
Evaluation of Genomic Prediction for Fusarium Head Blight Resistance with a Multi-Parental Population

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Disease Inoculation and Phenotyping
2.3. Genotyping
2.4. Evaluation of GS
2.4.1. Prediction Models
2.4.2. Optimization TP
2.4.3. Bayesian Multi-Trait Multi-Environment (BMTME)
2.4.4. Cross Validations of the Prediction Accuracy (PA)
3. Results
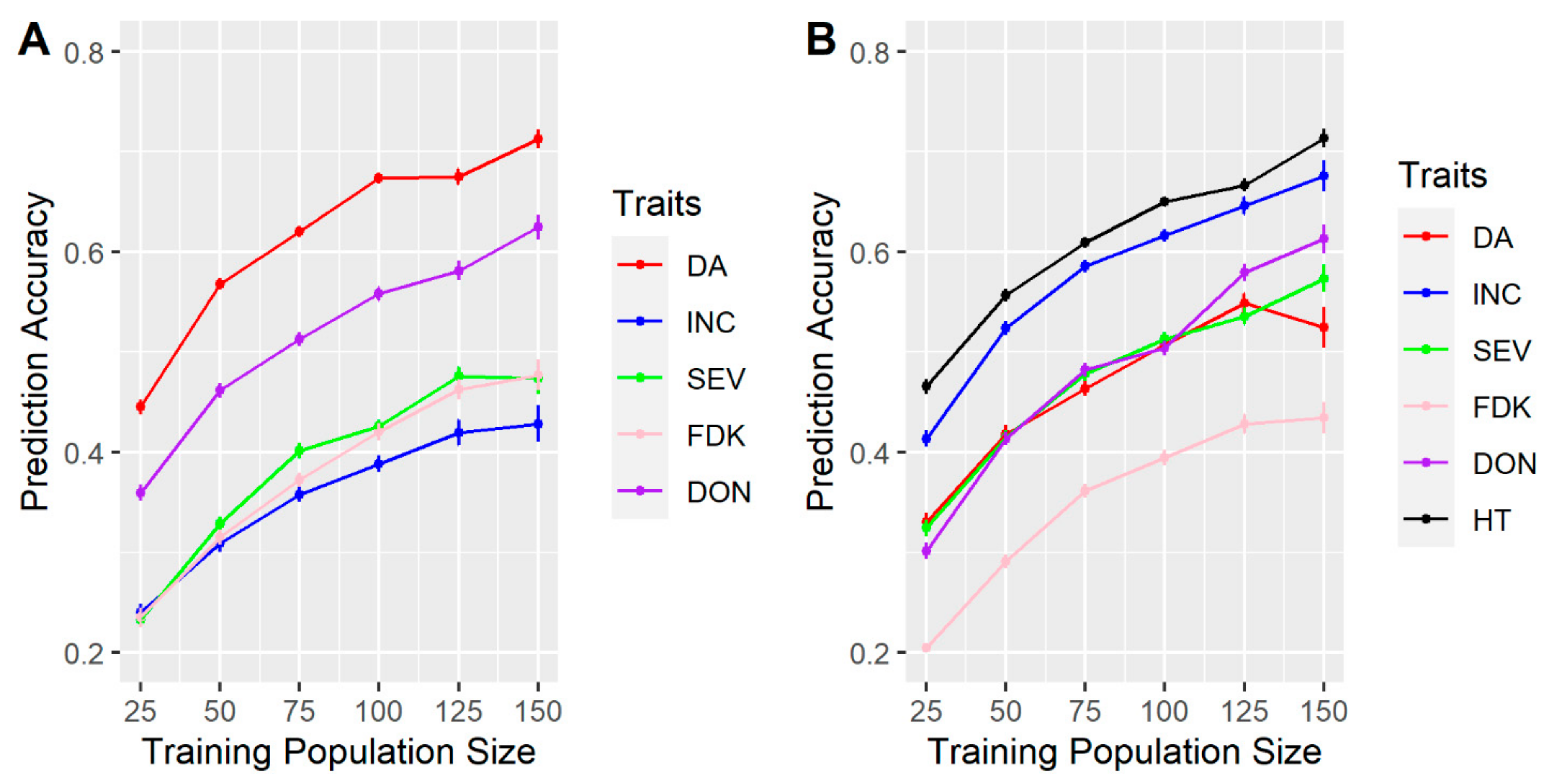
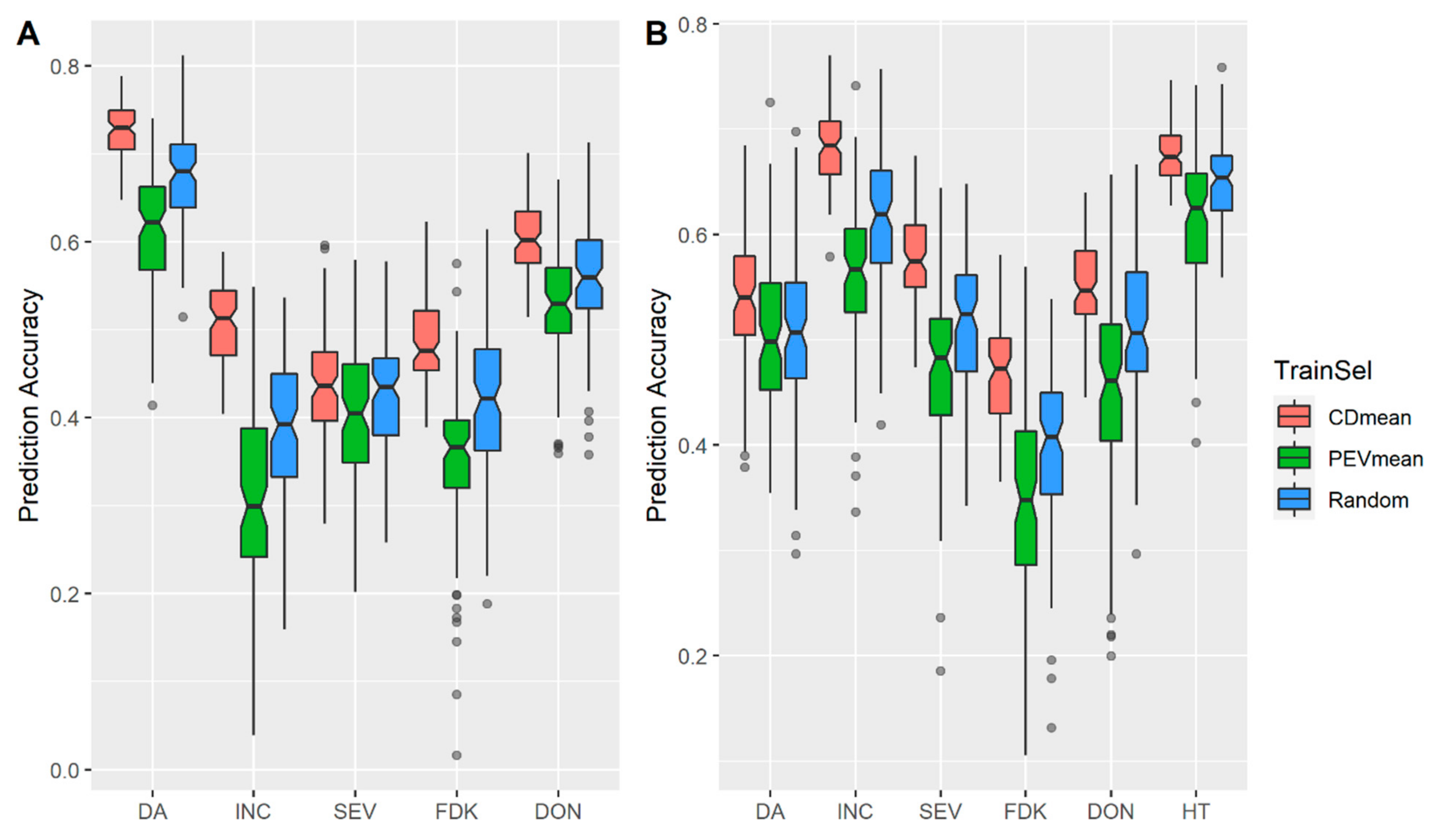
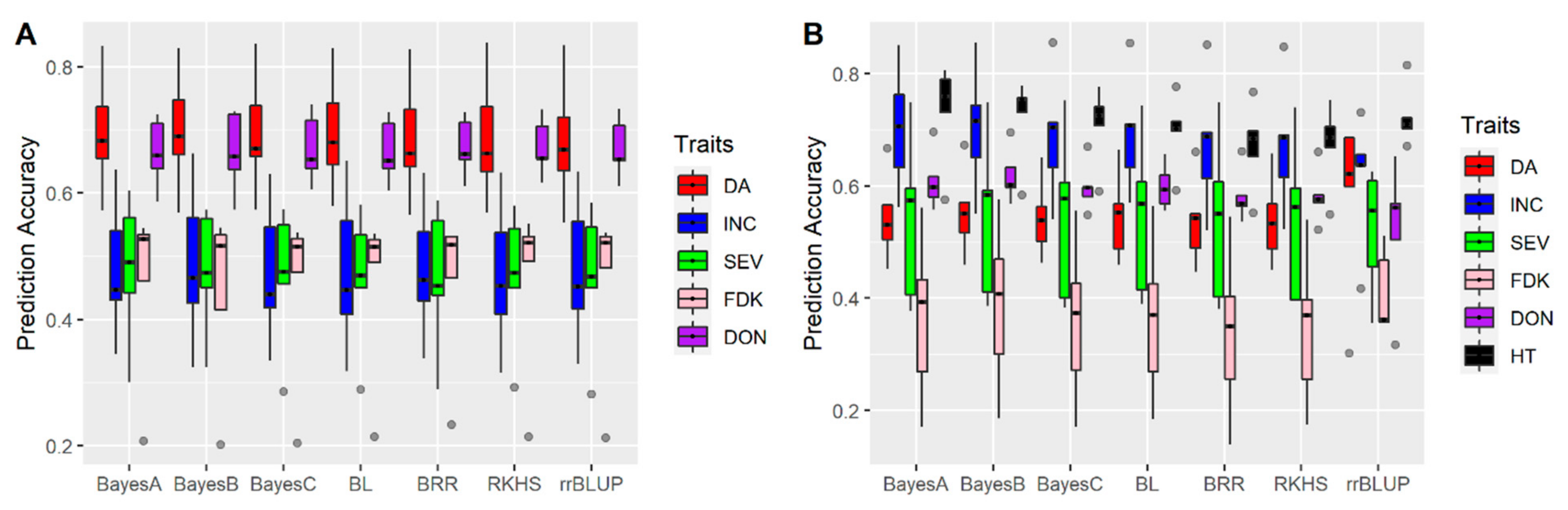
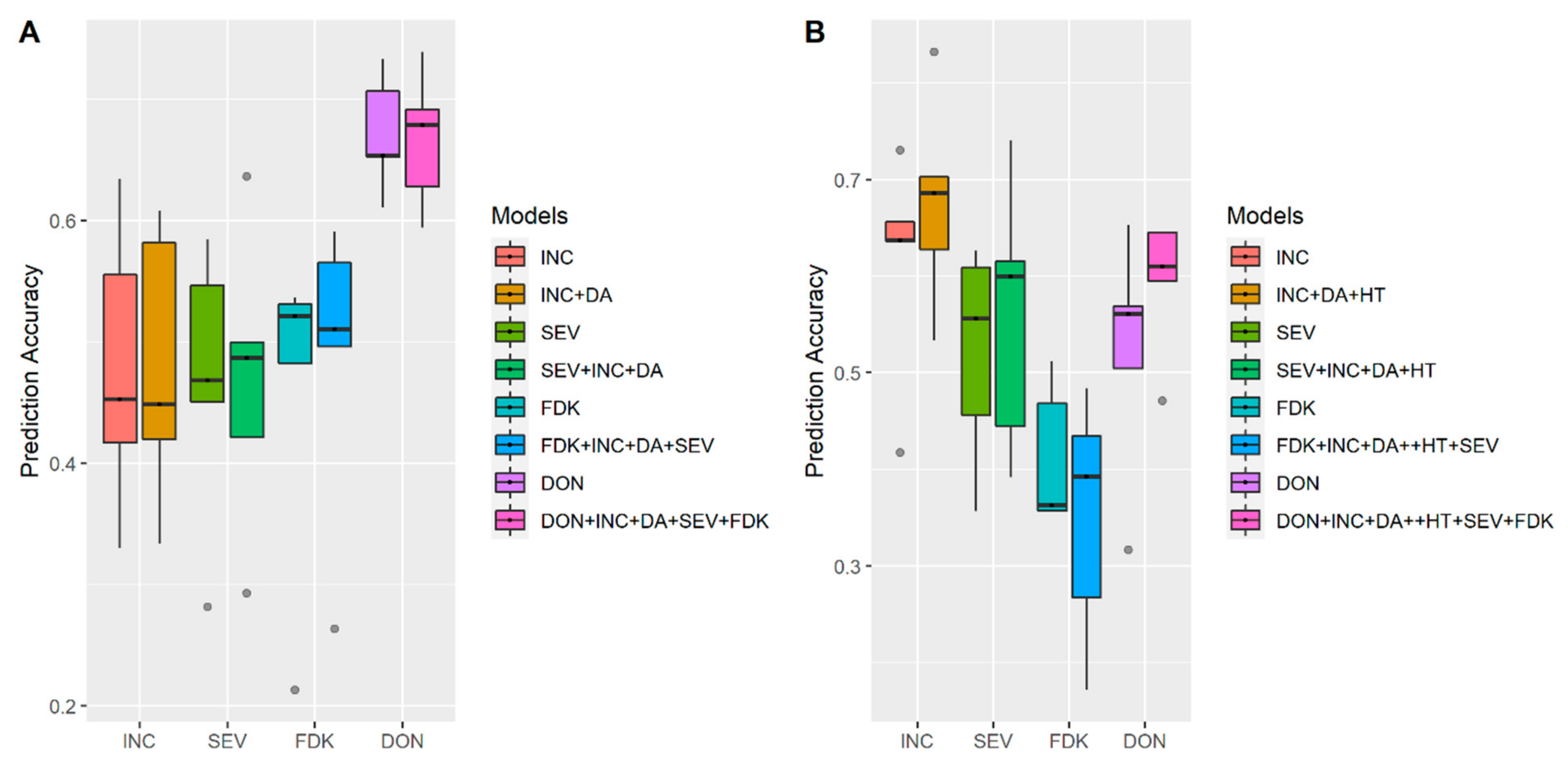
3.1. Effects of Training Population (TP) Size, TP Optimization and Models of Prediction
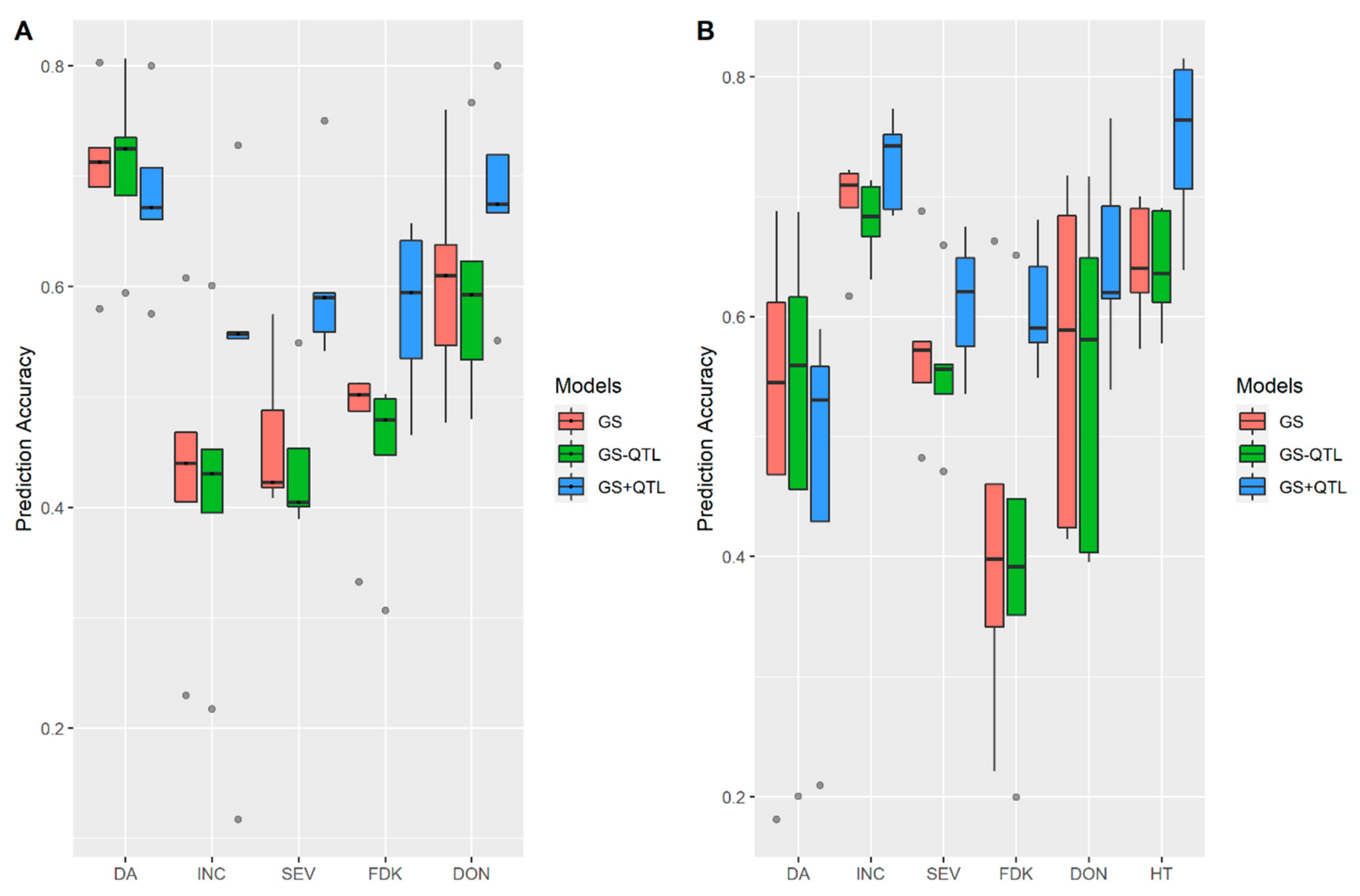
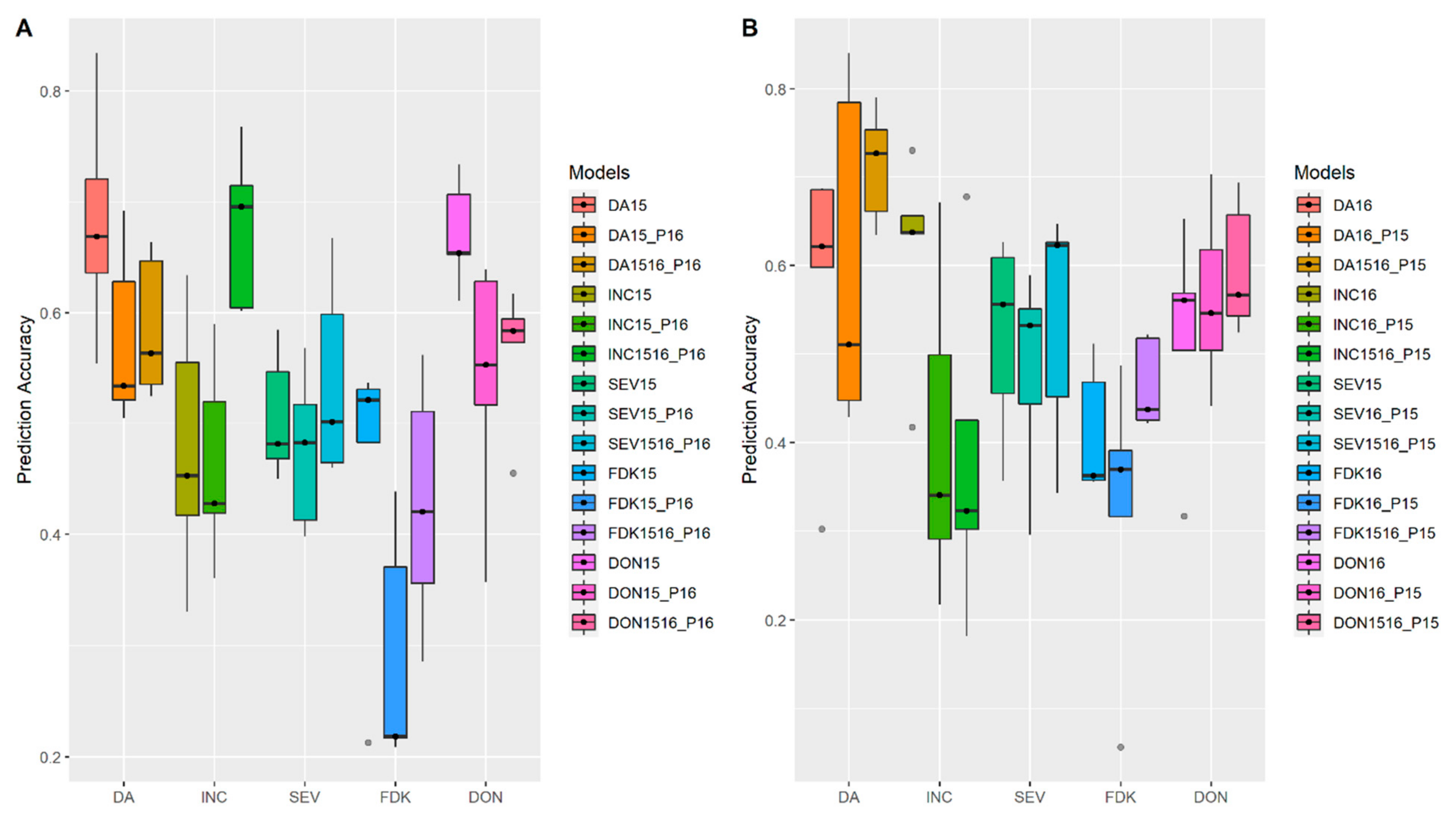
3.2. Incorporation of Prior Known QTL into the Genomic Prediction Model
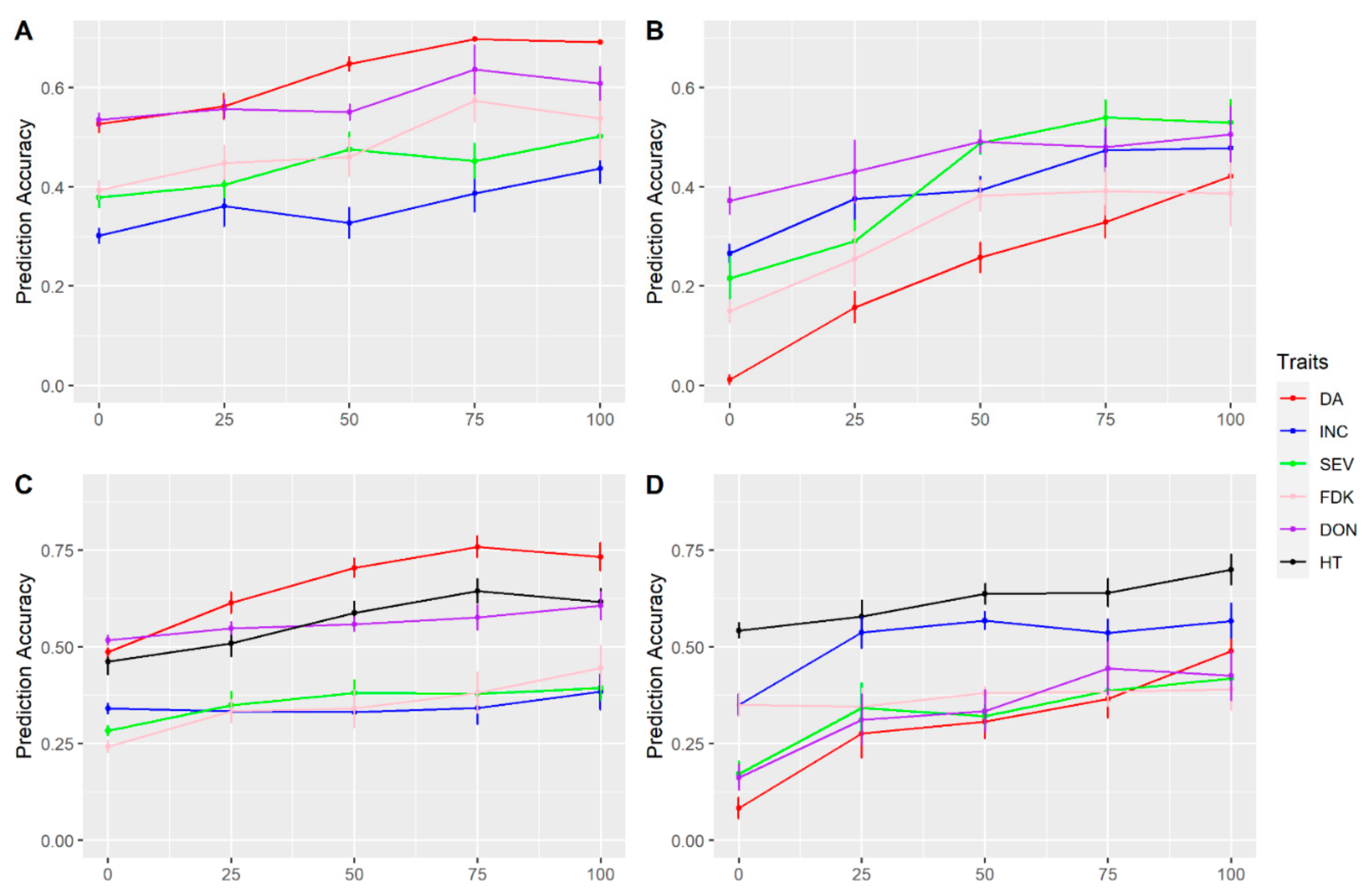
3.3. Multi-Traits and Multi-Environment Prediction
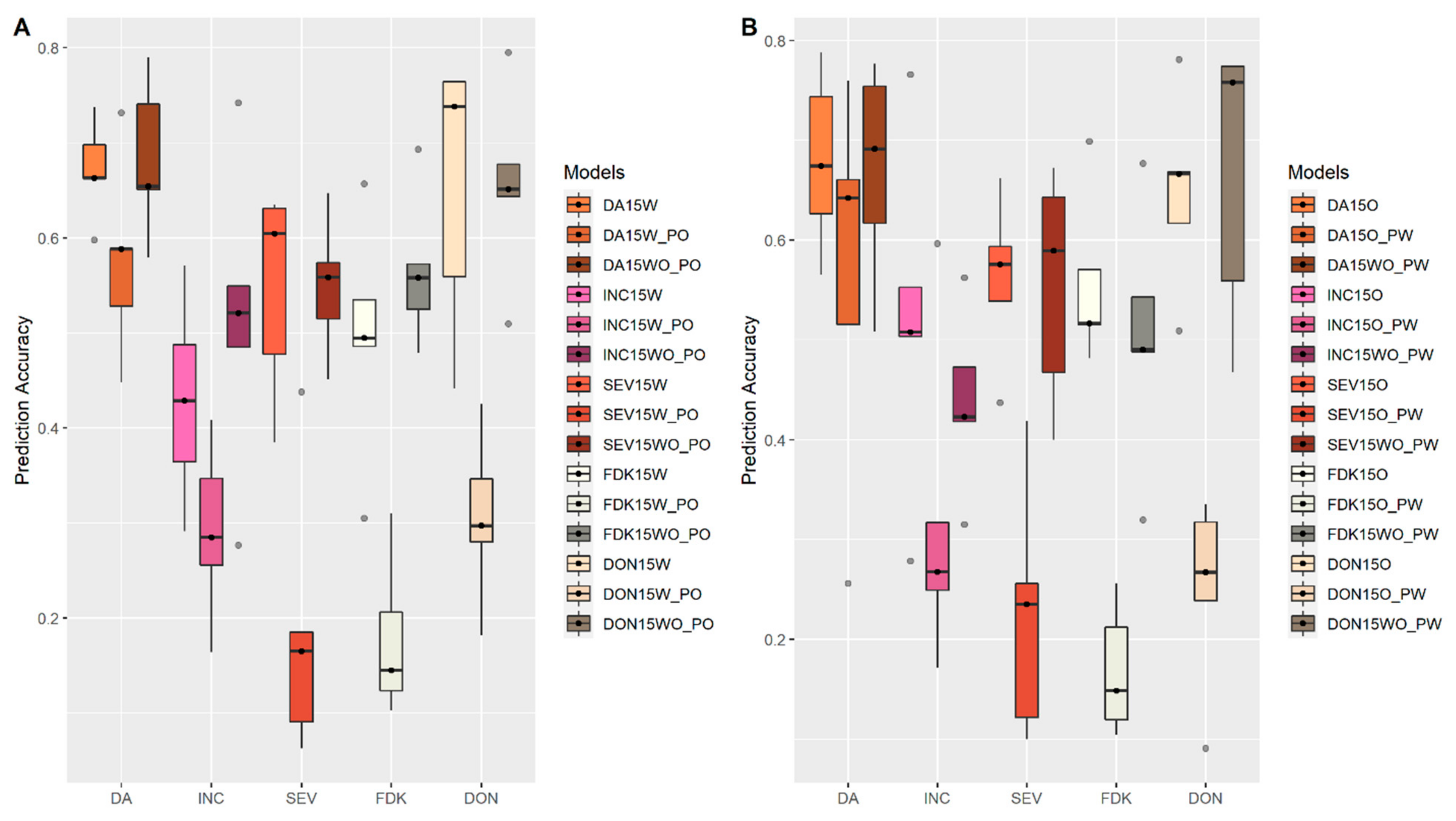
3.4. Prediction within and across Populations
4. Discussion
4.1. TP Size
4.2. Design TP with Optimization Algorithms
4.3. Models of Prediction
4.4. Incorporation of Prior Known QTL
4.5. Predictability with Multi-Trait and Multi-Environment Models
4.6. Predictability within and across Populations
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilbert, J.; Tekauz, A. Review: Recent developments in research on fusarium head blight of wheat in Canada. Can. J. Plant Pathol. 2000, 22, 1–8. [Google Scholar] [CrossRef]
- Bai, G.; Shaner, G. Management and resistance in wheat and barley to Fusarium head blight. Annu. Rev. Phytopathol. 2004, 42, 135–161. [Google Scholar] [CrossRef]
- McMullen, M.; Bergstrom, G.; De Wolf, E.; Dill-Macky, R.; Hershman, D.; Shaner, G.; Van Sanford, D. A unified effort to fight an enemy of wheat and barley: Fusarium head blight. Plant Dis. 2012, 96, 1712–1728. [Google Scholar] [CrossRef] [PubMed]
- Haile, J.K.; N’Diaye, A.; Walkowiak, S.; Nilsen, K.T.; Clarke, J.M.; Kutcher, H.R.; Steiner, B.; Buerstmayr, H.; Pozniak, C.J. Fusarium head blight in durum wheat: Recent status, breeding directions, and future research prospects. Phytopathology 2019, 109, 1664–1675. [Google Scholar] [CrossRef]
- Wilson, W.; Dahl, B.; Nganje, W. Economic Costs of Fusarium Head Blight, Scab and Deoxynivalenol. World Mycotoxin J. 2018, 11, 291–302. [Google Scholar] [CrossRef]
- Gilbert, J.; Haber, S. Overview of some recent research developments in Fusarium head blight of wheat. Can. J. Plant Pathol. 2013, 35, 149–174. [Google Scholar] [CrossRef]
- Prat, N.; Buerstmayr, M.; Steiner, B.; Robert, O.; Buerstmayr, H. Current knowledge on resistance to Fusarium head blight in tetraploid wheat. Mol. Breed. 2014, 34, 1689–1699. [Google Scholar] [CrossRef]
- Liu, S.; Hall, M.D.; Griffey, C.A.; McKendry, A.L. Meta-analysis of QTL associated with Fusarium Head Blight resistance in wheat. Crop Sci. 2009, 49, 1955–1968. [Google Scholar] [CrossRef]
- Löffler, M.; Schön, C.C.; Miedaner, T. Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol. Breed. 2009, 23, 473–488. [Google Scholar] [CrossRef]
- Venske, E.; Dos Santos, R.S.; Farias, D.D.R.; Rother, V.; Maia, L.C.D.; Pegoraro, C.; Costa De Oliveira, A. Meta-analysis of the QTLome of Fusarium head blight resistance in bread wheat: Refining the current puzzle. Front. Plant Sci. 2019, 10, 727. [Google Scholar] [CrossRef]
- Miller, J.D.; Young, J.C.; Sampson, D.R. Deoxynivalenol and Fusarium head blight resistance in spring cereals. J. Phytopathol. 1985, 113, 359–367. [Google Scholar] [CrossRef]
- Mesterhazy, A. Types and components of resistance to Fusarium head blight of wheat. Plant. Breed. 1995, 114, 377–386. [Google Scholar] [CrossRef]
- Mesterhazy, A.; Bartok, T.; Mirocha, C.G.; Komoroczy, R. Nature of wheat resistance to Fusarium head blight and the role of deoxynivalenol for breeding. Plant. Breed. 1999, 118, 97–110. [Google Scholar] [CrossRef]
- Steiner, B.; Buerstmayr, M.; Michel, S.; Schweiger, W.; Lemmens, M.; Buerstmayr, H. Breeding strategies and advances in line selection for Fusarium head blight resistance in wheat. Trop. Plant. Pathol. 2017, 42, 165–174. [Google Scholar] [CrossRef]
- Steiner, B.; Lemmens, M.; Griesser, M.; Scholz, U.; Schondelmaier, J.; Buerstmayr, H. Molecular mapping of resistance to Fusarium head blight in the spring wheat cultivar Frontana. Theor. Appl. Genet. 2004, 109, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Song, Y.; Gao, L.; Yao, Q.; Zhou, R.; Xu, R.; Jia, J. Integration of QTL detection and marker assisted selection for improving resistance to Fusarium head blight and important agronomic traits in wheat. Crop J. 2014, 2, 70–78. [Google Scholar] [CrossRef]
- Zhang, W.; Boyle, K.; Brûlé-Babel, A.L.; Fedak, G.; Gao, P.; Robleh Djama, Z.; Polley, B.; Cuthbert, R.; Randhawa, H.; Jiang, F.Y.; et al. Genetic characterization of multiple components contributing to Fusarium head blight resistance of FL62R1, a Canadian bread wheat developed using systemic breeding. Front. Plant Sci. 2020, 26, 580833. [Google Scholar] [CrossRef]
- Gervais, L.; Dedryver, F.; Morlais, J.Y.; Bodusseau, V.; Negre, S.; Bilous, M.; Groos, C.; Trottet, M. Mapping of quantitative trait loci for field resistance to Fusarium head blight in a European winter wheat. Theor. Appl. Genet. 2003, 106, 961–970. [Google Scholar] [CrossRef]
- Srinivasachary, G.N.; Steed, A.; Faure, S.; Bayles, R.; Jennings, P.; Nicholson, P. Mapping of QTL associated with Fusarium head blight in spring wheat RL4137. Czech J. Genet. Plant. 2008, 44, 147–159. [Google Scholar] [CrossRef]
- Srinivasachary, G.N.; Steed, A.; Hollins, T.; Bayles, R.; Jennings, P.; Nicholson, P. Semi-dwarfing Rht-B1 and Rht-D1 loci of wheat differ significantly in their influence on resistance to Fusarium head blight. Theor. Appl. Genet. 2009, 118, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Buerstmayr, H.; Ban, T.; Anderson, J.A. QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: A review. Plant Breed. 2009, 128, 1–26. [Google Scholar] [CrossRef]
- Skinnes, H.; Semagn, K.; Tarkegne, Y.; Maroy, A.G.; Bjornstad, A. The inheritance of anther extrusion in hexaploid wheat and its relationship to Fusarium head blight resistance and deoxynivalenol content. Plant Breed. 2010, 129, 149–155. [Google Scholar] [CrossRef]
- Lu, Q.X.; Lillemo, M.; Skinnes, H.; He, X.Y.; Shi, J.R.; Ji, F.; Dong, Y.H.; Bjornstad, A. Anther extrusion and plant height are associated with Type I resistance to Fusarium head blight in bread wheat line ‘Shanghai-3/ Catbird’. Theor. Appl. Genet. 2013, 126, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Szabo-Hever, A.; Lehoczki-Krsjak, S.; Varga, M.; Purnhauser, L.; Pauk, J.; Lantos, C.; Mesterházy, Á. Differential influence of QTL linked to Fusarium head blight, Fusarium-damaged kernel, deoxynivalenol contents and associated morphological traits in a Frontana-derived wheat population. Euphytica 2014, 200, 9–26. [Google Scholar] [CrossRef]
- Buerstmayr, M.; Buerstmayr, H. The semidwarfing alleles Rht-D1b and Rht-B1b show marked differences in their associations with anther-retention in wheat heads and with Fusarium head blight susceptibility. Phytopathology 2016, 106, 1544–1552. [Google Scholar] [CrossRef]
- Buerstmayr, M.; Steiner, B.; Buerstmayr, H. Breeding for Fusarium head blight resistance in wheat—Progress and challenges. Plant Breed. 2020, 139, 429–454. [Google Scholar] [CrossRef]
- Van Sanford, D.; Anderson, J.; Campbell, K.; Costa, J.; Cregan, P.; Griffey, C.; Hayes, P.; Ward, R. Discovery and deployment of molecular markers linked to fusarium head blight resistance: An integrated system for wheat and barley. Crop Sci. 2001, 41, 638–644. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Smith, K.P.; Jannink, J.L. Potential and optimization of genomic selection for Fusarium head blight resistance in six-row barley. Crop Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Rutkoski, J.; Benson, J.; Jia, Y.; Brown-Guedira, G.; Jannink, J.-L.; Sorrells, M. Evaluation of Genomic Prediction Methods for Fusarium Head Blight Resistance in Wheat. Plant Genome 2012, 5, 51–61. [Google Scholar] [CrossRef]
- Arruda, M.P.; Brown, P.J.; Lipka, A.E.; Krill, A.M.; Thurber, C.; Kolb, F.L. Genomic selection for predicting head blight resistance in a wheat breeding program. Plant Genome 2015, 8, 1–12. [Google Scholar] [CrossRef]
- Steiner, B.; Michel, S.; Maccaferri, M.; Lemmens, M.; Tuberosa, R.; Buerstmayr, H. Exploring and exploiting the genetic variation of Fusarium head blight resistance for genomic-assisted breeding in the elite durum wheat gene pool. Theor. Appl. Genet. 2019, 132, 969–988. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Voss-Fels, K.P.; Cooper, M.; Hayes, B.J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 2019, 132, 669–686. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing genetic gain through genomic selection: From livestock to plants. Plant Commun. 2020, 1, 100005. [Google Scholar] [CrossRef]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.C.; Hussain, T.; Jones, E.S.; Graham, G.; Podlich, D.; Wall, S.; Williams, M. Use of doubled haploids in maize breeding: Implications for intellectual property protection and genetic diversity in hybrid crops. Mol. Breed. 2008, 22, 51–59. [Google Scholar] [CrossRef]
- Lorenzana, R.E.; Bernardo, R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef]
- Battenfield, S.D.; Guzman, C.; Gaynor, R.C.; Singh, R.P.; Pena, R.J.; Dreisigaker, S.; Poland, J.A. Genomic selection for processing and end-use quality traits in the CIMMYT spring bread wheat breeding program. Plant Genome 2016, 9, 1–12. [Google Scholar] [CrossRef]
- Xavier, A.; Muir, W.M.; Rainey, K.M. Assessing predictive properties of genome-wide selection in soybeans. G3 2016, 6, 2611–2616. [Google Scholar] [CrossRef]
- Isidro, J.; Jannink, J.L.; Akdemir, D.; Poland, J.A.; Heslot, N.; Sorrels, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E. Accuracy of breeding values of ‘unrelated’ individuals predicted by dense SNP genotyping. Genet. Sel. Evol. 2009, 41, 35. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic Selection Accuracy Using Multifamily Prediction Models in a Wheat Breeding Program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Akdemir, D.; Sanchez, J.I.; Jannink, J.L. Optimization of genomic selection training populations with a genetic algorithm. Genet. Sel. Evol. 2015, 47, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Akdemir, D.; Isidro-Sanchez, J. Design of training populations for selective genotyping in genomic prediction. Sci. Rep. 2019, 9, 1446. [Google Scholar] [CrossRef]
- Rincent, R.; Laloë, D.; Nicolas, S.; Altmann, T.; Brunel, D.; Revilla, P.; Rodriguez, V.M.; Moreno-Gonzales, J.; Melchinger, A.E.; Bauer, E. Maximizing the reliability of genomic selection by optimizing the calibration set of reference individuals: Comparison of methods in two diverse groups of maize inbreds (Zea mays L.). Genetics 2012, 192, 715–728. [Google Scholar] [CrossRef]
- Tayeh, N.; Klein, A.; Le Paslier, M.C.; Jacquin, F.; Houtin, H.; Rond, C.; Chabert-Martinello, M.; Magnin-Robert, J.B.; Marget, P.; Aubert, G.; et al. Genomic prediction in pea: Effect of marker density and training population size and composition on prediction accuracy. Front. Plant Sci. 2015, 6, 941. [Google Scholar] [CrossRef] [PubMed]
- Bastiaansen, J.; Coster, A.; Calus, M.; Van Arendonk, J.; Bovenhuis, H. Long-term response to genomic selection: Effects of estimation method and reference population structure for different genetic architectures. Genet. Sel. Evol. 2012, 44, 3. [Google Scholar] [CrossRef]
- Dekkers, J.C.M. Application of genomics tools to animal breeding. Curr. Genomics 2012, 13, 207–212. [Google Scholar] [CrossRef] [PubMed]
- De los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jannink, J.L. Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 2012, 192, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-López, O.A.; Montesinos-López, A.; Pérez-Rodríguez, P.; de los Campos, G.; Eskridge, K.; Cross, J. Threshold models for genome-enabled prediction of ordinal categorical traits in plant breeding. G3 Genes Genomes Genet. 2015, 5, 291–300. [Google Scholar] [CrossRef]
- Bernardo, R. Genome wide selection when major genes are known. Crop Sci. 2014, 54, 68–75. [Google Scholar] [CrossRef]
- Spindel, J.E.; Begum, H.; Akdemir, D.; Collard, B.; Redoña, E.; Jannink, J.; McCouch, S. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Holland, J.B. Enhancing genomic prediction with genome-wide association studies in multiparental maize populations. Heredity 2017, 118, 585–593. [Google Scholar] [CrossRef]
- Larkin, D.L.; Holder, A.L.; Mason, R.E.; Moon, D.E.; Brown-Guedira, G.; Price, P.P.; Dong, Y. Genome-wide analysis and prediction of fusarium head blight resistance in soft red winter wheat. Crop Sci. 2020, 60, 2882–2900. [Google Scholar] [CrossRef]
- Herter, C.P.; Ebmeyer, E.; Kollers, S.; Korzun, V.; Würschum, T.; Miedaner, T. Accuracy of within-and among-family genomic prediction for Fusarium head blight and Septoria tritici blotch in winter wheat. Theor. Appl. Genet. 2019, 132, 1121–1135. [Google Scholar] [CrossRef]
- Comeau, A.; Langevin, F.; Caetano, V.R.; Haber, S.; Savard, M.E.; Voldeng, H.; Fedak, F.; Dion, Y.; Rioux, S.; Gilbert, J.; et al. A systemic approach for the development of FHB resistant germplasm accelerates genetic progress. Cereal Res. Commun. 2008, 36, 5–9. [Google Scholar] [CrossRef]
- DePauw, R.M.; Knox, R.E.; Clarke, F.R.; Clarke, J.M.; McCaig, T.N. Stettler hard red spring wheat. Can. J. Plant Sci. 2009, 89, 945–951. [Google Scholar] [CrossRef]
- DePauw, R.M.; Knox, R.E.; McCaig, T.N.; Clarke, F.R.; Clarke, J.M. Muchmore hard red spring wheat. Can. J. Plant Sci. 2011, 91, 1–7. [Google Scholar] [CrossRef][Green Version]
- Graf, R.; Beres, B.L.; Laroche, A.; Gaudet, D.A.; Eudes, F.; Pandeya, R.S.; Badea, A.; Randhawa, H.S. Emerson hard red winter wheat. Can. J. Plant Sci. 2013, 93, 741–748. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genome diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Akdemir, D.; Rio, S.; Isidro y Sánchez, J. TrainSel: An R Package for Selection of Training Populations. Front. Genet. 2021, 12, 655287. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-López, O.; Montesinos-López, A.; Luna-Vázquez, F.J.; Toledo, F.H.; Pérez-Rodríguez, P.; Lillemo, M.; Crossa, J. An R package for Bayesian analysis of multi-environment and multi-trait multi-environment data for genome-based prediction. G3 Genes Genomes Genet. 2019, 9, 1355–1369. [Google Scholar] [CrossRef]
- Van Raden, P.M. Efficient method to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Maulana, F.; Kim, K.S.; Anderson, J.D.; Sorrells, M.E.; Butler, T.J.; Liu, S.Y.; Baenziger, P.S.; Patrick, F.B.; Ma, X.F. Genomic selection of forage agronomic traits in winter wheat. Crop Sci. 2020, 60, 2882–2900. [Google Scholar] [CrossRef]
- Zhang, A.; Wang, H.; Beyene, Y.; Semagn, K.; Liu, Y.; Cao, S.; Cui, Z.; Ruan, Y.; Burgueño, J.; Vicente, F.S.; et al. Effect of Trait Heritability, Training Population Size and Marker Density on Genomic Prediction Accuracy Estimation in 22 bi-parental Tropical Maize Populations. Front. Plant Sci. 2017, 8, 1916. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. Genomic selection using low-density marker panels. Genetics 2009, 182, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Tiede, T.; Smith, K.P. Evaluation and retrospective optimization of genomic selection for yield and disease resistance in spring barley. Mol. Breed. 2018, 38, 55. [Google Scholar] [CrossRef]
- Desta, Z.A.; Ortiz, R. Genomic selection: Genome-wide prediction in plant improvement. Trends. Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef]
- Rutkoski, J.; Poland, J.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Barbier, H.; Rouse, M.N.; Jannink, J.; Sorrells, M.E. Genomic Selection for Quantitative Adult Plant Stem Rust Resistance in Wheat. Plant Genome 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Arruda, M.P.; Lipka, A.E.; Brown, P.J.; Krill, A.M.; Thurber, C.; Brown-Guedira, G.; Dong, Y.; Foresman, B.J.; Kolb, F.L. Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum). Mol. Breed. 2016, 36, 1–11. [Google Scholar] [CrossRef]
- Liabeuf, D.; Sim, S.; Francis, D. Comparison of marker-based genomic estimated breeding values and phenotypic evaluation for selection of bacterial spot resistance in tomato. Phytopathology 2018, 108, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rice, B.; Lipka, A.E. Evaluation of RR-BLUP genomic selection models that incorporate peak genome-wide association study signals in maize and sorghum. Plant Genome 2019, 12, 180052. [Google Scholar] [CrossRef]
- Haile, J.K.; N’Diaye, A.; Clarke, F.; Clarke, J.; Knox, R.; Rutkoski, J.; Bassi, F.M.; Pozniak, C.J. Genomic Selection for Grain Yield and Quality Traits in Durum Wheat. Mol. Breed. 2018, 38, 75. [Google Scholar] [CrossRef]
- Lado, B.; Vázquez, D.; Quincke, M.; Silva, P.; Aguilar, I.; Gutiérrez, L. Resource Allocation Optimization with Multi-Trait Genomic Prediction for Bread Wheat (Triticum aestivum L.) Baking Quality. Theor. Appl. Genet. 2018, 131, 2719–2731. [Google Scholar] [CrossRef] [PubMed]
- Lado, B.; Barrios, P.G.; Quincke, M.; Silva, P.; Gutiérrez, L. Modeling genotype × environment interaction for genomic selection with unbalanced data from a wheat breeding program. Crop Sci. 2016, 56, 1–15. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Endelman, J.B.; Stange, M.; Sorrells, M.E.; Jannink, J.L.; Melchinger, A.E. Genomic predictability of interconnected bi-parental maize populations. Genetics 2013, 194, 493–503. [Google Scholar] [CrossRef]
- Zaim, M.; Kabbaj, H.; Kehel, Z.; Gorjanc, G.; Filali, M.A.; Belkadi, B.; Nachit, M.M.; Bassi, F.M. Combining QTL analysis and genomic predictions for four durum wheat populations under drought conditions. Front. Genet. 2020, 11, 316. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Boyle, K.; Brule-Babel, A.; Fedak, G.; Gao, P.; Djama, Z.R.; Polley, B.; Cuthbert, R.; Randhawa, H.; Graf, R.; et al. Evaluation of Genomic Prediction for Fusarium Head Blight Resistance with a Multi-Parental Population. Biology 2021, 10, 756. https://doi.org/10.3390/biology10080756
Zhang W, Boyle K, Brule-Babel A, Fedak G, Gao P, Djama ZR, Polley B, Cuthbert R, Randhawa H, Graf R, et al. Evaluation of Genomic Prediction for Fusarium Head Blight Resistance with a Multi-Parental Population. Biology. 2021; 10(8):756. https://doi.org/10.3390/biology10080756
Chicago/Turabian StyleZhang, Wentao, Kerry Boyle, Anita Brule-Babel, George Fedak, Peng Gao, Zeinab Robleh Djama, Brittany Polley, Richard Cuthbert, Harpinder Randhawa, Robert Graf, and et al. 2021. "Evaluation of Genomic Prediction for Fusarium Head Blight Resistance with a Multi-Parental Population" Biology 10, no. 8: 756. https://doi.org/10.3390/biology10080756
APA StyleZhang, W., Boyle, K., Brule-Babel, A., Fedak, G., Gao, P., Djama, Z. R., Polley, B., Cuthbert, R., Randhawa, H., Graf, R., Jiang, F., Eudes, F., & Fobert, P. R. (2021). Evaluation of Genomic Prediction for Fusarium Head Blight Resistance with a Multi-Parental Population. Biology, 10(8), 756. https://doi.org/10.3390/biology10080756