
Male Differentiation in the Marine Copepod Oithona nana Reveals the Development of a New Nervous Ganglion and Lin12-Notch-Repeat Protein-Associated Proteolysis

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sex Ratio in the Toulon Little Bay
2.2. Immunofluorescence Analysis
2.3. Biological Materials and Rna-Seq Experiments
2.4. Sex-Determination System Identification by Rna-Seq
2.5. Copepod Phylogenetic Tree
2.6. Gene Annotation
2.7. Hmm Search for Ldpgs Identification
2.8. Phylogeny Tree of Oithona nana Lnr Domains
2.9. Differential Expression Analysis
2.10. Protein–Protein Interaction Assays by Yeast Two-Hybrid Screening
3. Results
3.1. Female-Biased Sex Ratio of Oithona nana
3.2. Central Nervous System Labelling by Immunofluorescence
3.3. Transcriptomic Support for Oithona nana Male Homogamety
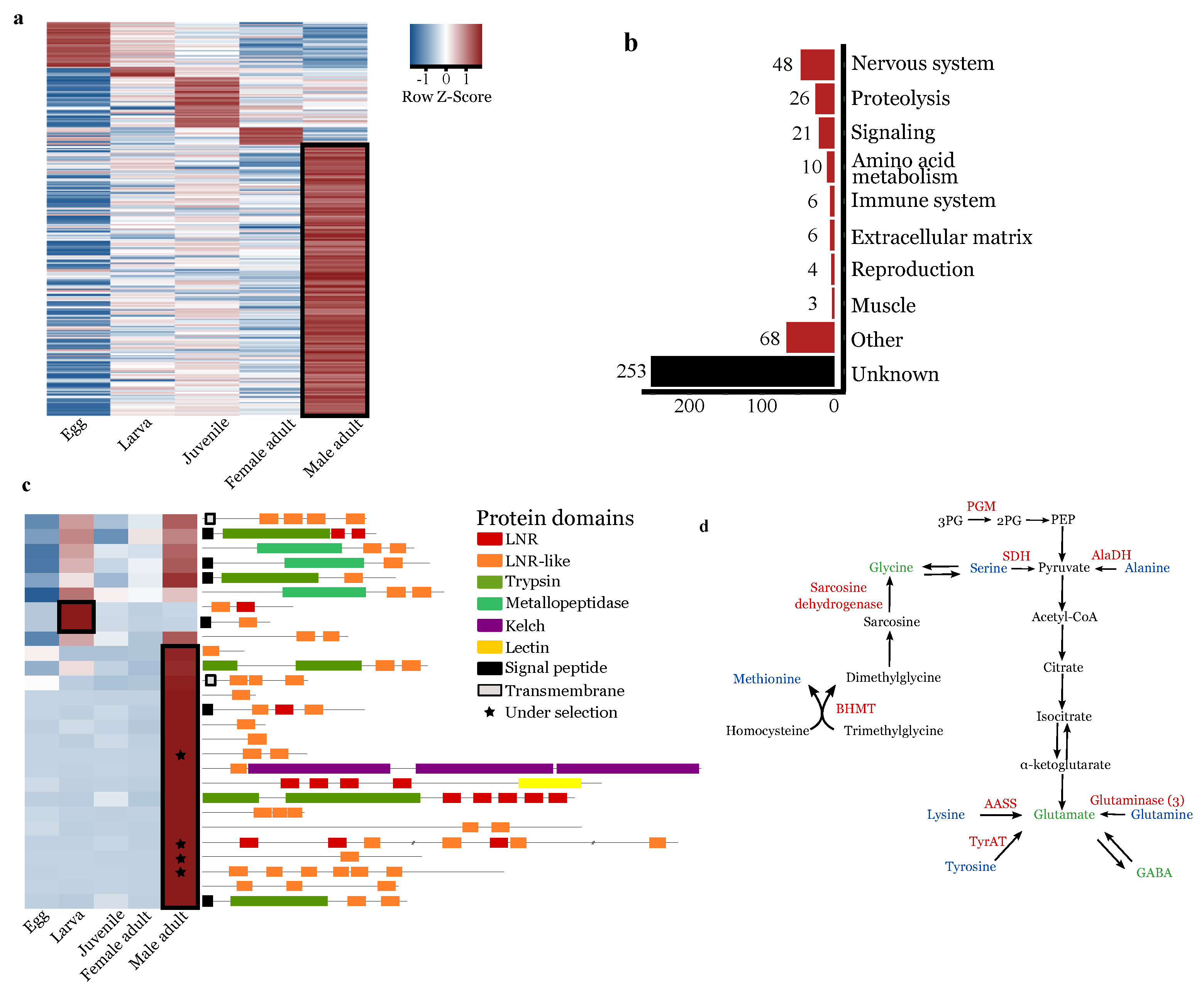
3.4. Lnr Domains Burst in the O. nana Proteome
3.5. Oithona nana Male Gene Expression
3.5.1. Upregulation of Lnr-Coding and Proteolytic Genes in Males
3.5.2. Upregulation of Nervous-System-Associated Genes in Adult Males
3.5.3. Upregulation of Amino-Acid Conversion into Neurotransmitters in Male Adults
3.5.4. Downregulation of Food Uptake Regulation in Male Adults
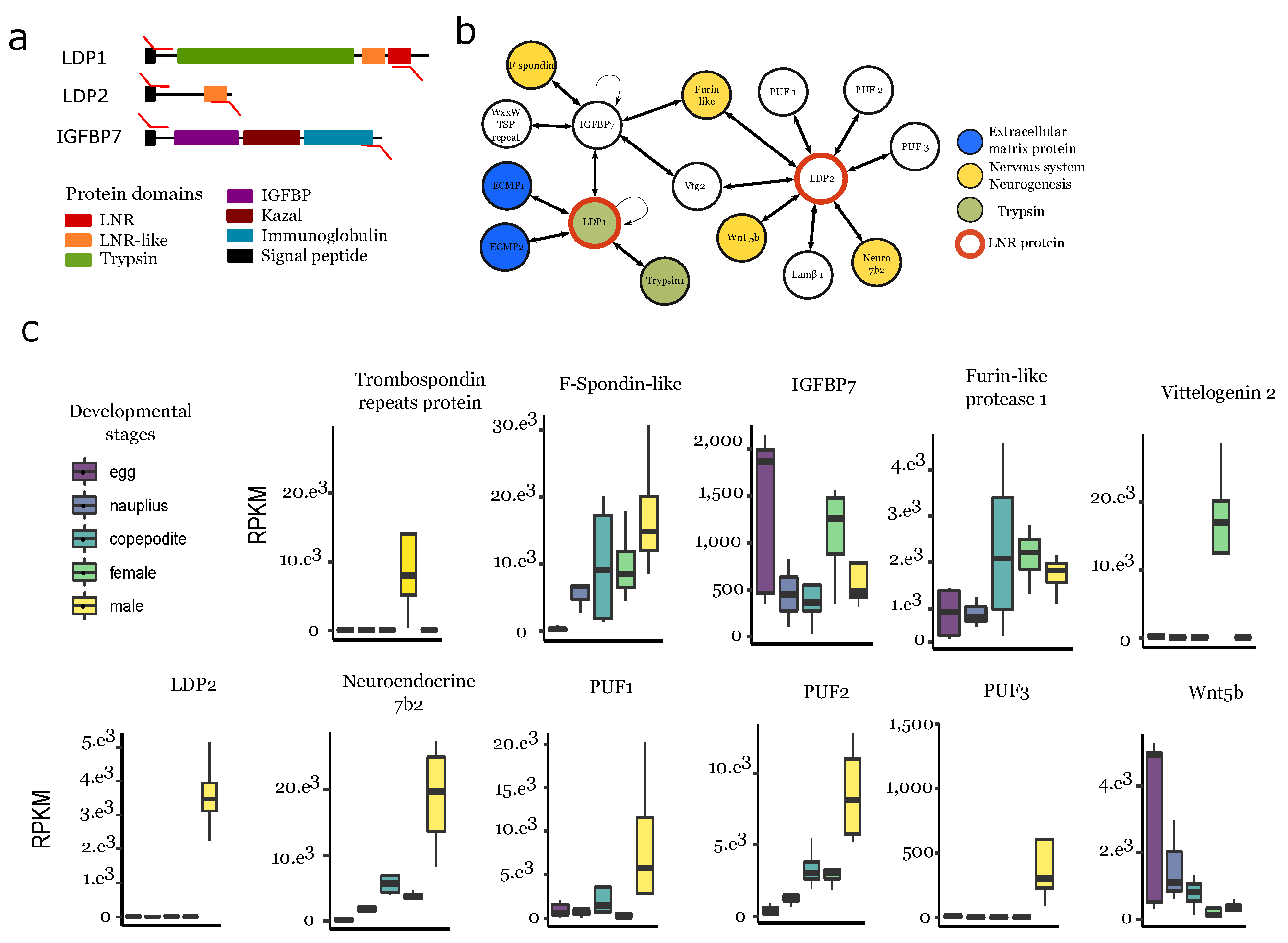
3.6. Protein–Protein Interaction Network Involving Ldps and Igfbp
4. Discussion
4.1. Zw Sexual System and High Mortality Rate of O. nana Males
4.2. The Development of a Male-Specific Nervous Ganglion Is Supported by Nervous System-Gene Expression
4.3. Ldp-Driven Proteolysis Is Potentially Linked to the Male Ganglion Formation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| LDPG | Lin12-Notch Domain-Containing Protein coding Gene |
| LDP | Lin12-Notch Domain-Containing Protein |
References
- Huys, R.; Boxshall, G.A. Copepod Evolution; Ray Society: London, UK, 1991; p. 468. ISBN 0-903-87421-0. [Google Scholar]
- Kiørboe, T. What makes pelagic copepods so successful? J. Plankton Res. 2011, 33, 677–685. [Google Scholar] [CrossRef]
- Gallienne, C.P.; Robins, D.B. Is Oithona the most important copepod in the world’s oceans? J. Plankton Res. 2001, 23, 1421–1432. [Google Scholar] [CrossRef]
- Nishida, S. Taxonomy and distribution of the family oithonidae (Copepoda, Cyclopoida) in the Pacific and Indian. Bull. Ocean Res. Inst. 1985, 20, 1–167. [Google Scholar]
- Steinberg, D.K.; Landry, M.R. Zooplankton and the Ocean Carbon Cycle. Ann. Rev. Mar. Sci. 2017, 9, 413–444. [Google Scholar] [CrossRef]
- Cornils, A.; Wend-Heckmann, B.; Held, C. Global phylogeography of Oithona similis s.l. (Crustacea, Copepoda, Oithonidae)—A cosmopolitan plankton species or a complex of cryptic lineages? Mol. Phylogenet. Evol. 2017, 107, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Dvoretsky, V.G.; Dvoretsky, A.G. Life cycle of Oithona similis (Copepoda: Cyclopoida) in Kola Bay (Barents Sea). Mar. Biol. 2009, 156, 1433–1446. [Google Scholar] [CrossRef]
- Kiørboe, T. Mate finding, mating, and population dynamics in a planktonic copepod Oithona davisae: There are too few males. Limnol. Oceanogr. 2007, 52, 1511–1522. [Google Scholar] [CrossRef]
- Madoui, M.A.; Poulain, J.; Sugier, K.; Wessner, M.; Noel, B.; Berline, L.; Labadie, K.; Cornils, A.; Blanco-Bercial, L.; Stemmann, L.; et al. New insights into global biogeography, population structure and natural selection from the genome of the epipelagic copepod Oithona. Mol. Ecol. 2017, 26, 4467–4482. [Google Scholar] [CrossRef]
- Mironova, E.; Pasternak, A. Female gonad morphology of small copepods Oithona similis and Microsetella norvegica. Polar Biol. 2017, 40, 685–696. [Google Scholar] [CrossRef]
- Paffenhöfer, G.A. On the ecology of marine cyclopoid copepods (Crustacea, Copepoda). J. Plankton Res. 1993, 15, 37–55. [Google Scholar] [CrossRef]
- Sugier, K.; Vacherie, B.; Cornils, A.; Wincker, P.; Jamet, J.L.; Madoui, M.A. Chitin distribution in the Oithona digestive and reproductive systems revealed by fluorescence microscopy. PeerJ 2018, 2018. [Google Scholar] [CrossRef]
- Zamora-Terol, S.; Kjellerup, S.; Swalethorp, R.; Saiz, E.; Nielsen, T.G. Population dynamics and production of the small copepod Oithona spp. in a subarctic fjord of West Greenland. Polar Biol. 2014, 37, 953–965. [Google Scholar] [CrossRef]
- Richard, S.; Jamet, J.l. An Unusual Distribution of Oithona nana GIESBRECHT (1892) (Crustacea: Cyclopoida) in a Bay: The Case of Toulon Bay (France, Miditerranean Sea). J. Coast. Res. 2001, 17, 957–963. [Google Scholar]
- Temperoni, B.; Viñas, M.D.; Diovisalvi, N.; Negri, R.; Vinas, M.D.; Diovisalvi, N.; Negri, R. Seasonal production of Oithona nana Giesbrecht, 1893 (Copepoda: Cyclopoida) in temperate coastal waters off Argentina. J. Plankton Res. 2011, 33, 729–740. [Google Scholar] [CrossRef]
- Hirst, A.G.; Bonnet, D.; Conway, D.V.P.; Kiørboe, T. Does predation control adult sex ratios and longevities in marine pelagic copepods? Limnol. Oceanogr. 2010, 55, 2193–2206. [Google Scholar] [CrossRef]
- Kiørboe, T. Sex, sex-ratios, and the dynamics of pelagic copepod populations. Oecologia 2006, 148, 40–50. [Google Scholar] [CrossRef]
- Voordouw, M.J.; Anholt, B.R. Environmental sex determination in a splash pool copepod. Biol. J. Linn. Soc. 2002, 76, 511–520. [Google Scholar] [CrossRef][Green Version]
- Arif, M.; Gauthier, J.; Sugier, K.; Iudicone, D.; Jaillon, O.; Wincker, P.; Peterlongo, P.; Madoui, M.M.A.M. Discovering Millions of Plankton Genomic Markers from the Atlantic Ocean and the Mediterranean Sea. Mol. Ecol. Resour. 2019, 19, 526–535. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Käfer, J.; Lartillot, N.; Marais, G.A.B.; Picard, F. Detecting sex-linked genes using genotyped individuals sampled in natural populations. Genetics 2021, 218, iyab053. [Google Scholar] [CrossRef]
- Muyle, A.; Käfer, J.; Zemp, N.; Mousset, S.; Picard, F.; Marais, G.A. Sex-detector: A probabilistic approach to study sex chromosomes in non-model organisms. Genome Biol. Evol. 2016, 8, 2530–2543. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.S.; Call, G.B.; Guptan, P.; Cespedes, A.; Marshall, J.; Yackle, K.; Owusu-Ansah, E.; Mandal, S.; Fang, Q.A.; Goodstein, G.L.; et al. An efficient genetic screen in Drosophila to identify nuclear-encoded genes with mitochondrial function. Genetics 2006, 174, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Rosen, O.; Sagi, A. From the discovery of the crustacean androgenic gland to the insulin-like hormone in six decades. Gen. Comp. Endocrinol. 2011, 173, 381–388. [Google Scholar] [CrossRef]
- Frase, T.; Richter, S. The brain and the corresponding sense organs in calanoid copepods—Evidence of vestiges of compound eyes. Arthropod Struct. Dev. 2020, 54, 100902. [Google Scholar] [CrossRef] [PubMed]
- Andrew, D.R.; Brown, S.M.; Strausfeld, N.J. The minute brain of the copepod Tigriopus californicus supports a complex ancestral ground pattern of the tetraconate cerebral nervous systems. J. Comp. Neurol. 2012, 520, 3446–3470. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Fetter, R.D.; Bargmann, C.I. Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell 2004, 116, 869–881. [Google Scholar] [CrossRef]
- Zou, Y. Wnt signaling in axon guidance. Trends Neurosci. 2004, 27, 528–532. [Google Scholar] [CrossRef]
- Randlett, O.; Poggi, L.; Zolessi, F.R.; Harris, W.A. The oriented emergence of axons from retinal ganglion cells is directed by laminin contact. Neuron 2011, 70, 266–280. [Google Scholar] [CrossRef]
- Boldt, H.B.; Conover, C.A. Pregnancy-associated plasma protein-A (PAPP-A): A local regulator of IGF bioavailability through cleavage of IGFBPs. Growth Horm. IGF Res. 2007, 17, 10–18. [Google Scholar] [CrossRef]
- Sanchez-Irizarry, C.; Carpenter, A.C.; Weng, A.P.; Pear, W.S.; Aster, J.C.; Blacklow, S.C. Notch Subunit Heterodimerization and Prevention of Ligand-Independent Proteolytic Activation Depend, Respectively, on a Novel Domain and the LNR Repeats. Mol. Cell. Biol. 2004, 24, 9265–9273. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugier, K.; Laso-Jadart, R.; Vacherie, B.; Käfer, J.; Bertrand, L.; Labadie, K.; Martins, N.; Orvain, C.; Petit, E.; Wincker, P.; et al. Male Differentiation in the Marine Copepod Oithona nana Reveals the Development of a New Nervous Ganglion and Lin12-Notch-Repeat Protein-Associated Proteolysis. Biology 2021, 10, 657. https://doi.org/10.3390/biology10070657
Sugier K, Laso-Jadart R, Vacherie B, Käfer J, Bertrand L, Labadie K, Martins N, Orvain C, Petit E, Wincker P, et al. Male Differentiation in the Marine Copepod Oithona nana Reveals the Development of a New Nervous Ganglion and Lin12-Notch-Repeat Protein-Associated Proteolysis. Biology. 2021; 10(7):657. https://doi.org/10.3390/biology10070657
Chicago/Turabian StyleSugier, Kevin, Romuald Laso-Jadart, Benoît Vacherie, Jos Käfer, Laurie Bertrand, Karine Labadie, Nathalie Martins, Céline Orvain, Emmanuelle Petit, Patrick Wincker, and et al. 2021. "Male Differentiation in the Marine Copepod Oithona nana Reveals the Development of a New Nervous Ganglion and Lin12-Notch-Repeat Protein-Associated Proteolysis" Biology 10, no. 7: 657. https://doi.org/10.3390/biology10070657
APA StyleSugier, K., Laso-Jadart, R., Vacherie, B., Käfer, J., Bertrand, L., Labadie, K., Martins, N., Orvain, C., Petit, E., Wincker, P., Jamet, J.-L., Alberti, A., & Madoui, M.-A. (2021). Male Differentiation in the Marine Copepod Oithona nana Reveals the Development of a New Nervous Ganglion and Lin12-Notch-Repeat Protein-Associated Proteolysis. Biology, 10(7), 657. https://doi.org/10.3390/biology10070657