglia, D.; Pappas, G.J.P., Jr. Patterns of DNA methylation changes in elite Eucalyptus clones across contrasting environments. For. Ecol. Manag. 2020, 474, 118319. [Google Scholar] [CrossRef]
- Xie, H.; Konate, M.; Sai, N.; Tesfamicael, K.G.; Cavagnaro, T.; Gilliham, M.; Breen, J.; Metcalfe, A.; Stephen, J.R.; De Bei, R.; et al. Global DNA Methylation Patterns Can Play a Role in Defining Terroir in Grapevine (Vitis vinifera cv. Shiraz). Front. Plant. Sci. 2017, 8, 1860. [Google Scholar] [CrossRef]
- Baránková, K.; Nebish, A.; Tříska, J.; Raddová, J.; Baránek, M. Comparison of DNA methylation landscape between Czech and Armenian vineyards show their unique character and increased diversity. Czech. J. Genet. Plant. Breed. 2021, 57, 67–75. [Google Scholar] [CrossRef]
- Varela, A.; Ibañez, V.N.; Alonso, R.; Zavallo, D.; Asurmendi, S.; Talquenca, S.G.; Marfil, C.F.; Berli, F.J. Vineyard environments influence Malbec grapevine phenotypic traits and DNA methylation patterns in a clone-dependent way. Plant. Cell Rep. 2020, 40, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant. Sci. 2010, 15, 57–71. [Google Scholar] [CrossRef]
- Schnable, P.S.; Springer, N.M. Progress Toward Understanding Heterosis in Crop Plants. Annu. Rev. Plant. Biol. 2013, 64, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Greaves, I.; Groszmann, M.; Ying, H.; Taylor, J.; Peacock, W.J.; Dennis, E.S. Trans Chromosomal Methylation in Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y.; et al. Genome-Wide Analysis of DNA Methylation and Gene Expression Changes in Two Arabidopsis Ecotypes and Their Reciprocal Hybrids. Plant. Cell 2012, 24, 875–892. [Google Scholar] [CrossRef]
- Rigal, M.; Becker, C.; Pélissier, T.; Pogorelcnik, R.; Devos, J.; Ikeda, Y.; Weigel, D.; Mathieu, O. Epigenome confrontation triggers immediate reprogramming of DNA methylation and transposon silencing in Arabidopsis thaliana F1 epihybrids. Proc. Natl. Acad. Sci. USA 2016, 113, E2083–E2092. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, D.; Lang, Z.; He, L.; Yang, L.; Zeng, L.; Li, Y.; Zhao, C.; Huang, H.; Zhang, H.; et al. Methylation interactions in Arabidopsis hybrids require RNA-directed DNA methylation and are influenced by genetic variation. Proc. Natl. Acad. Sci. USA 2016, 113, E4248–E4256. [Google Scholar] [CrossRef]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global Epigenetic and Transcriptional Trends among Two Rice Subspecies and Their Reciprocal Hybrids. Plant. Cell 2010, 22, 17–33. [Google Scholar] [CrossRef]
- Ma, X.; Xing, F.; Jia, Q.; Zhang, Q.; Hu, T.; Wu, B.; Shao, L.; Zhao, Y.; Zhang, Q.; Zhou, D.-X. Parental variation in CHG methylation is associated with allelic-specific expression in elite hybrid rice. Plant. Physiol. 2021, 186, 1025–1041. [Google Scholar] [CrossRef]
- Sinha, P.; Singh, V.; Saxena, R.K.; Kale, S.M.; Li, Y.; Garg, V.; Meifang, T.; Khan, A.W.; Kim, K.D.; Chitikineni, A.; et al. Genome-wide analysis of epigenetic and transcriptional changes associated with heterosis in pigeonpea. Plant. Biotechnol. J. 2020, 18, 1697–1710. [Google Scholar] [CrossRef]
- Li, H.; Yuan, J.; Wu, M.; Han, Z.; Li, L.; Jiang, H.; Jia, Y.; Han, X.; Liu, M.; Sun, D.; et al. Transcriptome and DNA methylome reveal insights into yield heterosis in the curds of broccoli (Brassica oleracea L var. italic). BMC Plant. Biol. 2018, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Sun, S.; Hua, S.; Shen, E.; Ye, C.; Cai, D.; Timko, M.P.; Zhu, Q.; Fan, L. Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant. J. 2017, 91, 874–893. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.-L.; Meyers, B.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Xu, T.; Srivastava, A.K.; Wang, D.; Zeng, L.; Yang, L.; He, L.; Zhang, H.; Zheng, Z.; et al. The chromatin remodeler DDM1 promotes hybrid vigor by regulating salicylic acid metabolism. Cell Discov. 2016, 2, 16027. [Google Scholar] [CrossRef] [PubMed]
- Groszmann, M.; Greaves, I.; Albert, N.; Fujimoto, R.; Helliwell, C.; Dennis, E.; Peacock, W.J. Epigenetics in plants—Vernalisation and hybrid vigour. Biochim. Biophys. Acta BBA Bioenerg. 2011, 1809, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.; Thiemann, A.; Schrag, T.A.; Rybka, D.; Melchinger, A.E.; Frisch, M.; Scholten, S. Small RNA-based prediction of hybrid performance in maize. BMC Genom. 2018, 19, 371. [Google Scholar] [CrossRef]
- Crisp, P.A.; Hammond, R.; Zhou, P.; Vaillancourt, B.; Lipzen, A.; Daum, C.; Barry, K.; De Leon, N.; Buell, C.R.; Kaeppler, S.M.; et al. Variation and Inheritance of Small RNAs in Maize Inbreds and F1 Hybrids. Plant. Physiol. 2019, 182, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, P.V.; Dunn, R.M.; Santos, B.; Bassett, A.; Baulcombe, D. Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J. 2011, 31, 257–266. [Google Scholar] [CrossRef]
- Kenan-Eichler, M.; Leshkowitz, D.; Tal, L.; Noor, E.; Melamed-Bessudo, C.; Feldman, M.; Levy, A.A. Wheat Hybridization and Polyploidization Results in Deregulation of Small RNAs. Genetics 2011, 188, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Peng, Y.; Wei, X.; Dai, Y.; Yuan, D.; Lu, Y.; Pan, Y.; Zhu, Z. Small RNAs as important regulators for the hybrid vigour of super-hybrid rice. J. Exp. Bot. 2014, 65, 5989–6002. [Google Scholar] [CrossRef]
- Li, Y.; Varala, K.; Moose, S.P.; Hudson, M.E. The Inheritance Pattern of 24 nt siRNA Clusters in Arabidopsis Hybrids Is Influenced by Proximity to Transposable Elements. PLoS ONE 2012, 7, e47043. [Google Scholar] [CrossRef]
- He, G.; Chen, B.; Wang, X.; Li, X.; Li, J.; He, H.; Yang, M.; Lu, L.; Qi, Y.; Wang, X.; et al. Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol. 2013, 14, R57. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Lu, J.; Tian, L.; Ramachandran, V.; Kasschau, K.D.; Chapman, E.J.; Carrington, J.C.; Chen, X.; Wang, X.-J.; Chen, Z.J. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. USA 2009, 106, 17835–17840. [Google Scholar] [CrossRef]
- Groszmann, M.; Greaves, I.; Fujimoto, R.; Peacock, W.J.; Dennis, E.S. The role of epigenetics in hybrid vigour. Trends Genet. 2013, 29, 684–690. [Google Scholar] [CrossRef]
- Groszmann, M.; Gonzalez-Bayon, R.; Lyons, R.L.; Greaves, I.; Kazan, K.; Peacock, W.J.; Dennis, E.S. Hormone-regulated defense and stress response networks contribute to heterosis in Arabidopsis F1 hybrids. Proc. Natl. Acad. Sci. USA 2015, 112, E6397–E6406. [Google Scholar] [CrossRef] [PubMed]
- Groszmann, M.; Gonzalez-Bayon, R.; Greaves, I.; Wang, L.; Huen, A.K.; Peacock, W.J.; Dennis, E.S. Intraspecific Arabidopsis Hybrids Show Different Patterns of Heterosis Despite the Close Relatedness of the Parental Genomes. Plant. Physiol. 2014, 166, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Hauben, M.; Haesendonckx, B.; Standaert, E.; Van Der Kelen, K.; Azmi, A.; Akpo, H.; Van Breusegem, F.; Guisez, Y.; Bots, M.; Lambert, B.; et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. USA 2009, 106, 20109–20114. [Google Scholar] [CrossRef]
- Seifert, F.; Thiemann, A.; Grant-Downton, R.; Edelmann, S.; Rybka, D.; Schrag, T.A.; Frisch, M.; Dickinson, H.G.; Melchinger, A.E.; Scholten, S. Parental Expression Variation of Small RNAs Is Negatively Correlated with Grain Yield Heterosis in a Maize Breeding Population. Front. Plant. Sci. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.-S.; Osborne, E.J.; Casale, F.P.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjalmsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Xia, W.; Li, R.; Wang, J.; Shao, M.; Feng, J.; King, G.; Meng, J. Epigenetic QTL Mapping in Brassica napus. Genetics 2011, 189, 1093–1102. [Google Scholar] [CrossRef]
- Duan, Y.; Qian, J.; Sun, Y.; Yi, Z.; Yan, M. Construction of methylation linkage map based on MSAP and SSR markers in Sorghum bicolor (L.). IUBMB Life 2009, 61, 663–669. [Google Scholar] [CrossRef]
- Alonso, C.; Medrano, M.; Pérez, R.; Canto, A.; Parra-Tabla, V.; Herrera, C.M. Interspecific variation across angiosperms in global DNA methylation: Phylogeny, ecology and plant features in tropical and Mediterranean communities. N. Phytol. 2019, 224, 949–960. [Google Scholar] [CrossRef]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic Predictor of Age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- McCartney, D.L.; Hillary, R.F.; Stevenson, A.J.; Ritchie, S.J.; Walker, R.M.; Zhang, Q.; Morris, S.W.; Bermingham, M.L.; Campbell, A.; Murray, A.D.; et al. Epigenetic prediction of complex traits and death. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Hu, Y.; Morota, G.; Rosa, G.J.M.; Gianola, D. Prediction of Plant Height in Arabidopsis thaliana Using DNA Methylation Data. Genetics 2015, 201, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Regulski, M.; Lu, Z.; Kendall, J.; Donoghue, M.T.; Reinders, J.; Llaca, V.; Deschamps, S.; Smith, A.; Levy, D.; McCombie, W.R.; et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013, 23, 1651–1662. [Google Scholar] [CrossRef]
- Sung, S.; Amasino, R.M. Vernalization in Arabidopsis thaliana is mediated by the PHD finger protein VIN3. Nat. Cell Biol. 2004, 427, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Trap-Gentil, M.-V.; Hébrard, C.; Lafon-Placette, C.; Delaunay, A.; Hagège, D.; Joseph, C.; Brignolas, F.; Lefebvre, M.; Barnes, S.; Maury, S. Time course and amplitude of DNA methylation in the shoot apical meristem are critical points for bolting induction in sugar beet and bolting tolerance between genotypes. J. Exp. Bot. 2011, 62, 2585–2597. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Makarevich, G. Epigenetic mechanisms governing seed development in plants. EMBO Rep. 2006, 7, 1223–1227. [Google Scholar] [CrossRef]
- Satgé, C.; Moreau, S.; Sallet, E.; Lefort, G.; Auriac, M.-C.; Remblière, C.; Cottret, L.; Gallardo, K.; Noirot, C.; Jardinaud, M.-F.; et al. Reprogramming of DNA methylation is critical for nodule development in Medicago truncatula. Nat. Plants 2016, 2, 16166. [Google Scholar] [CrossRef]
- Angel, A.; Song, J.; Yang, H.; Questa, J.; Dean, C.; Howard, M. Vernalizing cold is registered digitally at FLC. Proc. Natl. Acad. Sci. USA 2015, 112, 4146–4151. [Google Scholar] [CrossRef]
- Gallusci, P.; Dai, Z.; Génard, M.; Gauffretau, A.; Leblanc-Fournier, N.; Richard-Molard, C.; Vile, D.; Brunel-Muguet, S. Epigenetics for Plant Improvement: Current Knowledge and Modeling Avenues. Trends Plant. Sci. 2017, 22, 610–623. [Google Scholar] [CrossRef]
- Wang, E.; Robertson, M.; Hammer, G.; Carberry, P.; Holzworth, D.; Meinke, H.; Chapman, S.; Hargreaves, J.; Huth, N.; McLean, G. Development of a generic crop model template in the cropping system model APSIM. Eur. J. Agron. 2002, 18, 121–140. [Google Scholar] [CrossRef]
- Asseng, S.; Zhu, Y.; Wang, E.; Zhang, W. Chapter 20—Crop modeling for climate change impact and adaptation. In Crop Physiology, 2nd ed.; Sadras, V.O., Calderini, D.F., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 505–546. ISBN 9780124171046. [Google Scholar]
- Martre, P.; Wallach, D.; Asseng, S.; Ewert, F.; Jones, J.W.; Rötter, R.; Boote, K.; Ruane, A.C.; Thorburn, P.J.; Cammarano, D.; et al. Multimodel ensembles of wheat growth: Many models are better than one. Glob. Chang. Biol. 2014, 21, 911–925. [Google Scholar] [CrossRef]
- Chenu, K.; Porter, J.R.; Martre, P.; Basso, B.; Chapman, S.; Ewert, F.; Bindi, M.; Asseng, S. Contribution of Crop Models to Adaptation in Wheat. Trends Plant. Sci. 2017, 22, 472–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, F.; Jiang, D. Priming: A promising strategy for crop production in response to future climate. J. Integr. Agric. 2017, 16, 2709–2716. [Google Scholar] [CrossRef]
- Tao, F.; Rötter, R.; Palosuo, T.; Díaz-Ambrona, C.G.H.; Minguez, M.I.; Semenov, M.; Kersebaum, K.C.; Nendel, C.; Specka, X.; Hoffmann, H.; et al. Contribution of crop model structure, parameters and climate projections to uncertainty in climate change impact assessments. Glob. Chang. Biol. 2017, 24, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Martre, P.; Ewert, F.; Porter, J.R.; Challinor, A.J.; Müller, C.; Ruane, A.C.; Waha, K.; Thorburn, P.J.; Aggarwal, P.K.; et al. Global wheat production with 1.5 and 2.0 °C above pre-industrial warming. Glob. Chang. Biol. 2018, 25, 1428–1444. [Google Scholar] [CrossRef] [PubMed]
- Holzworth, D.P.; Huth, N.I.; Devoil, P.G.; Zurcher, E.; Herrmann, N.; McLean, G.; Chenu, K.; van Oosterom, E.J.; Snow, V.; Murphy, C.; et al. APSIM—Evolution towards a new generation of agricultural systems simulation. Environ. Model. Softw. 2014, 62, 327–350. [Google Scholar] [CrossRef]
- Brisson, N.; Gary, C.; Justes, E.; Roche, R.; Mary, B.; Ripoche, D.; Zimmer, D.; Sierra, J.; Bertuzzi, P.; Burger, P.; et al. An overview of the crop model stics. Eur. J. Agron. 2003, 18, 309–332. [Google Scholar] [CrossRef]
- Penning de Vries, F.W.; Van Laar, H.H. Simulation of Plant. Growth and Crop. Production. Simulation Monographs, Pudoc, Wageningen; Centre for Agricultural Publishing and Documentation: Wageningen, The Netherlands, 1982; Volume 308. [Google Scholar]
- Jones, J.W.; Tsuji, G.Y.; Hoogenboom, G.; Hunt, L.A.; Thornton, P.K.; Wilkens, P.W.; Imamura, D.T.; Bowen, W.T.; Singh, U. Decision support system for agrotechnology transfer: DSSAT v3. In Understanding Options for Agricultural Production; Tsuji, G.Y., Hoogenboom, G., Thornton, P.K., Eds.; Systems Approaches for Sustainable Agricultural Development; Springer: Dordrecht, The Netherlands, 1998; pp. 157–177. ISBN 9789401736244. [Google Scholar]
- Boote, K.J.; Jones, J.W.; Hoogenboom, G.; Pickering, N.B. The CROPGRO model for grain legumes. In Understanding Options for Agricultural Production; Tsuji, G.Y., Hoogenboom, G., Thornton, P.K., Eds.; Systems Approaches for Sustainable Agricultural Development; Springer: Dordrecht, The Netherlands, 1998; pp. 99–128. ISBN 9789401736244. [Google Scholar]
- King, G.J.; Amoah, S.; Kurup, S. Exploring and exploiting epigenetic variation in crops. Genome 2010, 53, 856–868. [Google Scholar] [CrossRef]
- Vriet, C.; Hennig, L.; Laloi, C. Stress-induced chromatin changes in plants: Of memories, metabolites and crop improvement. Cell. Mol. Life Sci. 2015, 72, 1261–1273. [Google Scholar] [CrossRef]
- Giovannoni, J. Harnessing epigenome modifications for better crops. J. Exp. Bot. 2016, 67, 2535–2537. [Google Scholar] [CrossRef] [PubMed]
- Sundström, J.F.; Albihn, A.; Boqvist, S.; Ljungvall, K.; Marstorp, H.; Martiin, C.; Nyberg, K.; Vågsholm, I.; Yuen, J.; Magnusson, U. Future threats to agricultural food production posed by environmental degradation, climate change, and animal and plant diseases—A risk analysis in three economic and climate settings. Food Secur. 2014, 6, 201–215. [Google Scholar] [CrossRef]
- Kumar, M. Impact of climate change on crop yield and role of model for achieving food security. Environ. Monit. Assess. 2016, 188. [Google Scholar] [CrossRef] [PubMed]
- Rötter, R.; Carter, T.R.; Olesen, J.E.; Porter, J.R. Crop–climate models need an overhaul. Nat. Clim. Chang. 2011, 1, 175–177. [Google Scholar] [CrossRef]
- Brunel-Muguet, S.; D’Hooghe, P.; Bataillã, M.-P.; Larrã, C.; Kim, T.-H.; Trouverie, J.; Avice, J.-C.; Etienne, P.; DãRr, C.; Bataillé, M.-P.; et al. Heat stress during seed filling interferes with sulfur restriction on grain composition and seed germination in oilseed rape (Brassica napus L.). Front. Plant. Sci. 2015, 6, 213. [Google Scholar] [CrossRef]
- Hatzig, S.V.; Nuppenau, J.-N.; Snowdon, R.J.; Schießl, S.V. Drought stress has transgenerational effects on seeds and seedlings in winter oilseed rape (Brassica napus L.). BMC Plant. Biol. 2018, 18, 297. [Google Scholar] [CrossRef]
- Gevers, C.; Van Rijswick, H.F.; Swart, J. Peasant Seeds in France: Fostering A More Resilient Agriculture. Sustainability 2019, 11, 3014. [Google Scholar] [CrossRef]
- Dürr, C.; Aubertot, J.-N.; Richard, G.; Dubrulle, P.; Duval, Y.; Boiffin, J. Simple. Soil Sci. Soc. Am. J. 2001, 65, 414–423. [Google Scholar] [CrossRef]
- Bradford, K.J. Applications of hydrothermal time to quantifying and modeling seed germination and dormancy. Weed Sci. 2002, 50, 248–260. [Google Scholar] [CrossRef]
- Eichten, S.R.; Schmitz, R.J.; Springer, N.M. Epigenetics: Beyond Chromatin Modifications and Complex Genetic Regulation. Plant. Physiol. 2014, 165, 933–947. [Google Scholar] [CrossRef]
- Díaz-Sala, C. Molecular Dissection of the Regenerative Capacity of Forest Tree Species: Special Focus on Conifers. Front. Plant. Sci. 2019, 9, 1943. [Google Scholar] [CrossRef]
- Ibáñez, S.; Carneros, E.; Testillano, P.; Pérez-Pérez, J. Advances in Plant Regeneration: Shake, Rattle and Roll. Plants 2020, 9, 897. [Google Scholar] [CrossRef]
- Ghosh, A.; Igamberdiev, A.U.; Debnath, S.C. Tissue culture-induced DNA methylation in crop plants: A review. Mol. Biol. Rep. 2021, 48, 823–841. [Google Scholar] [CrossRef]
- Fehér, A. Somatic embryogenesis—Stress-induced remodeling of plant cell fate. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1849, 385–402. [Google Scholar] [CrossRef]
- Testillano, P.S. Microspore embryogenesis: Targeting the determinant factors of stress-induced cell reprogramming for crop improvement. J. Exp. Bot. 2019, 70, 2965–2978. [Google Scholar] [CrossRef]
- Maluszynski, M.; Kasha, K.; Forster, B.P.; Szarejko, I. Doubled Haploid Production in Crop Plants: A Manual; Springer Science & Business Media: Cham, Switzerland, 2003; ISBN 9781402015441. [Google Scholar]
- Yakovlev, I.A.; Lee, Y.; Rotter, B.; Olsen, J.E.; Skrøppa, T.; Johnsen, Ø.; Fossdal, C.G. Temperature-dependent differential transcriptomes during formation of an epigenetic memory in Norway spruce embryogenesis. Tree Genet. Genomes 2014, 10, 355–366. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Carneros, E.; Lee, Y.; Olsen, J.E.; Fossdal, C.G. Transcriptional profiling of epigenetic regulators in somatic embryos during temperature induced formation of an epigenetic memory in Norway spruce. Planta 2016, 243, 1237–1249. [Google Scholar] [CrossRef]
- Ede-La-Peña, C.; Nic-Can, G.I.; Galaz-Ávalos, R.M.; Eavilez-Montalvo, R.; Loyola-Vargas, V.M. The role of chromatin modifications in somatic embryogenesis in plants. Front. Plant. Sci. 2015, 6, 635. [Google Scholar] [CrossRef]
- Corredoira, E.; Cano, V.; Bárány, I.; Solís, M.-T.; Rodríguez, H.; Vieitez, A.-M.; Risueño, M.C.; Testillano, P.S. Initiation of leaf somatic embryogenesis involves high pectin esterification, auxin accumulation and DNA demethylation in Quercus alba. J. Plant. Physiol. 2017, 213, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Testillano, P.S.; Solís, M.-T.; Risueño, M.C. The 5-methyl-deoxy-cytidine (5mdC) localization to reveal in situ the dynamics of DNA methylation chromatin pattern in a variety of plant organ and tissue cells during development. Physiol. Plant. 2012, 149, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Testillano, P.S.; Risueño, M.C. Detection of Epigenetic Modifications During Microspore Embryogenesis: Analysis of DNA Methylation Patterns Dynamics. In In Vitro Embryogenesis in Higher Plants; Germana, M.A., Lambardi, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 491–502. ISBN 9781493930616. [Google Scholar]
- El-Tantawy, A.A.; Solís, M.-T.; Risueño, M.C.; Testillano, P.S. Changes in DNA Methylation Levels and Nuclear Distribution Patterns after Microspore Reprogramming to Embryogenesis in Barley. Cytogenet. Genome Res. 2014, 143, 200–208. [Google Scholar] [CrossRef]
- Berenguer, E.; Bárány, I.; Solís, M.-T.; Pérez-Pérez, Y.; Risueño, M.C.; Testillano, P.S. Inhibition of Histone H3K9 Methylation by BIX-01294 Promotes Stress-Induced Microspore Totipotency and Enhances Embryogenesis Initiation. Front. Plant. Sci. 2017, 8, 1161. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sanz, H.; Moreno-Romero, J.; Solís, M.-T.; Köhler, C.; Risueño, M.C.; Testillano, P.S. Changes in Histone Methylation and Acetylation during Microspore Reprogramming to Embryogenesis Occur Concomitantly with BnHKMT and BnHAT Expression and Are Associated with Cell Totipotency, Proliferation, and Differentiation in Brassica napus. Cytogenet. Genome Res. 2014, 143, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Soriano, M.; Cordewener, J.; Muino, J.M.; Riksen, T.; Fukuoka, H.; Angenent, G.C.; Boutilier, K. The Histone Deacetylase Inhibitor Trichostatin A Promotes Totipotency in the Male Gametophyte. Plant. Cell 2014, 26, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Onder, T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.; Cahan, P.; Mancarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nat. Cell Biol. 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Horstman, A.; Bemer, M.; Boutilier, K. A transcriptional view on somatic embryogenesis. Regeneration 2017, 4, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, A.M.; Wójcikowska, B.; Gaj, M.D. Current Perspectives on the Auxin-Mediated Genetic Network that Controls the Induction of Somatic Embryogenesis in Plants. Int. J. Mol. Sci. 2020, 21, 1333. [Google Scholar] [CrossRef]
- Su, Y.H.; Tang, L.P.; Zhao, X.Y.; Zhang, X.S. Plant cell totipotency: Insights into cellular reprogramming. J. Integr. Plant. Biol. 2020, 63, 228–243. [Google Scholar] [CrossRef]
- Hauser, A.-T.; Robaa, D.; Jung, M. Epigenetic small molecule modulators of histone and DNA methylation. Curr. Opin. Chem. Biol. 2018, 45, 73–85. [Google Scholar] [CrossRef]
- Kim, Y.; Jeong, J.; Choi, D. Small-molecule-mediated reprogramming: A silver lining for regenerative medicine. Exp. Mol. Med. 2020, 52, 213–226. [Google Scholar] [CrossRef]
- Osorio-Montalvo, P.; Sáenz-Carbonell, L.; De-La-Peña, C. 5-Azacytidine: A Promoter of Epigenetic Changes in the Quest to Improve Plant Somatic Embryogenesis. Int. J. Mol. Sci. 2018, 19, 3182. [Google Scholar] [CrossRef] [PubMed]
- Carneros, E.; Pérez-Pérez, Y.; Ivett Bárány, I.; Pintos, B.; Gómez-Garay, A.; Gómez-Garay, M.; Testillano, P. Dynamics of DNA Methylation and Effects of De-Methylating Agents on Somatic Embryogenesis of Quercus Suber L. In Proceedings of the Clonal Trees in the Bioeconomy Age: Opportunities and Challenges, Coimbra, Portugal, 5 April 2019. [Google Scholar]
- Maxwell, M. Sanatech Seed Launches World’s First GE Tomato. Available online: http://www.fruitnet.com/eurofruit/article/184662/sanatech-seed-launches-worlds-first-ge-tomato (accessed on 28 July 2021).
- Gallego-Bartolome, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.-C.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell 2019, 176, 1068–1082.e19. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. N. Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nat. Cell Biol. 2014, 507, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Bartolome, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, D.-L.; Huang, H.; Zhang, G.; He, L.; Pang, J.; Lozano-Durán, R.; Lang, Z.; Zhu, J.-K. Epigenetic memory marks determine epiallele stability at loci targeted by de novo DNA methylation. Nat. Plants 2020, 6, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular Mechanism of Action of Plant DRM De Novo DNA Methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Steward, N.; Ito, M.; Yamaguchi, Y.; Koizumi, N.; Sano, H. Periodic DNA Methylation in Maize Nucleosomes and Demethylation by Environmental Stress. J. Biol. Chem. 2002, 277, 37741–37746. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhang, T.; Stelly, D.; Chen, Z.J. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, T.; Li, S.; He, Q.; Yang, Z.; Zhang, W.; Gan, Y.; Sun, P.; Xiang, G.; Zhang, H.; et al. The Methylation Patterns and Transcriptional Responses to Chilling Stress at the Seedling Stage in Rice. Int. J. Mol. Sci. 2019, 20, 5089. [Google Scholar] [CrossRef]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef]
- Qian, Y.; Hu, W.; Liao, J.; Zhang, J.; Ren, Q. The Dynamics of DNA methylation in the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Biochem. Biophys. Res. Commun. 2019, 512, 742–749. [Google Scholar] [CrossRef]
- Pandey, G.; Yadav, C.B.; Sahu, P.P.; Muthamilarasan, M.; Prasad, M. Salinity induced differential methylation patterns in contrasting cultivars of foxtail millet (Setaria italica L.). Plant. Cell Rep. 2016, 36, 759–772. [Google Scholar] [CrossRef]
- Komivi, D.; Marie, A.M.; Rong, Z.; Qi, Z.; Mei, Y.; Ndiaga, C.; Diaga, D.; Linhai, W.; Xiurong, Z. The contrasting response to drought and waterlogging is underpinned by divergent DNA methylation programs associated with transcript accumulation in sesame. Plant. Sci. 2018, 277, 207–217. [Google Scholar] [CrossRef]
- Kou, H.; Li, Y.; Song, X.; Ou, X.; Xing, S.; Ma, J.; Von Wettstein, D.; Liu, B. Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J. Plant. Physiol. 2011, 168, 1685–1693. [Google Scholar] [CrossRef]
- Wang, J.; Niu, B.; Huang, J.; Wang, H.; Yang, X.; Dong, A.; Makaroff, C.; Ma, H.; Wang, Y. The PHD Finger Protein MMD1/DUET Ensures the Progression of Male Meiotic Chromosome Condensation and Directly Regulates the Expression of the Condensin Gene CAP-D3. Plant. Cell 2016, 28, 1894–1909. [Google Scholar] [CrossRef] [PubMed]
- Catoni, M.; Griffiths, J.; Becker, C.; Zabet, N.R.; Bayon, C.; Dapp, M.; Lieberman-Lazarovich, M.; Weigel, D.; Paszkowski, J. DNA sequence properties that predict susceptibility to epiallelic switching. EMBO J. 2017, 36, 617–628. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.-L.; Meyers, B.; Jacobsen, S.E. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2013, 2, e00354. [Google Scholar] [CrossRef] [PubMed]
- Dubrovina, A.S.; Kiselev, K.V. Exogenous RNAs for Gene Regulation and Plant Resistance. Int. J. Mol. Sci. 2019, 20, 2282. [Google Scholar] [CrossRef] [PubMed]
- Que, Q.; Chilton, M.-D.M.; Elumalai, S.; Zhong, H.; Dong, S.; Shi, L. Repurposing Macromolecule Delivery Tools for Plant Genetic Modification in the Era of Precision Genome Engineering. In Transgenic Plants: Methods and Protocols; Kumar, S., Barone, P., Smith, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1864, pp. 3–18. [Google Scholar] [CrossRef]
- Perrone, A.; Martinelli, F. Plant stress biology in epigenomic era. Plant. Sci. 2019, 294, 110376. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Ecker, J.R. Diversity and dynamics of DNA methylation: Epigenomic resources and tools for crop breeding. Breed. Sci. 2019, 69, 191–204. [Google Scholar] [CrossRef]
- Tirnaz, S.; Batley, J. Epigenetics: Potentials and Challenges in Crop Breeding. Mol. Plant. 2019, 12, 1309–1311. [Google Scholar] [CrossRef]
- Thieme, M.; Lanciano, S.; Balzergue, S.; Daccord, N.; Mirouze, M.; Bucher, E. Inhibition of RNA polymerase II allows controlled mobilisation of retrotransposons for plant breeding. Genome Biol. 2017, 18, 1–10. [Google Scholar] [CrossRef]
- Cai, Q.; He, B.; Kogel, K.-H.; Jin, H. Cross-kingdom RNA trafficking and environmental RNAi—Nature’s blueprint for modern crop protection strategies. Curr. Opin. Microbiol. 2018, 46, 58–64. [Google Scholar] [CrossRef]
- Muhammad, T.; Zhang, F.; Zhang, Y.; Liang, Y. RNA Interference: A Natural Immune System of Plants to Counteract Biotic Stressors. Cells 2019, 8, 38. [Google Scholar] [CrossRef]
- Champigny, M.J.; Unda, F.; Skyba, O.; Soolanayakanahally, R.Y.; Mansfield, S.D.; Campbell, M.M. Learning from methylomes: Epigenomic correlates of Populus balsamifera traits based on deep learning models of natural DNA methylation. Plant. Biotechnol. J. 2019, 18, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.-T.; Aufsatz, W.; Jonak, C.; Luschnig, C. Transgenerational epigenetic inheritance in plants. Biochim. Biophys. Acta BBA Bioenerg. 2011, 1809, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Plitta-Michalak, B.P.; Naskręt-Barciszewska, M.; Barciszewski, J.; Bujarska-Borkowska, B.; Chmielarz, P. Global 5-methylcytosine alterations in DNA during ageing of Quercus robur seeds. Ann. Bot. 2015, 116, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Plitta-Michalak, B.P.; Naskręt-Barciszewska, M.Z.; Kotlarski, S.; Tomaszewski, D.; Tylkowski, T.; Barciszewski, J.; Chmielarz, P.; Michalak, M. Changes in genomic 5-methylcytosine level mirror the response of orthodox (Acer Platanoides L.) and recalcitrant (Acer Pseudoplatanus L.) seeds to severe desiccation. Tree Physiol. 2017, 38, 617–629. [Google Scholar] [CrossRef]
- Luo, C.; Fernie, A.R.; Yan, J. Single-Cell Genomics and Epigenomics: Technologies and Applications in Plants. Trends Plant. Sci. 2020, 25, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Müller, F.; Rieu, I.; Winter, P. Epigenetic events in plant male germ cell heat stress responses. Plant. Reprod. 2015, 29, 21–29. [Google Scholar] [CrossRef]
- Correia, B.; Valledor, L.; Meijon, M.; Rodríguez, J.L.; Dias, M.C.; Santos, C.; Cañal, M.J.; Rodriguez, R.; Pinto, G. Is the Interplay between Epigenetic Markers Related to the Acclimation of Cork Oak Plants to High Temperatures? PLoS ONE 2013, 8, e53543. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Sugimoto, K.; Ramadan, A.; Arimura, G.-I. Memory of plant communications for priming anti-herbivore responses. Sci. Rep. 2013, 3, srep01872. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Ji, D.; Li, S.; Wang, P.; Li, Q.; Xiang, F. The Dynamic Changes of DNA Methylation and Histone Modifications of Salt Responsive Transcription Factor Genes in Soybean. PLoS ONE 2012, 7, e41274. [Google Scholar] [CrossRef]
- Choi, C.-S.; Sano, H. Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genom. 2007, 277, 589–600. [Google Scholar] [CrossRef]
- González, R.M.; Ricardi, M.M.; Iusem, N.D. Epigenetic marks in an adaptive water stress-responsive gene in tomato roots under normal and drought conditions. Epigenetics 2013, 8, 864–872. [Google Scholar] [CrossRef]
- Zhang, B.; Tieman, D.M.; Jiao, C.; Xu, Y.; Chen, K.; Fei, Z.; Giovannoni, J.J.; Klee, H.J. Chilling-induced tomato flavor loss is associated with altered volatile synthesis and transient changes in DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 12580–12585. [Google Scholar] [CrossRef]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.E.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef]
- Zuo, J.; Grierson, D.; Courtney, L.T.; Wang, Y.; Gao, L.; Zhao, X.; Zhu, B.; Luo, Y.; Wang, Q.; Giovannoni, J.J. Relationships between genome methylation, levels of non-coding RNAs, mRNAs and metabolites in ripening tomato fruit. Plant. J. 2020, 103, 980–994. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakoulidou, I.; Avramidou, E.V.; Baránek, M.; Brunel-Muguet, S.; Farrona, S.; Johannes, F.; Kaiserli, E.; Lieberman-Lazarovich, M.; Martinelli, F.; Mladenov, V.; et al. Epigenetics for Crop Improvement in Times of Global Change. Biology 2021, 10, 766. https://doi.org/10.3390/biology10080766
Kakoulidou I, Avramidou EV, Baránek M, Brunel-Muguet S, Farrona S, Johannes F, Kaiserli E, Lieberman-Lazarovich M, Martinelli F, Mladenov V, et al. Epigenetics for Crop Improvement in Times of Global Change. Biology. 2021; 10(8):766. https://doi.org/10.3390/biology10080766
Chicago/Turabian StyleKakoulidou, Ioanna, Evangelia V. Avramidou, Miroslav Baránek, Sophie Brunel-Muguet, Sara Farrona, Frank Johannes, Eirini Kaiserli, Michal Lieberman-Lazarovich, Federico Martinelli, Velimir Mladenov, and et al. 2021. "Epigenetics for Crop Improvement in Times of Global Change" Biology 10, no. 8: 766. https://doi.org/10.3390/biology10080766
APA StyleKakoulidou, I., Avramidou, E. V., Baránek, M., Brunel-Muguet, S., Farrona, S., Johannes, F., Kaiserli, E., Lieberman-Lazarovich, M., Martinelli, F., Mladenov, V., Testillano, P. S., Vassileva, V., & Maury, S. (2021). Epigenetics for Crop Improvement in Times of Global Change. Biology, 10(8), 766. https://doi.org/10.3390/biology10080766