
Integrated Analysis of lncRNA and mRNA in Subcutaneous Adipose Tissue of Ningxiang Pig

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animal and Sample Collection
2.3. RNA Extraction
2.4. Library Construction and RNA-Seq
2.5. Read Mapping and Transcriptome Assembly
2.6. Identification and Classification of LncRNAs
2.7. Functional Enrichment and Differential Expression Analysis
2.8. Time Series Analysis (Short Time-Series Expression Miner, STEM)
2.9. Co-Expression Network (Weight Correlation Network Analysis, WGCNA)
2.10. Real-Time PCR Quantification
3. Results
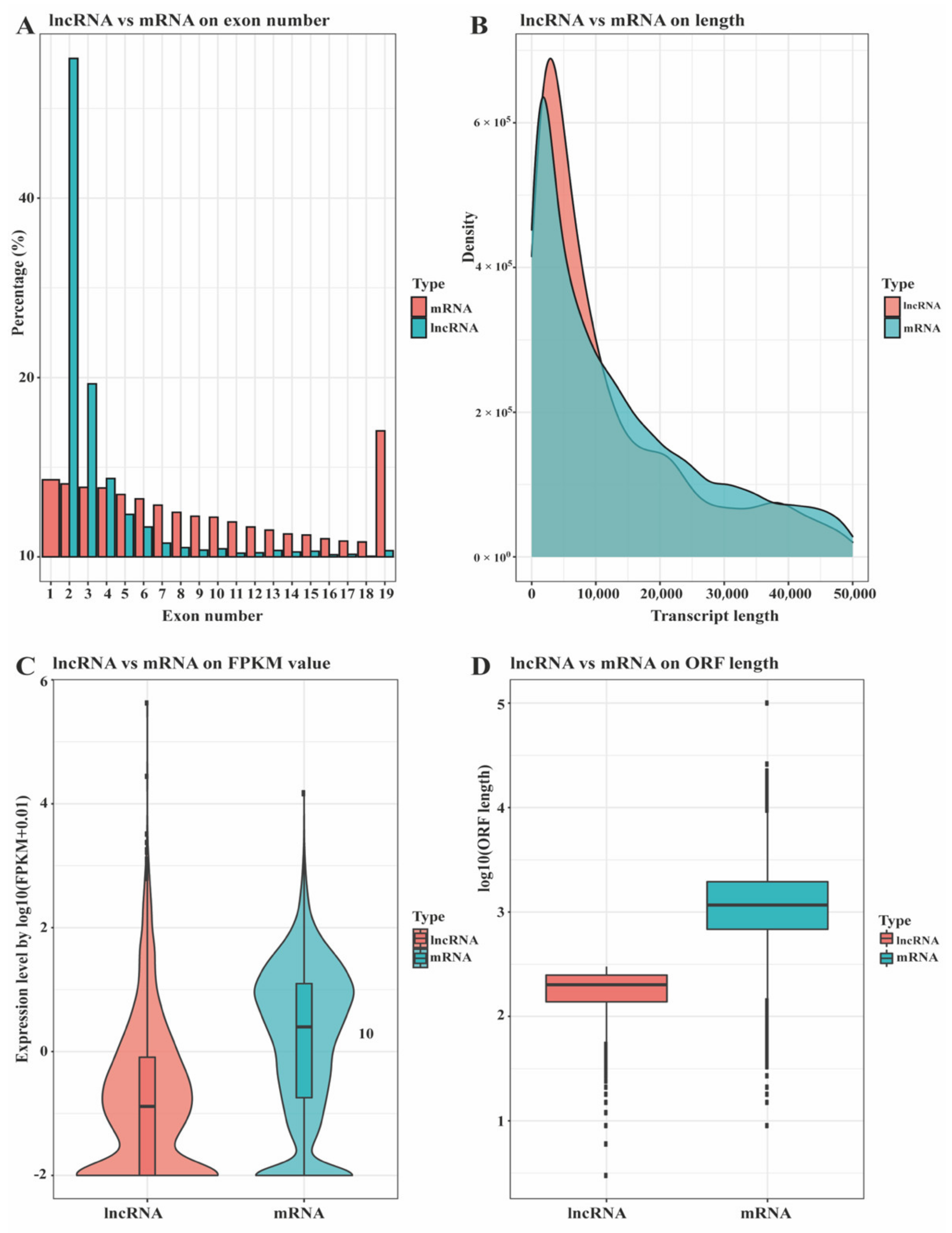
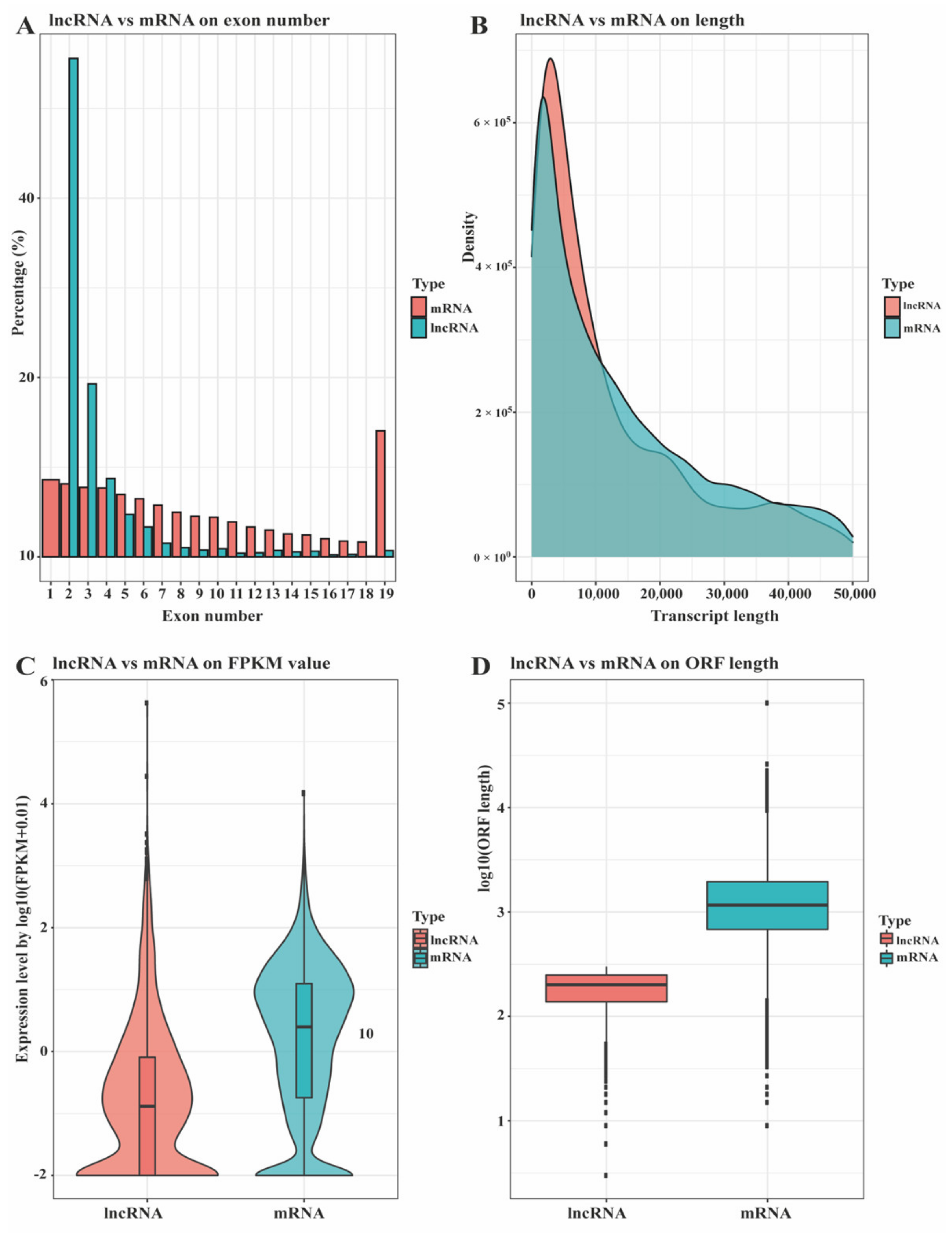
3.1. Identification and Classification of lncRNA in Ningxiang Pig Subcutaneous Adipose Tissue
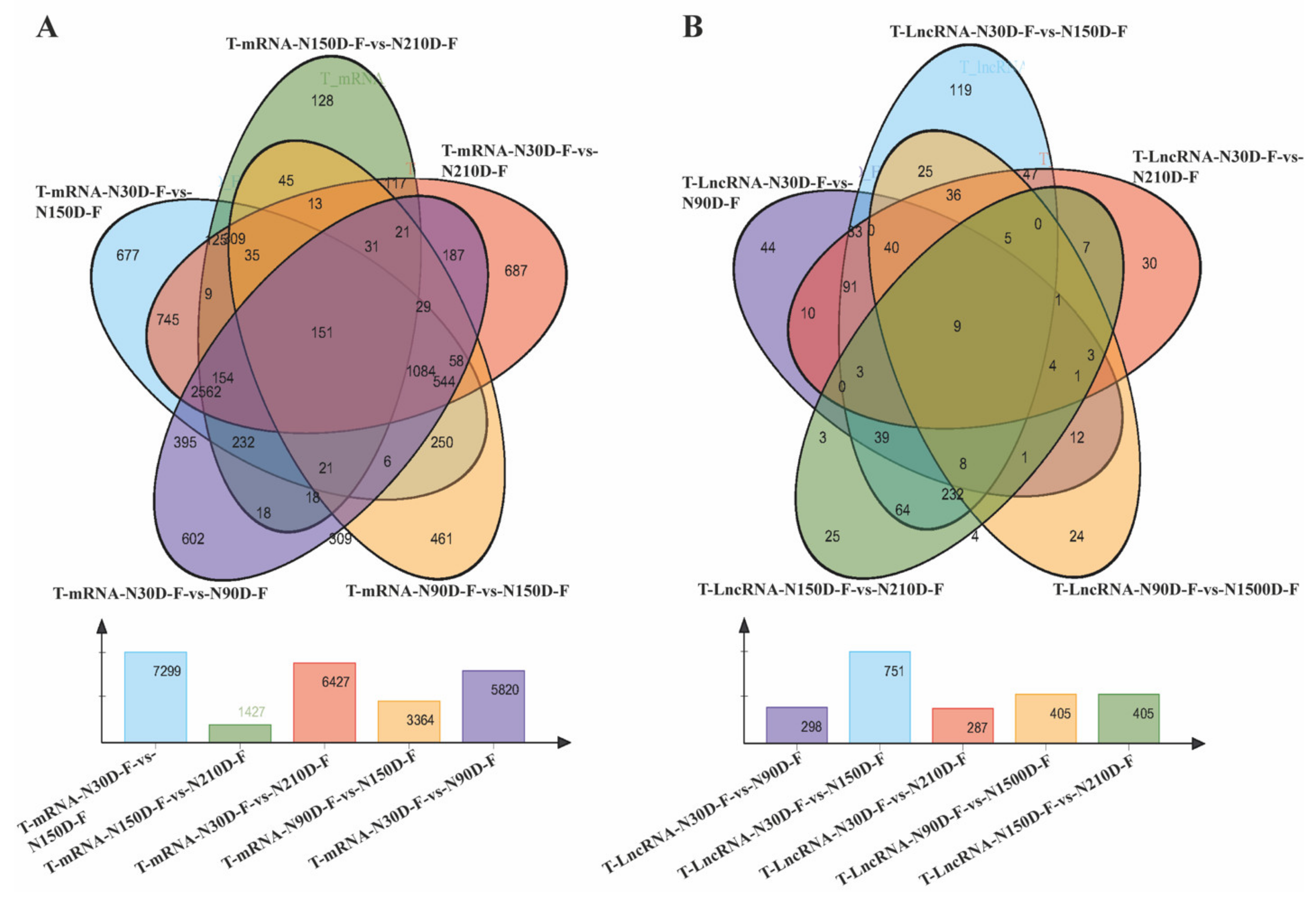
3.2. Analysis of Differentially Expressed mRNA and lncRNAs
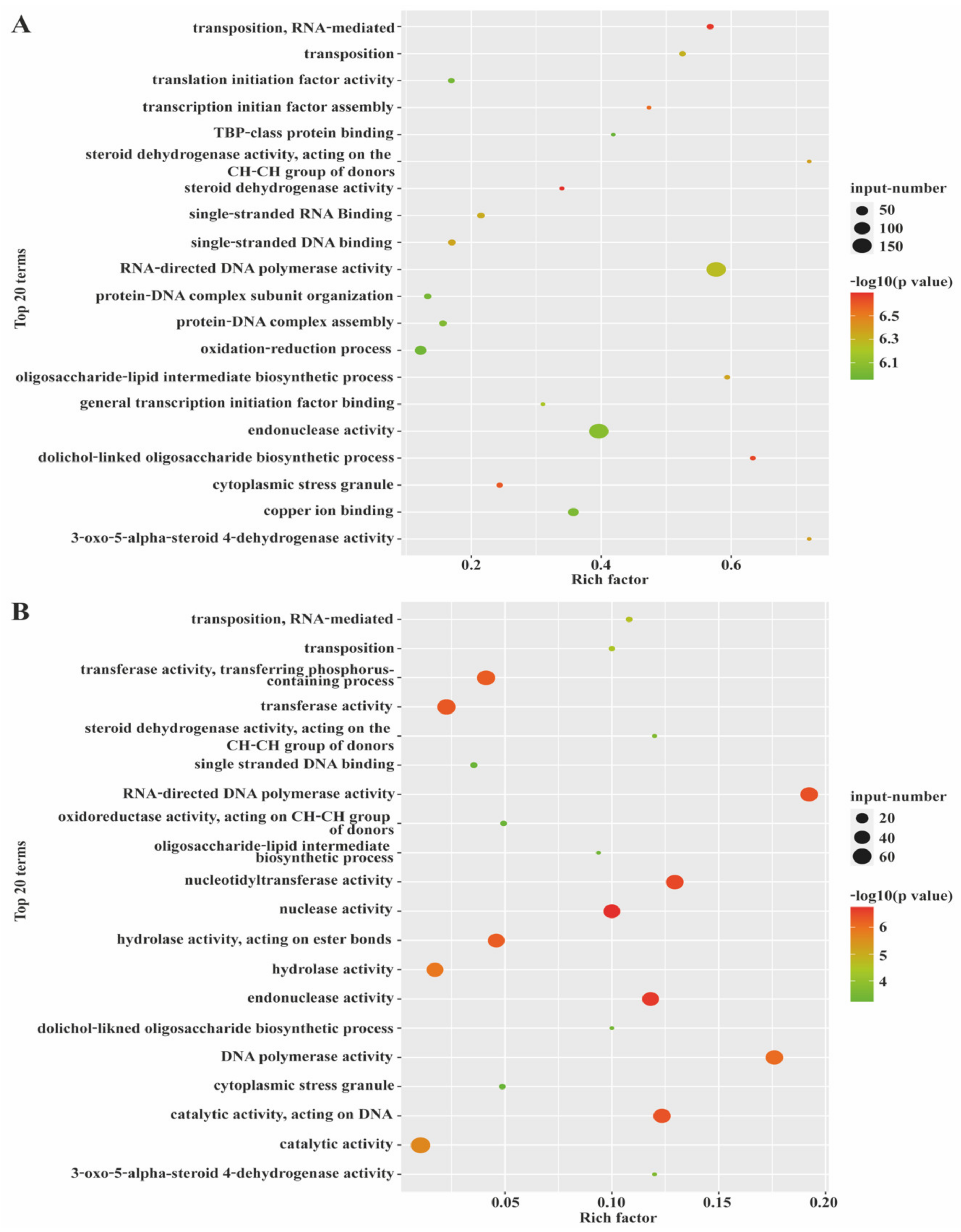
3.3. Expression Patterns and GO Enrichment Analysis of mRNAs and lncRNAs
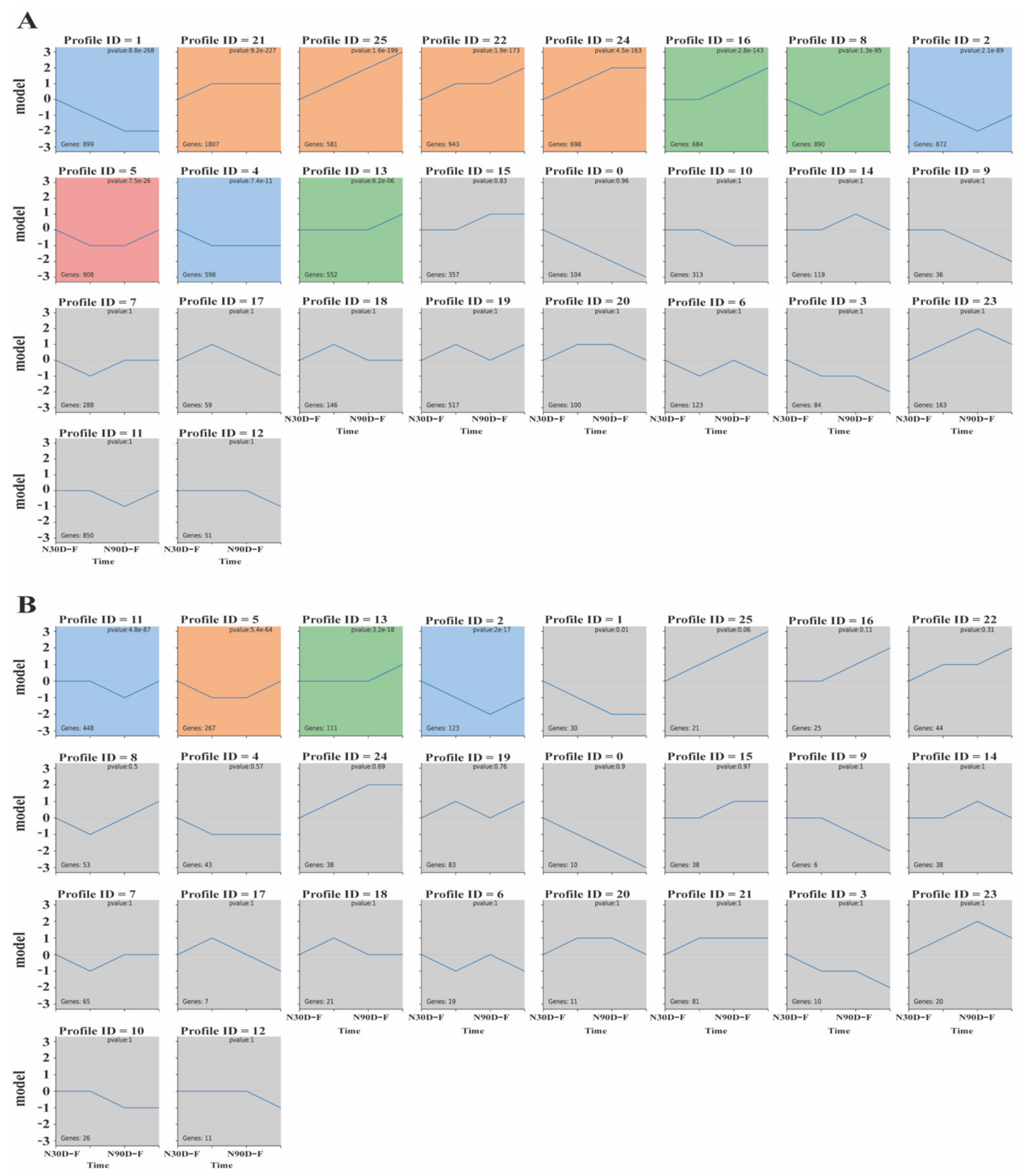
3.4. Time-Series Analysis (STEM) of mRNAs and LncRNAs
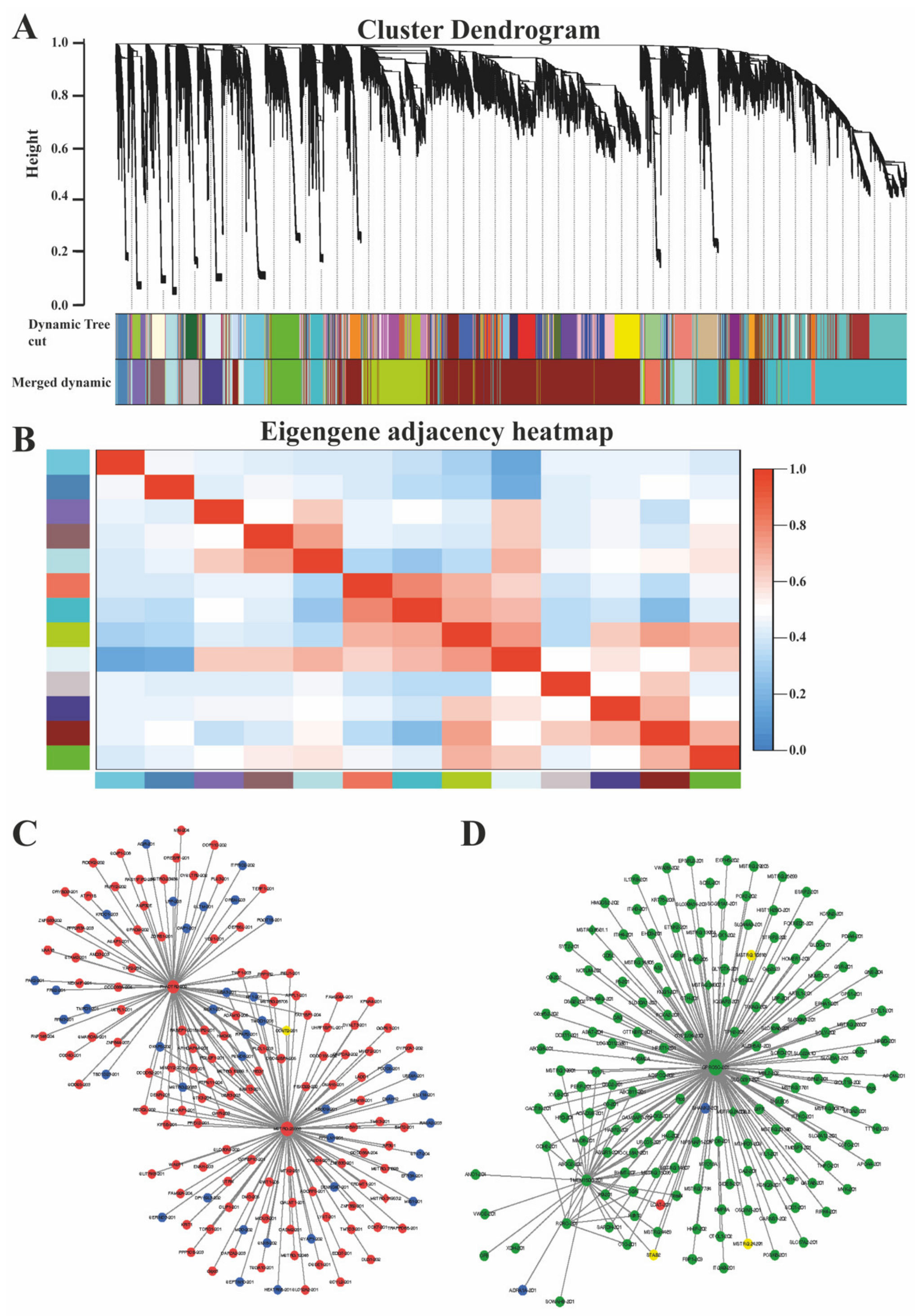
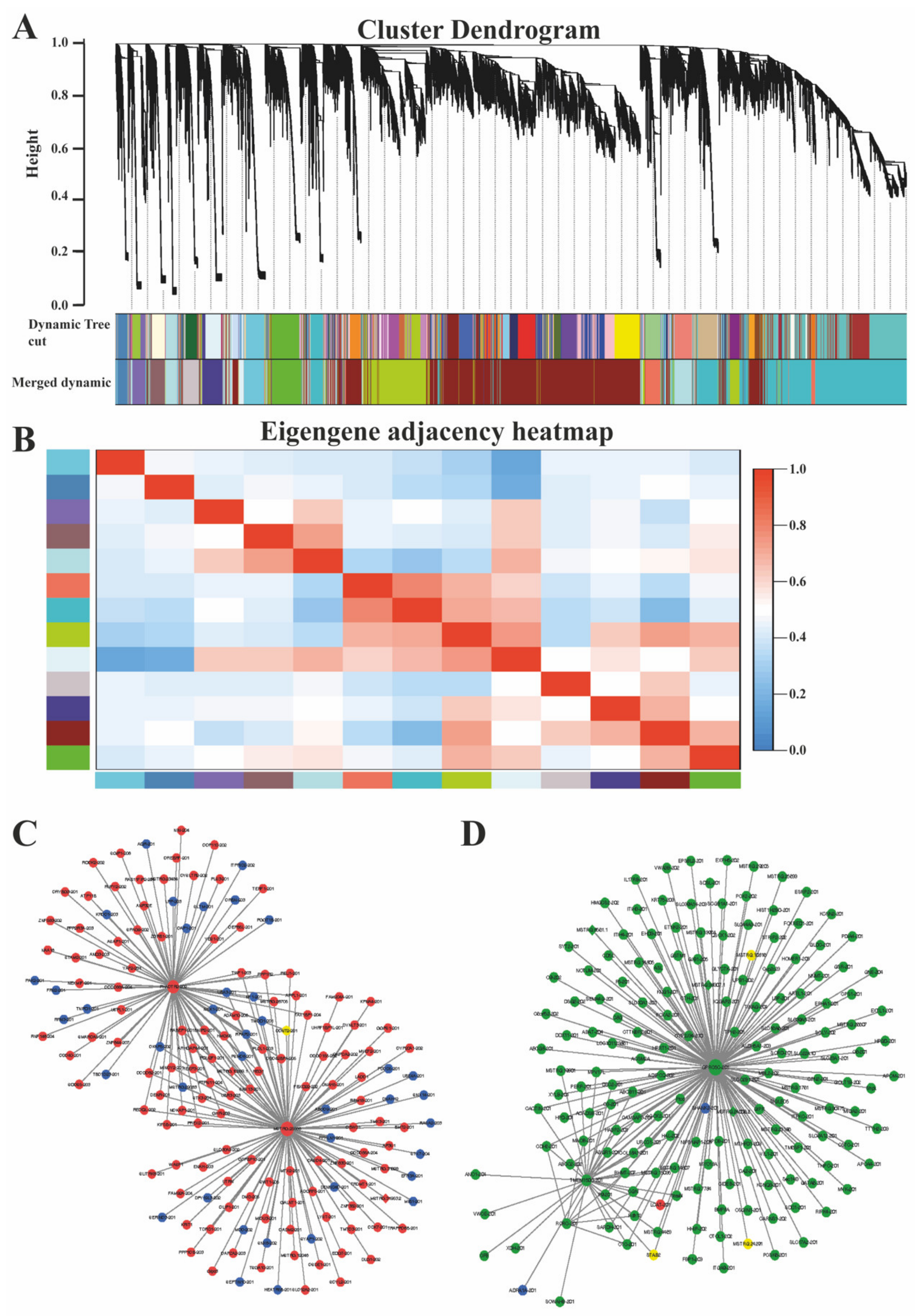
3.5. Co-Expression Network Analysis (WGCNA) of mRNA and LncRNAs
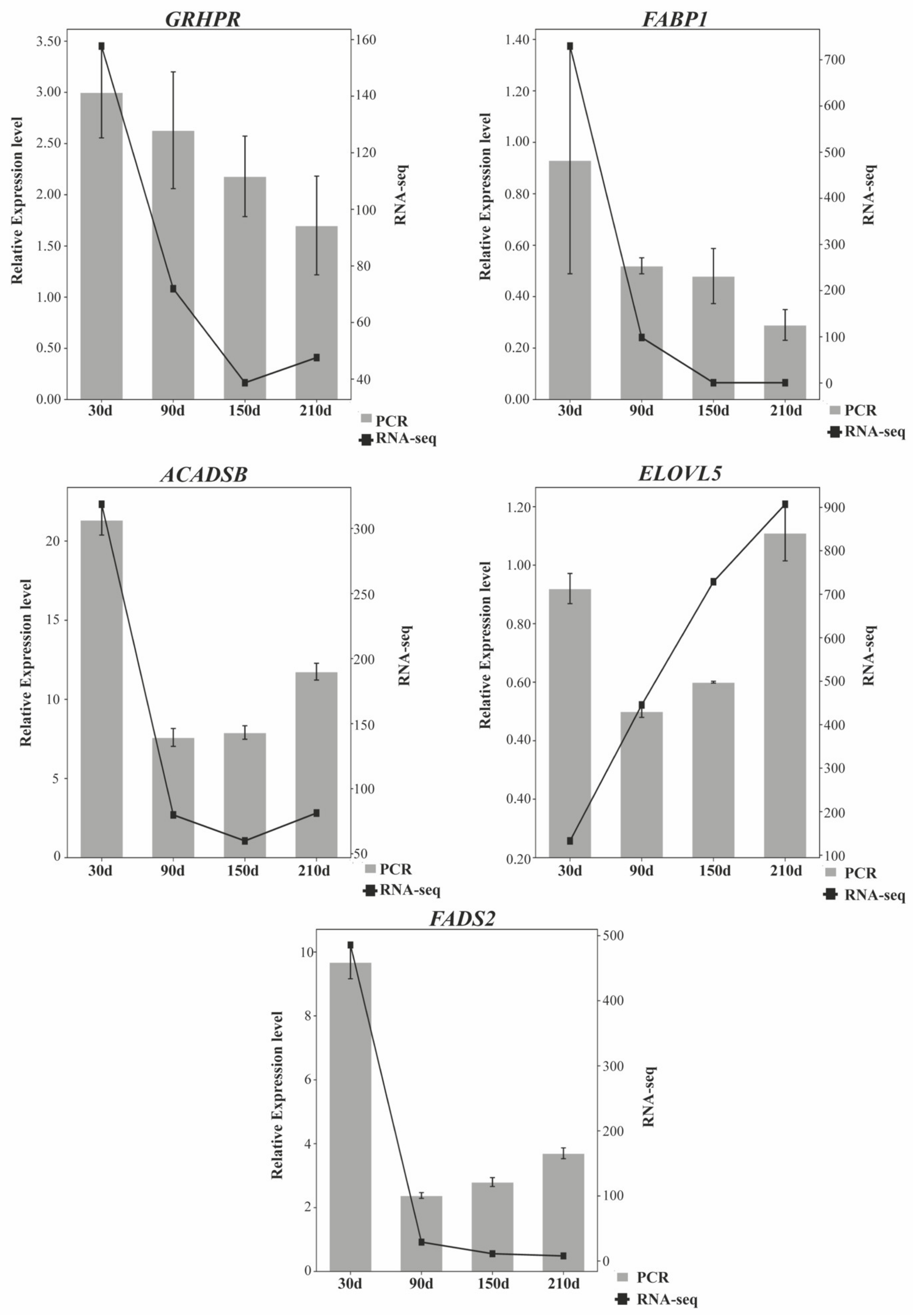
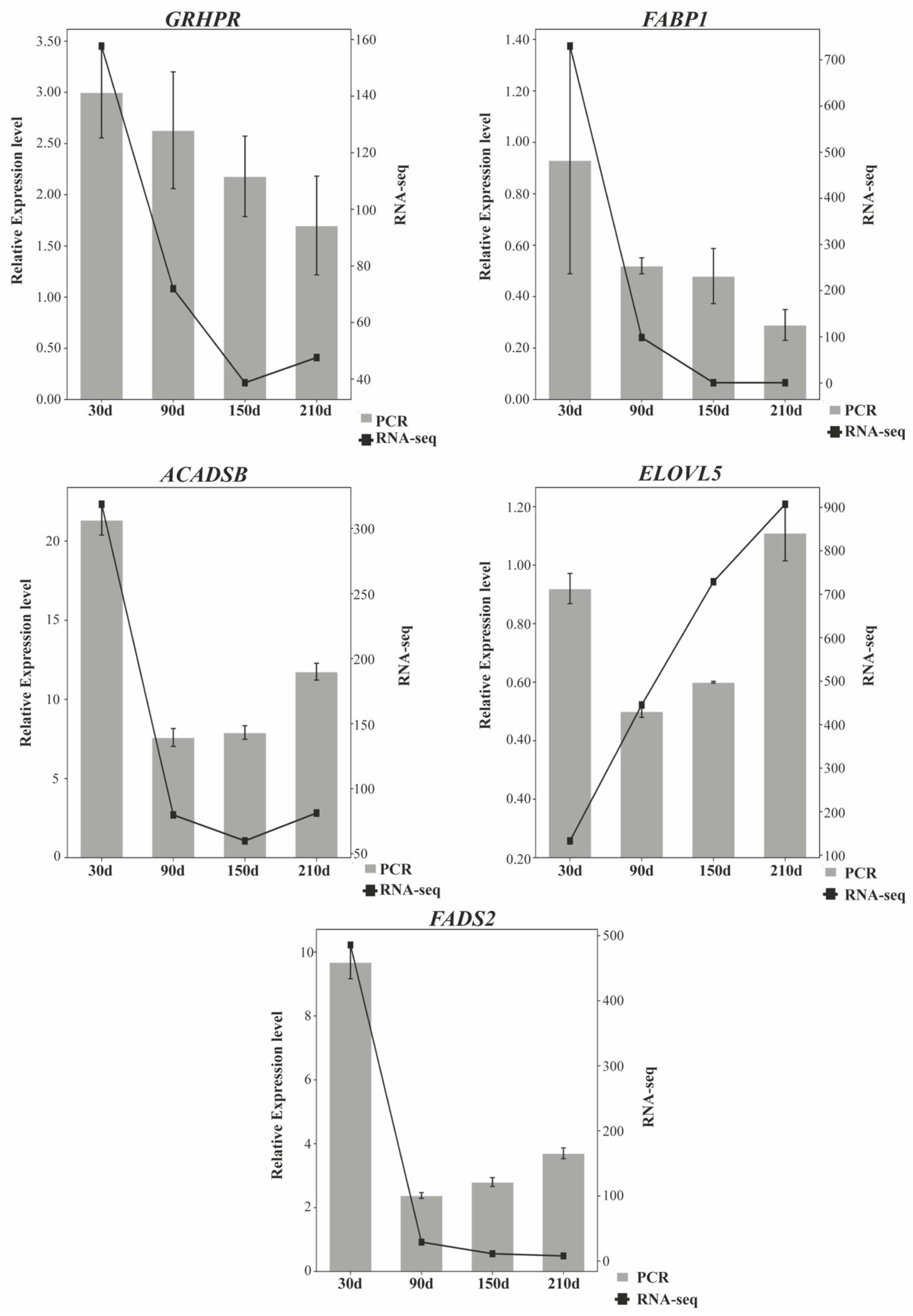
3.6. RT-qPCR Quantification of mRNAs and lncRNAs
4. Discussion
4.1. Differentially Expressed lncRNAs and mRNAs
4.2. Time-Series Analysis (STEM) of mRNAs and lncRNAs
4.3. Co-Expression Network Analysis of mRNAs and LncRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.; Yang, S.; Zhao, S.; Fan, B.; Yu, M.; Wang, H.; Li, M.; Liu, B.; Xiong, T.; Li, K. genetic diversity analyses of 10 indigenous Chinese pig populations based on 20 microsatellites. J. Anim. Sci. 2004, 82, 368–374. [Google Scholar] [CrossRef]
- Xing, X.W.; Hawthorne, W.J.; Yi, S.; Simond, D.M.; Dong, Q.; Ye, B.; Tong, Q.J.; Ye, Z.; Wang, W. Investigation of porcine endogenous retrovirus in the conservation population of Ningxiang pig. Transplant. Proc. 2009, 41, 4389–4393. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, C.; Yu, Y.; Xing, Y.; Xiao, D.; Zhang, B. Comparison of fatty acid profile of three adipose tissues in Ningxiang pigs. Anim. Nutr. 2018, 4, 256–259. [Google Scholar] [CrossRef]
- Lada, E.; Anna, M.; Patrik, M.; Zbynek, T.; Miroslav, J.; Hynek, M.; Richard, P.; Sarah, L.; Vaclav, L. Porcine liver anatomy applied to biomedicine. J. Surg. Res. 2020, 250, 70–79. [Google Scholar] [CrossRef]
- Jiang, A.; Chao, Z.; Weng, Q.; Li, B.; Yu, F.; Ning, C.; Cao, Y.; Zhao, M.; Wei, W.; Liu, H.; et al. Comparison of determination methods of porcine intramuscular fat content and correlation analysis with carcass and meat quality traits. J. Nanjing Agric. Univ. Sci. 2017, 40, 130–137. [Google Scholar]
- Zhu, J.; Peng, Y.; Li, S.; Sun, J.; Liu, X.; Yang, S. Study on genetic resources and germplasm characteristics of Ningxiang pigs. JOURNAL-HUNAN Agric. Univ. 2008, 34, 47. [Google Scholar]
- He, J.; Wu, X.L.; Zeng, Q.; Li, H.; Ma, H.; Jiang, J.; Rosa, G.J.M.; Gianola, D.; Tait, R.G.; Bauck, S. Genomic mating as sustainable breeding for Chinese indigenous Ningxiang pigs. PLoS One 2020, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, C. Establishing the triplet nature of the genetic code. Cell 2007, 128, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15 Spec No 1, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Clark, M.B.; Gascoigne, D.K.; Dinger, M.E.; Mattick, J.S. LncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res. 2011, 39, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mattick, J.S.; Mehler, M.F. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010, 1338, 20–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. Molecular biology: The transcriptional landscape of the mammalian genome. Science (80-. ). 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhao, Y.; Yang, S.; Zhang, H.; Chen, F. An integrated analysis of miRNA, lncRNA, and mRNA expression profiles. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Chen, K.; Baxter, T.; Muir, W.M.; Groenen, M.A.; Schook, L.B. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int. J. Biol. Sci. 2007, 3, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Zambonelli, P.; Gaffo, E.; Zappaterra, M.; Bortoluzzi, S.; Davoli, R. Transcriptional profiling of subcutaneous adipose tissue in Italian Large White pigs divergent for backfat thickness. Anim. Genet. 2016, 47, 306–323. [Google Scholar] [CrossRef]
- Li, X.J.; Yang, H.; Li, G.X.; Zhang, G.H.; Cheng, J.; Guan, H.; Yang, G.S. Transcriptome profile analysis of porcine adipose tissue by high-throughput sequencing. Anim. Genet. 2012, 43, 144–152. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, E.C.; Cheng, W.T.K.; Sun, H.S.; Mersmann, H.J.; Ding, S.T. Abundantly expressed genes in pig adipose tissue: An expressed sequence tag approach. J. Anim. Sci. 2006, 84, 2673–2683. [Google Scholar] [CrossRef]
- Chen, C.; Ai, H.; Ren, J.; Li, W.; Li, P.; Qiao, R.; Ouyang, J.; Yang, M.; Ma, J.; Huang, L. A global view of porcine transcriptome in three tissues from a full-sib pair with extreme phenotypes in growth and fat deposition by paired-end RNA sequencing. BMC Genomics 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Kern, C.; Wang, Y.; Chitwood, J.; Korf, I.; Delany, M.; Cheng, H.; Medrano, J.F.; Van Eenennaam, A.L.; Ernst, C.; Ross, P.; et al. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genom. 2018, 19, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. XLincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Li, L.; Huang, A.; Cui, L.; Zhang, Y.; Liu, Q.; Wang, X.; Wang, Y.; Liu, Z.; Yuan, Z.; et al. Molecular characterization and biological function of a novel lncRNA CRNG in swine. Front. Pharmacol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ren, J.; Huang, L. Characterization of the porcine insulin-like growth factor-binding protein, acid-labile subunit gene: Full-length cDNA and DNA sequence, polymorphisms and expression profile. J. Anim. Breed. Genet. 2007, 124, 133–138. [Google Scholar] [CrossRef]
- Muñoz, G.; Alcázar, E.; Fernández, A.; Barragán, C.; Carrasco, A.; de Pedro, E.; Silió, L.; Sánchez, J.L.; Rodríguez, M.C. Effects of porcine MC4R and LEPR polymorphisms, gender and Duroc sire line on economic traits in Duroc×Iberian crossbred pigs. Meat Sci. 2011, 88, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Jernås, M.; Olsson, B.; Arner, P.; Jacobson, P.; Sjöström, L.; Walley, A.; Froguel, P.; McTernan, P.G.; Hoffstedt, J.; Carlsson, L.M.S. Regulation of carboxylesterase 1 (CES1) in human adipose tissue. Biochem. Biophys. Res. Commun. 2009, 383, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Preitner, M.; Berney, X.; Thorens, B. Plac8 is required for white adipocyte differentiation in vitro and cell number control in vivo. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Preitner, M.; Berney, X.; Uldry, M.; Vitali, A.; Cinti, S.; Ledford, J.G.; Thorens, B. Plac8 is an inducer of C/EBPβ required for brown fat differentiation, thermoregulation, and control of body weight. Cell Metab. 2011, 14, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Bar-Joseph, Z.; Gitter, A.; Simon, I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 2012, 13, 552–564. [Google Scholar] [CrossRef]
- Nice, T.; Baldridge, M.; McCune, B.; Norman, J.; Lazear, H.; Artyomov, M.; Diamond, M.; Virgin, H. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science (80-. ). 2015, 347, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fu, F.; Xue, M.; Chen, W.; Liu, J.; Shi, H.; Chen, J.; Bu, Z.; Feng, L.; Liu, P. IFN-lambda preferably inhibits PEDV infection of porcine intestinal epithelial cells compared with IFN-alpha. Antiviral Res. 2017, 140, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Rautureau, Y. ADCY9 (Adenylate cyclase Type 9) inactivation protects from atherosclerosis only in the absence of CETP (Cholesteryl ester transfer protein). Circulation 2018, 138, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.V.; Zychlinsky, A.; Bardoel, B.W. Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus α-hemolysin-mediated toxicity. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Amills, M.; Vidal, O.; Varona, L.; Toma, A.; Gil, M.; Sa, A.; Noguera, J.L. Polymorphism of the pig 2,4-dienoyl CoA reductase 1 gene (DECR1) and its association with carcass and meat quality traits. Prism 2000, 493–498. [Google Scholar]
- Zhang, X.D.; Zhang, S.J.; Ding, Y.Y.; Feng, Y.F.; Zhu, H.Y.; Huang, L.; Wu, T.; Zhou, J.; Yin, Z.J. Association between ADSL, GARS-AIRS-GART, DGAT1, and DECR1 expression levels and pork meat quality traits. Genet. Mol. Res. 2015, 14, 14823–14830. [Google Scholar] [CrossRef]
- Wider, C.; Lincoln, S.J.; Heckman, M.G.; Diehl, N.N.; Stone, J.T.; Haugarvoll, K.; Aasly, J.O.; Gibson, J.M.; Lynch, T.; Rajput, A.; et al. Phactr2 and Parkinson’s disease. Neurosci. Lett. 2009, 453, 9–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amisten, S.; Al-Amily, I.M.; Soni, A.; Hawkes, R.; Atanes, P.; Persaud, S.J.; Rorsman, P.; Salehi, A. Anti-diabetic action of all-trans retinoic acid and the orphan G protein coupled receptor GPRC5C in pancreatic β-cells. Endocr. J. 2017, 64, 325–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, T.; Kim, Y.J.; Oshima, E.; Shimizu, C.; Kiyonari, H.; Abe, T.; Higashi, H.; Yamada, K.; Hirabayashi, Y. Comparative characterization of GPRC5B and GPRC5C LacZ knockin mice; behavioral abnormalities in GPRC5B-deficient mice. Biochem. Biophys. Res. Commun. 2011, 412, 460–465. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kim, Y. Roles of GPRC5 family proteins: Focusing on GPRC5B and lipid-mediated signaling. J. Biochem. 2020, 167, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.S.; Lee, B.; Oh, U. Evidence for mechanosensitive channel activity of tentonin 3/TMEM150C. Neuron 2017, 94, 271–273.e2. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.O.; Schneider, E.R.; Matson, J.D.; Gracheva, E.O.; Bagriantsev, S.N. TMEM150C/Tentonin3 is a regulator of mechano-gated ion channels. Cell Rep. 2018, 23, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.M.; Gao, X.; McKay, J.; McKay, R.; Salo, Z.; Graff, J.M. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006, 3, 25–34. [Google Scholar] [CrossRef] [Green Version]
- James, A.W.; Leucht, P.; Levi, B.; Carre, A.L.; Xu, Y.; Helms, J.A.; Longaker, M.T. Sonic hedgehog influences the balance of osteogenesis and adipogenesis in mouse adipose-derived stromal cells. Tissue Eng. Part A 2010, 16, 2605–2616. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Y.; Jiang, K.; Jia, J. Hedgehog signaling promotes lipolysis in adipose tissue through directly regulating Bmm/ATGL lipase. Dev. Biol. 2020, 457, 128–139. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardestani, A.; Lupse, B.; Maedler, K. Hippo signaling: Key emerging pathway in cellular and whole-body metabolism. Trends Endocrinol. Metab. 2018, 29, 492–509. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Kang, Q.; Zhao, Y.; Hu, X.; Li, N. Lats2 modulates adipocyte proliferation and differentiation via hippo signaling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W.; Setchell, K.D.R. Bile acid biosynthesis. Biochemistry 1992, 31, 4737–4749. [Google Scholar] [CrossRef] [PubMed]
- Broeders, E.P.M.; Nascimento, E.B.M.; Havekes, B.; Brans, B.; Roumans, K.H.M.; Tailleux, A.; Schaart, G.; Kouach, M.; Charton, J.; Deprez, B.; et al. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015, 22, 418–426. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5’ to 3’) |
|---|---|
| MSTRG.1053.3-F | ACTTGGGAAGAAAGCAATTTTAAGA |
| MSTRG.1053.3-R | TGTAGTCCCAGCTACTCGGG |
| MSTRG.11451.1-F | AGACATCCGAGCCTGGGATA |
| MSTRG.11451.1-R | CGTTTCAGAAAGCGTTGGAAGT |
| MSTRG.25924.3-F | AGACATCCGAGCCTGGGATA |
| MSTRG.25924.3-R | GTGTCGAGGGCTGACTTTCA |
| MSTRG.12045.1-F | CGCTGAGCTGTTGGGTATGA |
| MSTRG.12045.1-R | AGCGTTGGGAAGTGCTCTTT |
| MSTRG.1054.2-F | TGTAGTCCCAGCTACTCGGG |
| MSTRG.1054.2-R | ACAGGGTCTCGCTATGTTGC |
| GRHPR-F | GCAGTGGGATTCCGATGAG |
| GRHPR-R | CGTGGTGTCGCTTCTTGATTTC |
| FADS2-F | CAGCACGATTACGGCCATCT |
| FADS2-R | AGATGTTGGGCTTGGCATGA |
| FABP1-F | GTCTGCCCCGACGAACTCAT |
| FABP1-R | CTCATTCTGGACGACCTTGGA |
| ELOVL5-F | CATCCTGCGCAAGAACAACC |
| ELOVL5-R | GGGATGGATGACAGACCGTAG |
| ACADSB-F | GGAAGAGTCCAACGCATTCA |
| ACADSB-R | GAGGAGGTTGCTCTCTGACG |
| Groups | mRNAs | lncRNAs |
|---|---|---|
| 30d vs. 90d (upregulated) | -Tissue and organ morphology development -Epithelial and endothelial cell proliferation -Extracellular matrix -Cell adhesion -Calcium ion binding | -Zinc ion binding -Transition metal element ion binding -Transcription factor complex -Steroid hormone receptor activity |
| 30d vs. 150d (upregulated) | -Cell link assembly regulation -Smooth muscle cell proliferation regulation -Negative feedback regulation of angiogenesis -Collagen fiber tissue development -Protein folding | -Transcription factor complex -Ligand activated transcription factor activity -Steroid hormone receptor activity -Thyroid hormone-mediated signaling pathway -Nuclear receptor activity |
| 30d vs. 210d (upregulated) | -Endothelial cell proliferation -Negative regulation of vascular development -Regulation of cell junction assembly -Collagen fiber tissue -Adsorption of cells and substrates | -Lipid biosynthesis regulation -Fatty acid oxidation regulation -Fatty acid metabolism regulation -Acyl-CoA biosynthesis -Male gonadal development -Ion homeostasis |
| 30d vs. 90d (downregulated) | -Steroid dehydrogenase activity -Steroid metabolism -RNA-guided DNA polymerase activity -Organic acid biosynthesis -Cellular amino acid metabolism -Catalytic activity on DNA | -Catalytic activity -Hydrolase activity -Myosin filaments -Myosin complex |
| 30d vs. 150d (downregulated) | -Steroid hydroxylase and dehydrogenase activity -Cholesterol reversal transport -Lipoprotein complex -Lipoprotein particles | -DNA packaging complex -Protein-DNA complex -Nucleosome -Protein heterodimerization activity -21-carbon steroid hormone metabolism -Androgen dehydrogenase activity -Ketosteroid monooxygenase activity |
| 30d vs. 210d (downregulated) | -Triglyceride homeostasis -Sterol biosynthesis and transport -High-density lipoprotein particles -Dihydroxy acid metabolism process | -Protein-DNA complex -Protein heterodimerization activity -Nucleosome |
| Groups | mRNAs | lncRNAs |
|---|---|---|
| 30d vs. 90d (upregulated) | -Tissue and organ morphology development -Epithelial and endothelial cell proliferation -Extracellular matrix -Cell adhesion -Calcium ion binding | -Zinc ion binding -Transition metal element ion binding -Transcription factor complex -Steroid hormone receptor activity |
| 90d vs. 150d (upregulated) | -Protein folding -Chaperone-mediated protein folding | -RNA binding -Positive regulation of epithelial cell -proliferation -Male gonadal development -Cell response to peptides |
| 150d vs. 210d (upregulated) | -Sterol homeostasis and transport, -Lipoprotein particles, -High-density lipoprotein particles, -Cholesterol homeostasis and transport activity | -Transferase activity -DNA polymerase activity -Nucleotide transaminase activity -Nuclease activity -Endonuclease activity -Hydrolase activity acting on ester bonds |
| 30d vs. 90d (downregulated) | -Steroid dehydrogenase activity -Steroid metabolism -RNA-guided DNA polymerase activity -Organic acid biosynthesis -Cellular amino acid metabolism -Catalytic activity on DNA | -Catalytic activity -Hydrolase activity -Myosin filaments -Myosin complex |
| 90d vs. 150d (downregulated) | -Transferase activity -DNA polymerase activity -Nuclease activity -Nucleotide transferase activity -Hydrolase activity acting on ester bonds -Steroid dehydrogenase activity | -Protein-DNA complexes -Protein heterodimers and dimer activity -Nucleosomes -DNA packaging complexes and chromatin parts |
| 150d vs. 210d (downregulated) | -Protein folding -Chaperone-mediated protein folding -Protein stability regulation | -Thyroid development -Signal pathway mediated by thyroid hormone -Negative regulation of transcription initiation by RNA polymerase II promoter |
| mRNA | mRNA Functional Description | Associated lncRNAs |
|---|---|---|
| IFNLR1 | Encoding receptors for cytokine ligands IFNL2 and IFNL3 and mediating their antiviral activity | MSTRG.23976.1 MSTRG.12196.6 MSTRG.26481.9 MSTRG.42001.1 |
| TARBP1 | S-adenosyl-L-methionine-dependent methyltransferase can methylate RNA molecules, such as tRNA | MSTRG.9023.5 MSTRG.11929.3 |
| ADCY9 | Adenylate cyclase can catalyze the signal molecule cAMP to respond to the activation of G protein-coupled receptors. | MSTRG.18828.1 MSTRG.15237.1 |
| mRNA | mRNA Functional Description | Associated lncRNAs |
|---|---|---|
| SGMS1 | Encoding ceramide choline phosphotransferase, which catalyzes the reversible transfer of part of phosphocholine during the biosynthesis of sphingomyelin. In the forward reaction, the head-end group of phosphatidylcholine can be transferred to ceramide to form ceramide phosphorylcholine and diacylglycerol. In the reverse reaction, the product obtained is the opposite of the uninhibited reaction. The direction of the reaction is related to the levels of ceramide and diacylglycerol in the Golgi membrane | MSTRG.1058.1 |
| DECR1 | Encoding 2,4-dienoyl-CoA reductase 1, involved in β-oxidation and metabolism of unsaturated fatty enoyl-CoA esters. | MSTRG.24004.1 |
| HS3ST4 | Encoding heparan sulfate-glucosamine 3-sulfotransferase 4 | MSTRG.25084.1 MSTRG.8247.1 MSTRG.8043.1 MSTRG.7789.1 MSTRG.7789.1 MSTRG.7605.3 MSTRG.6591.1 MSTRG.45142.4 |
| ZNF709 | Encoding zinc finger protein 709 | MSTRG.38367.1 MSTRG.24004.1 MSTRG.25790.1 MSTRG.27767.1 MSTRG.23025.1 MSTRG.1895.1 MSTRG.24004.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; He, J.; Li, B.; Xiao, Y.; Zeng, Q.; Xu, K.; Duan, Y.; He, J.; Ma, H. Integrated Analysis of lncRNA and mRNA in Subcutaneous Adipose Tissue of Ningxiang Pig. Biology 2021, 10, 726. https://doi.org/10.3390/biology10080726
Gong Y, He J, Li B, Xiao Y, Zeng Q, Xu K, Duan Y, He J, Ma H. Integrated Analysis of lncRNA and mRNA in Subcutaneous Adipose Tissue of Ningxiang Pig. Biology. 2021; 10(8):726. https://doi.org/10.3390/biology10080726
Chicago/Turabian StyleGong, Yan, Jun He, Biao Li, Yu Xiao, Qinghua Zeng, Kang Xu, Yehui Duan, Jianhua He, and Haiming Ma. 2021. "Integrated Analysis of lncRNA and mRNA in Subcutaneous Adipose Tissue of Ningxiang Pig" Biology 10, no. 8: 726. https://doi.org/10.3390/biology10080726
APA StyleGong, Y., He, J., Li, B., Xiao, Y., Zeng, Q., Xu, K., Duan, Y., He, J., & Ma, H. (2021). Integrated Analysis of lncRNA and mRNA in Subcutaneous Adipose Tissue of Ningxiang Pig. Biology, 10(8), 726. https://doi.org/10.3390/biology10080726