
Plant-Based Phytochemical Screening by Targeting Main Protease of SARS-CoV-2 to Design Effective Potent Inhibitors

 ,
,  ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Ligand Preparation
2.3. Molecular Docking Study
2.4. ADMET
2.5. Molecular Dynamics Simulation
3. Results
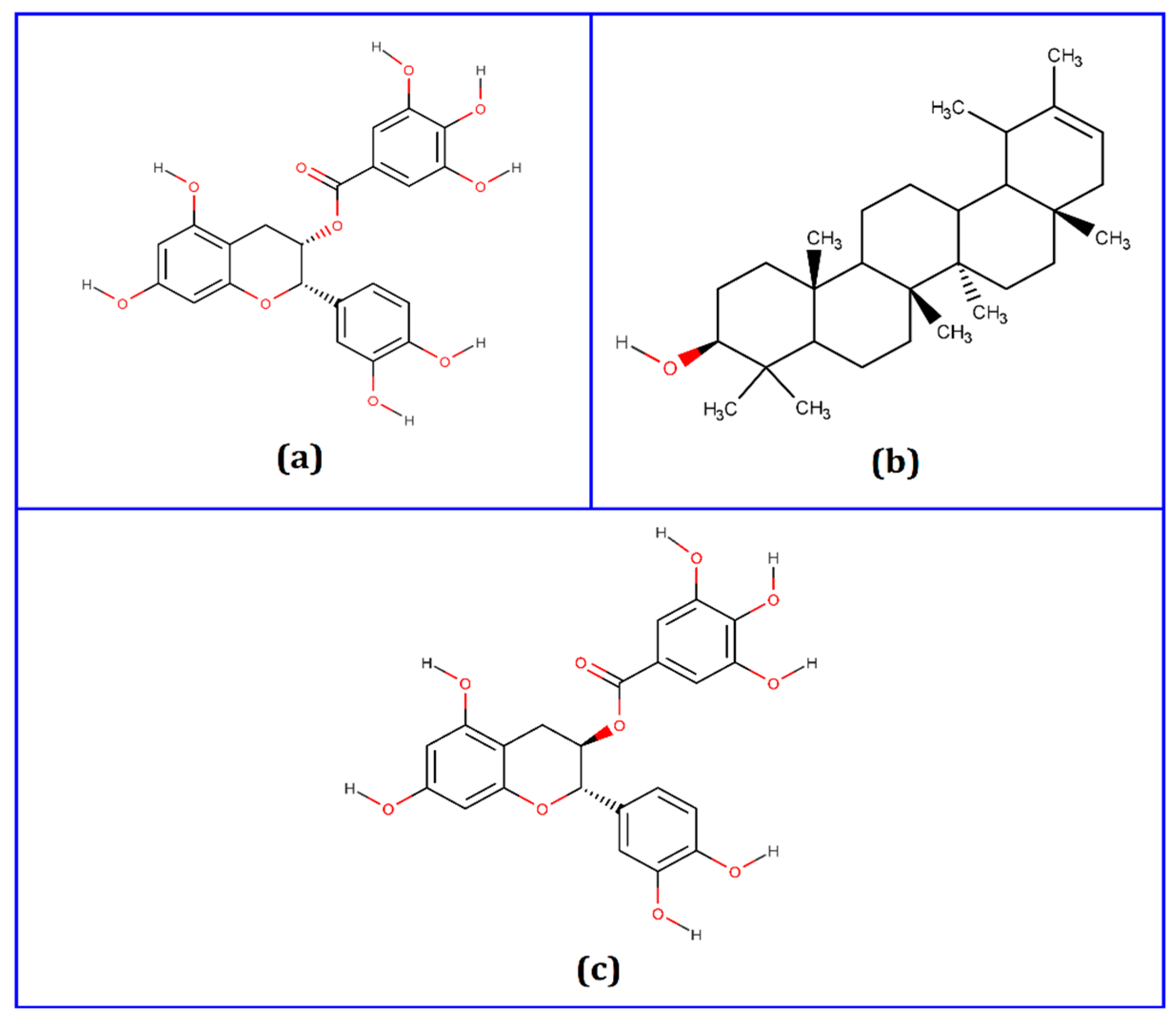
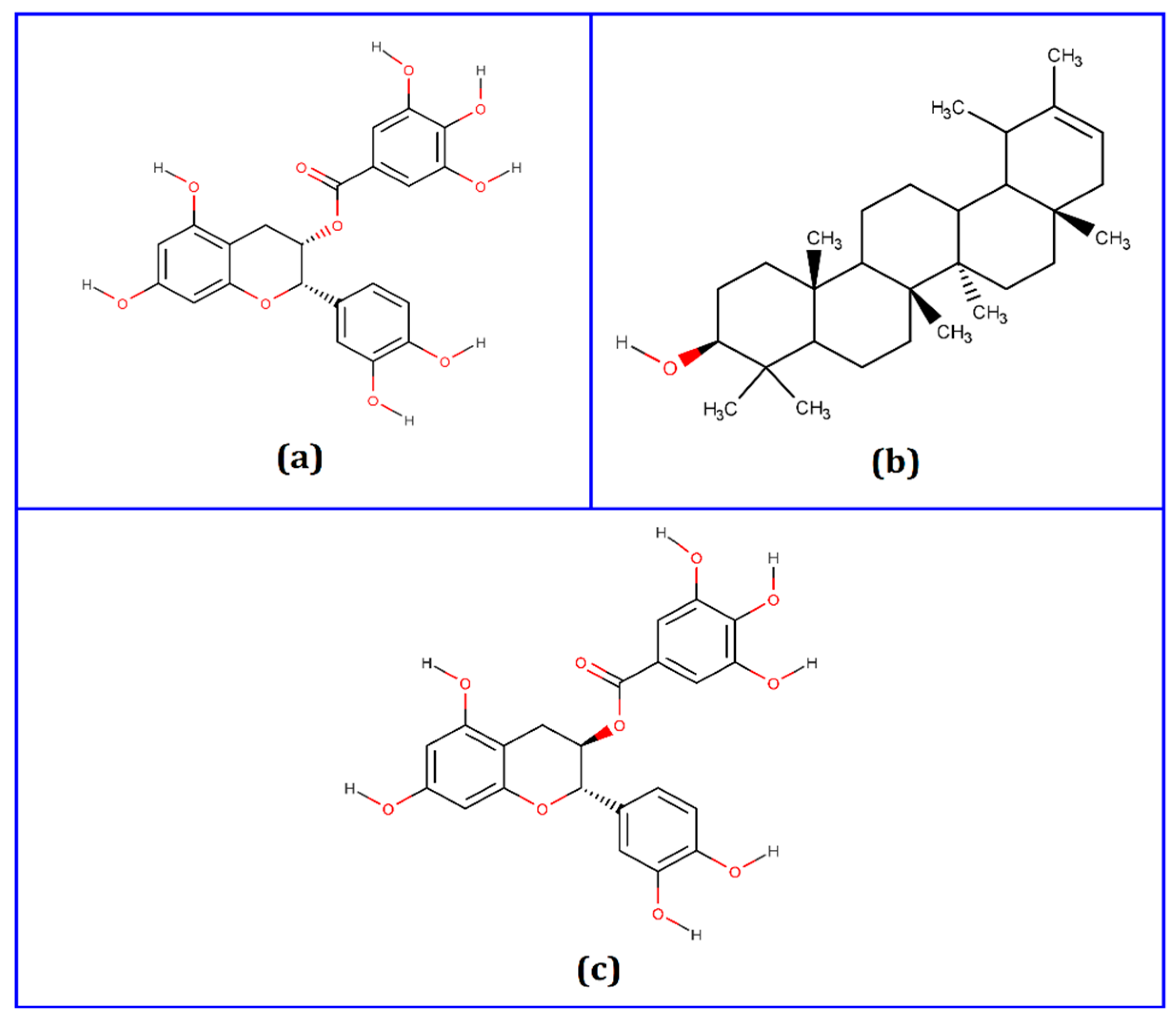
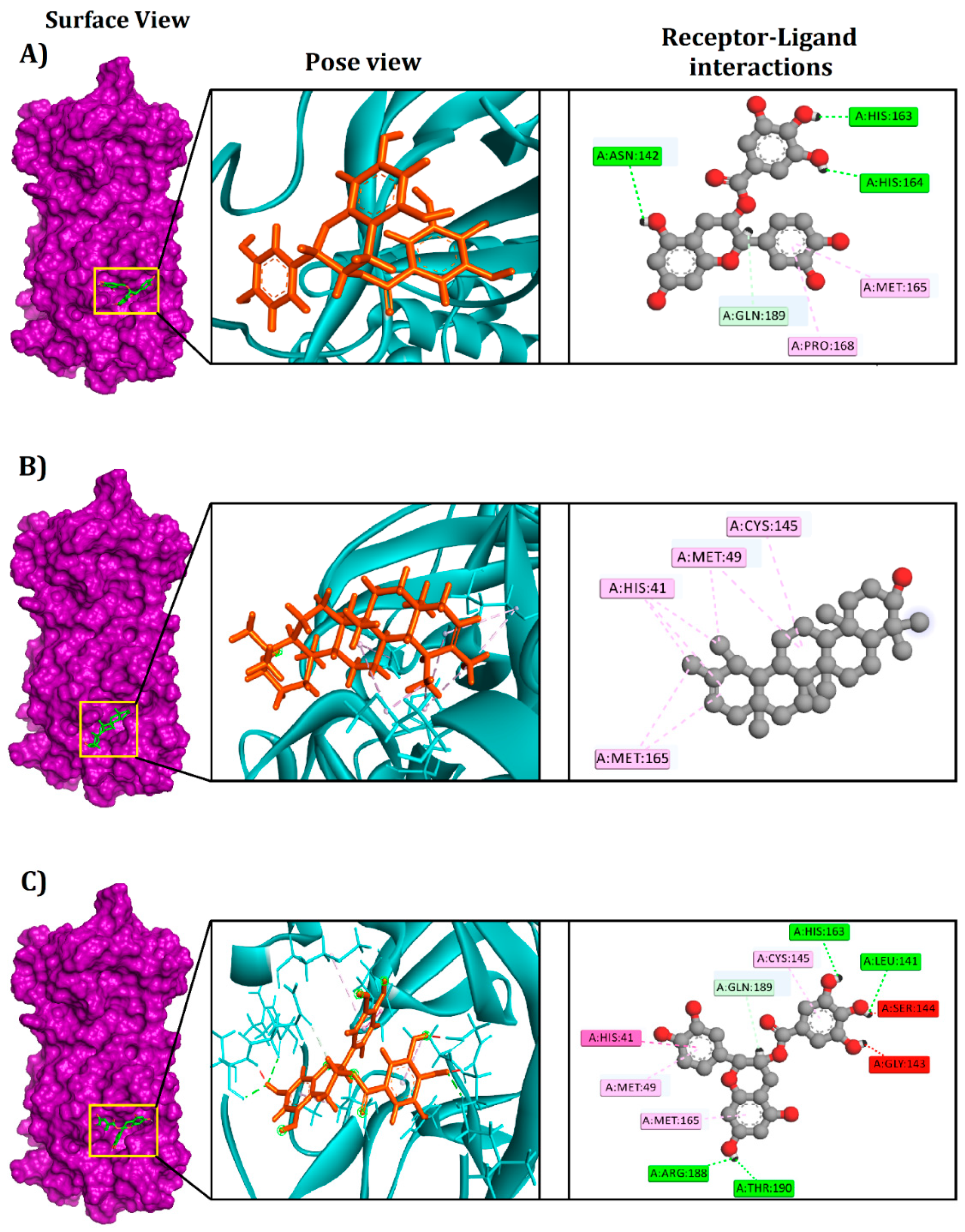
3.1. Molecular Docking Analysis
3.2. ADMET
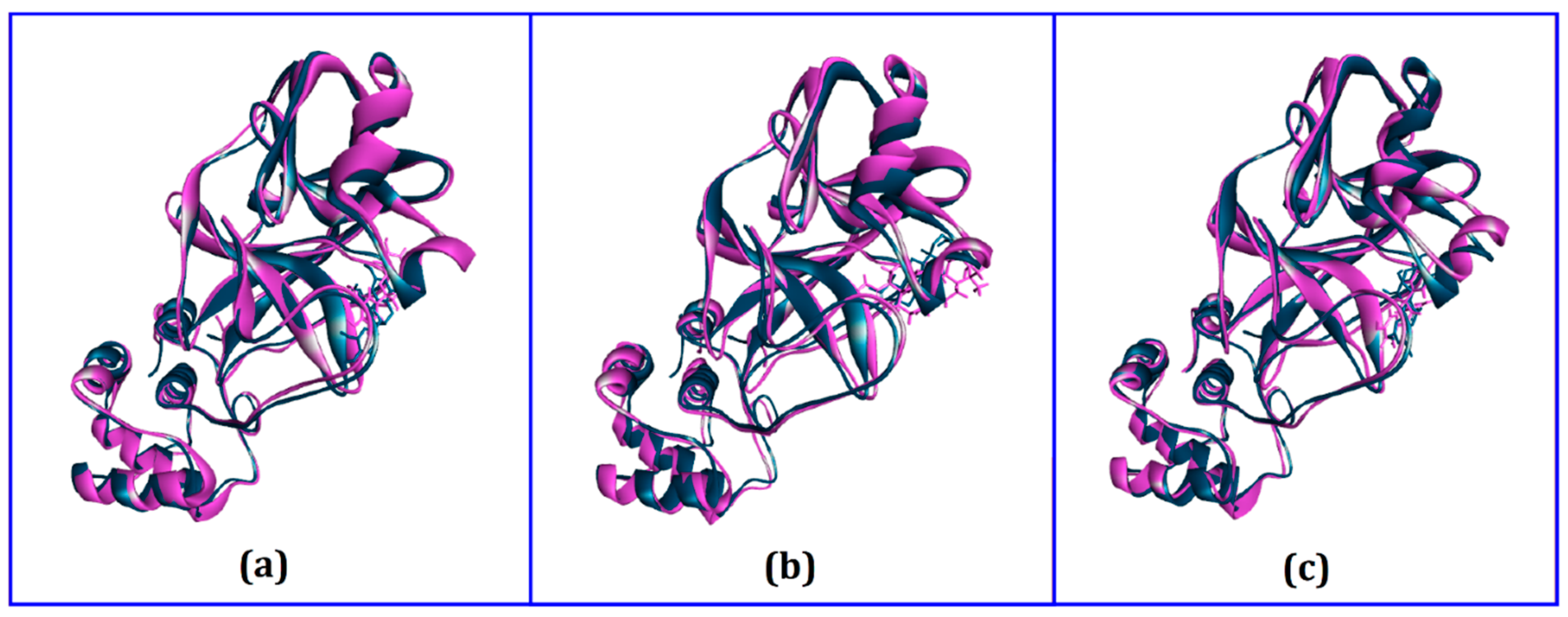
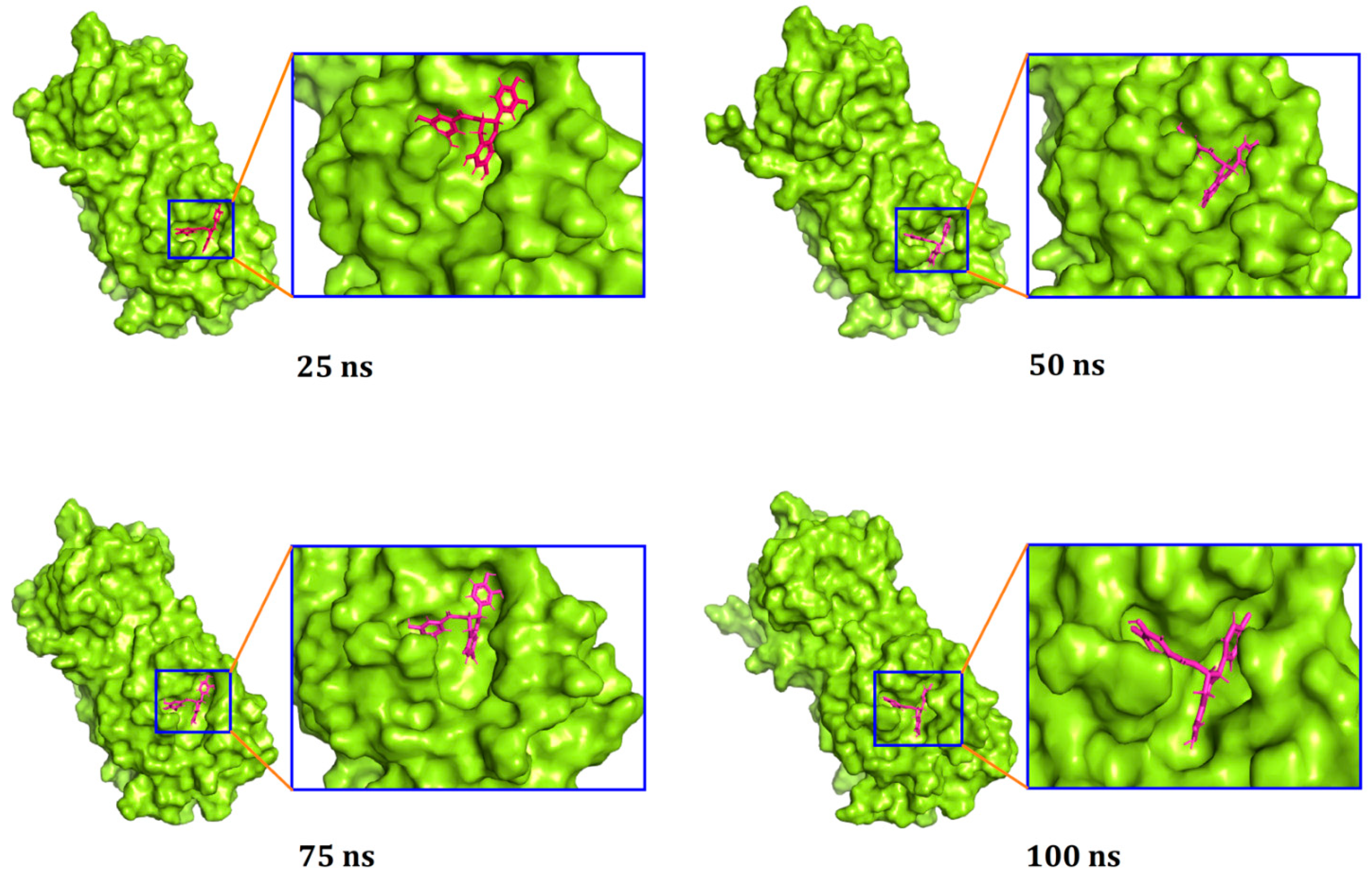
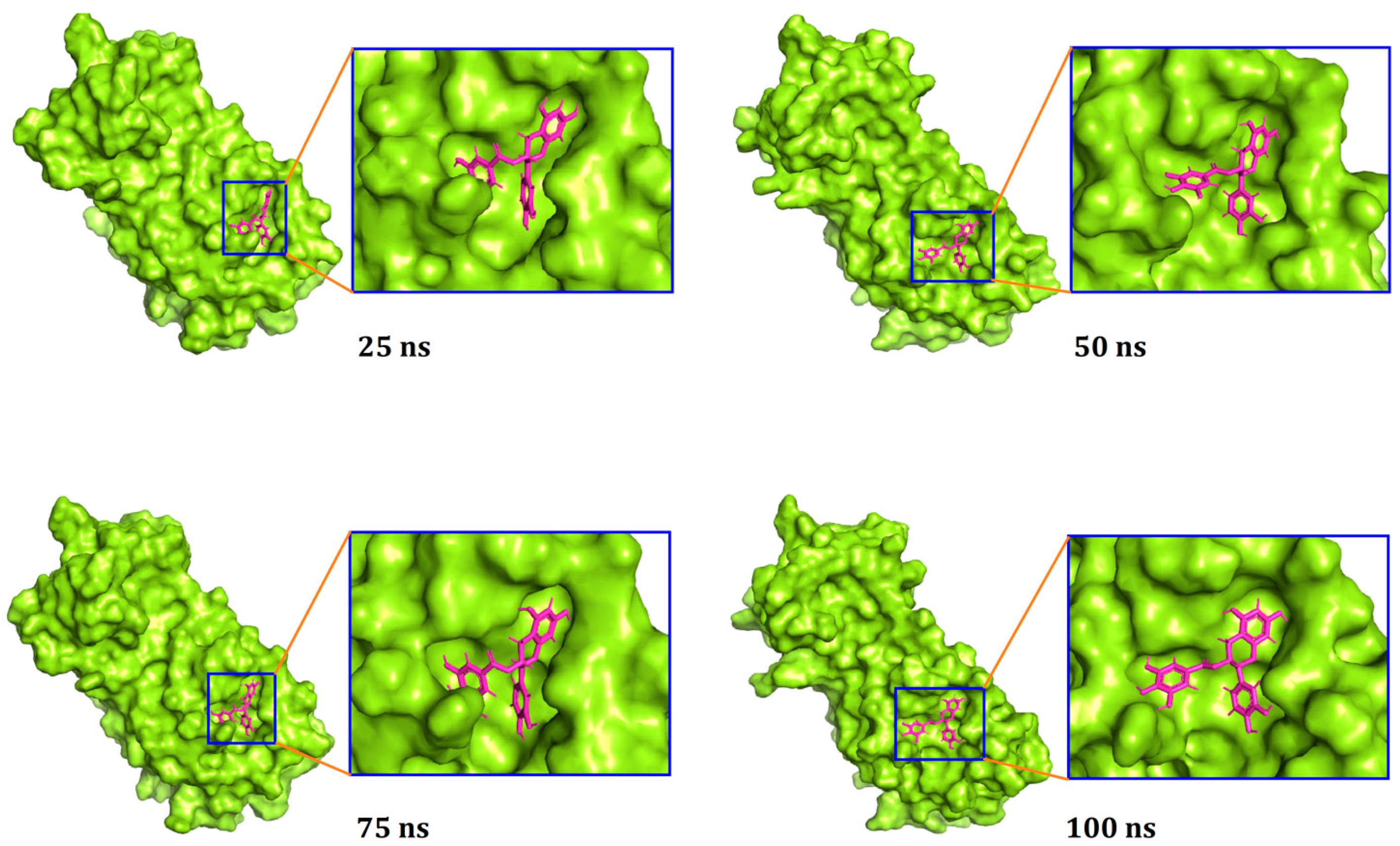
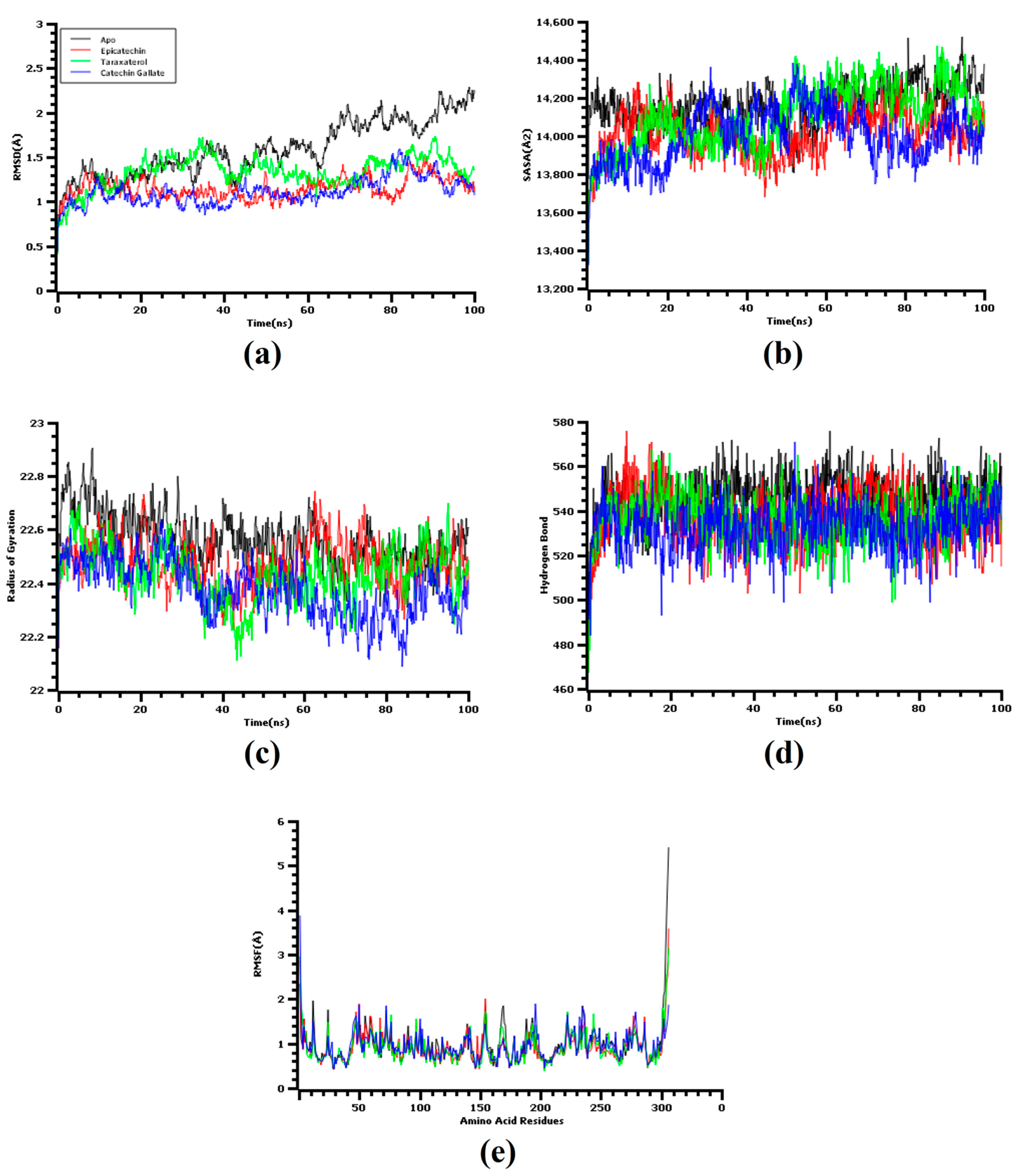
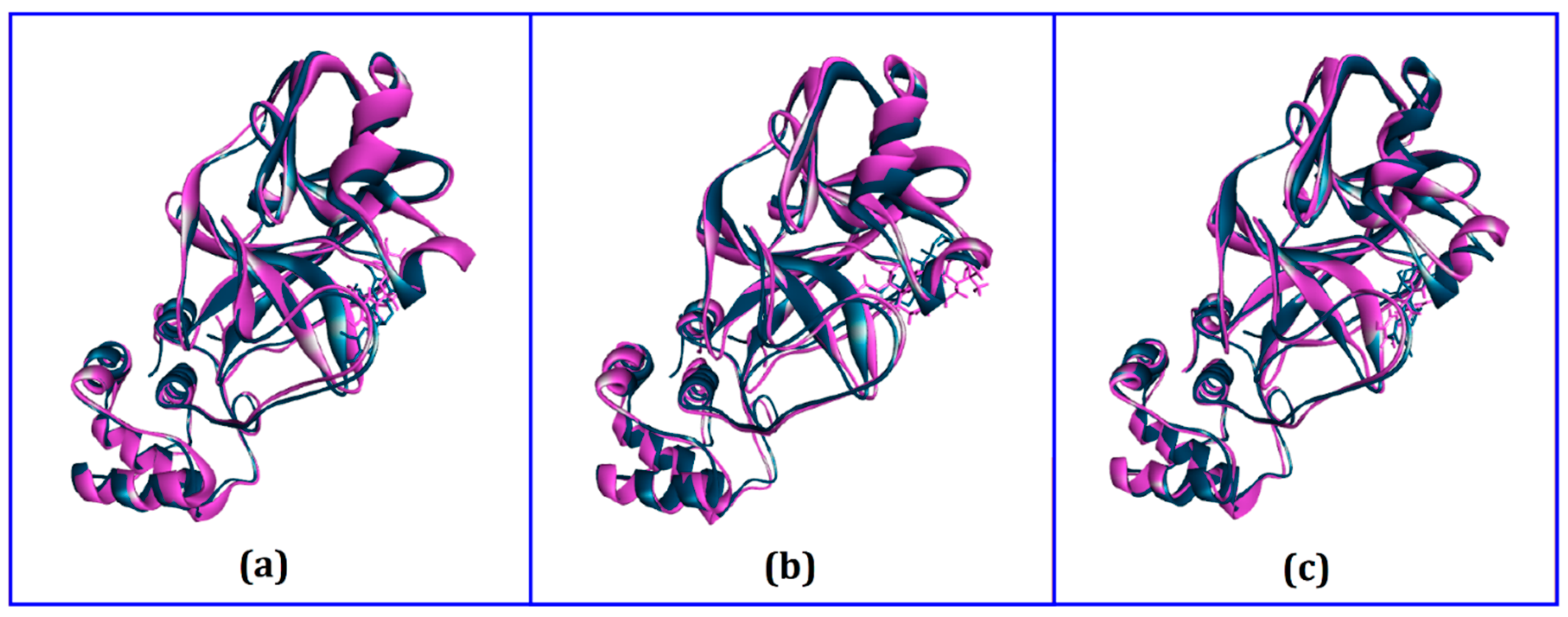
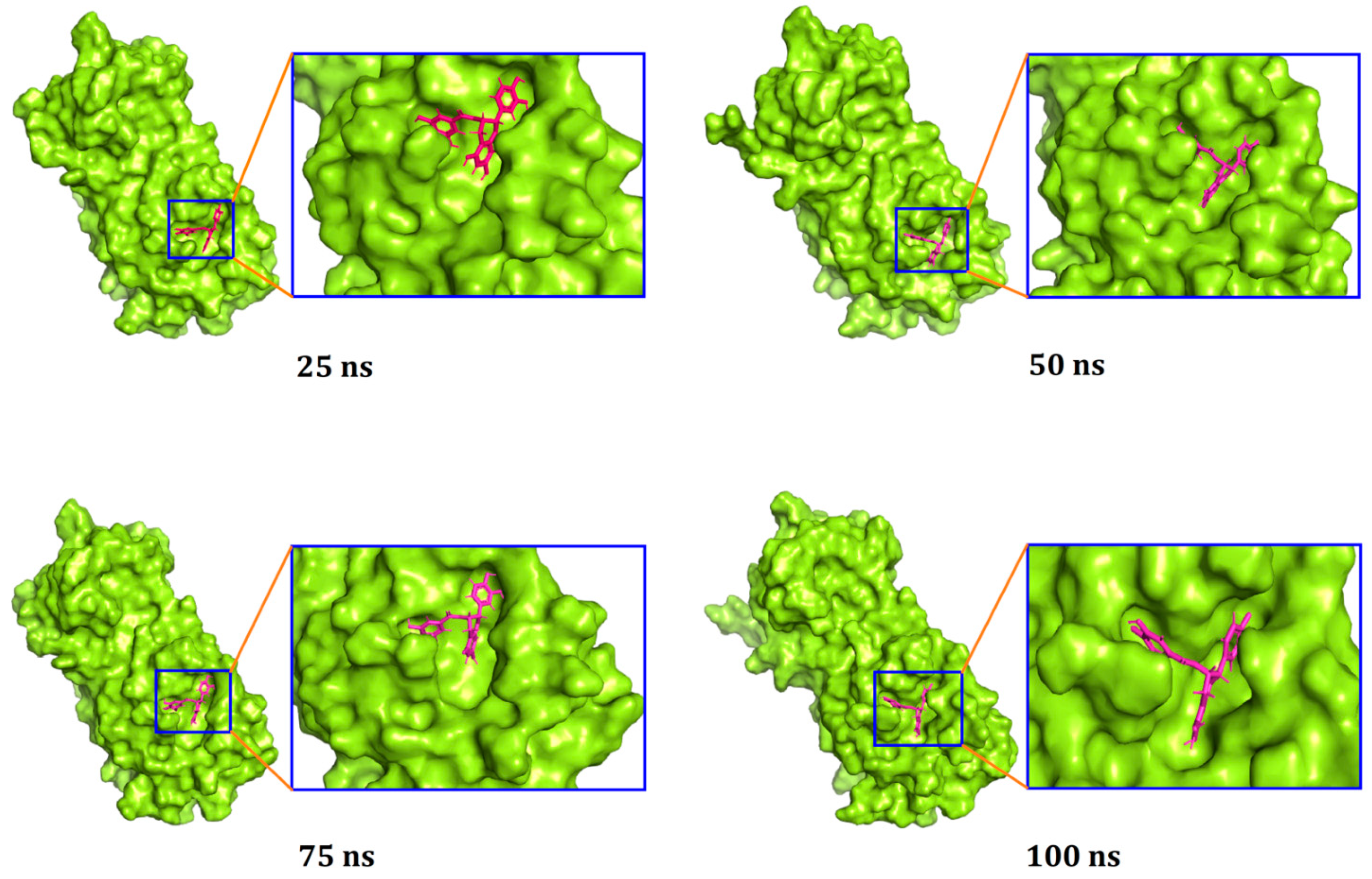
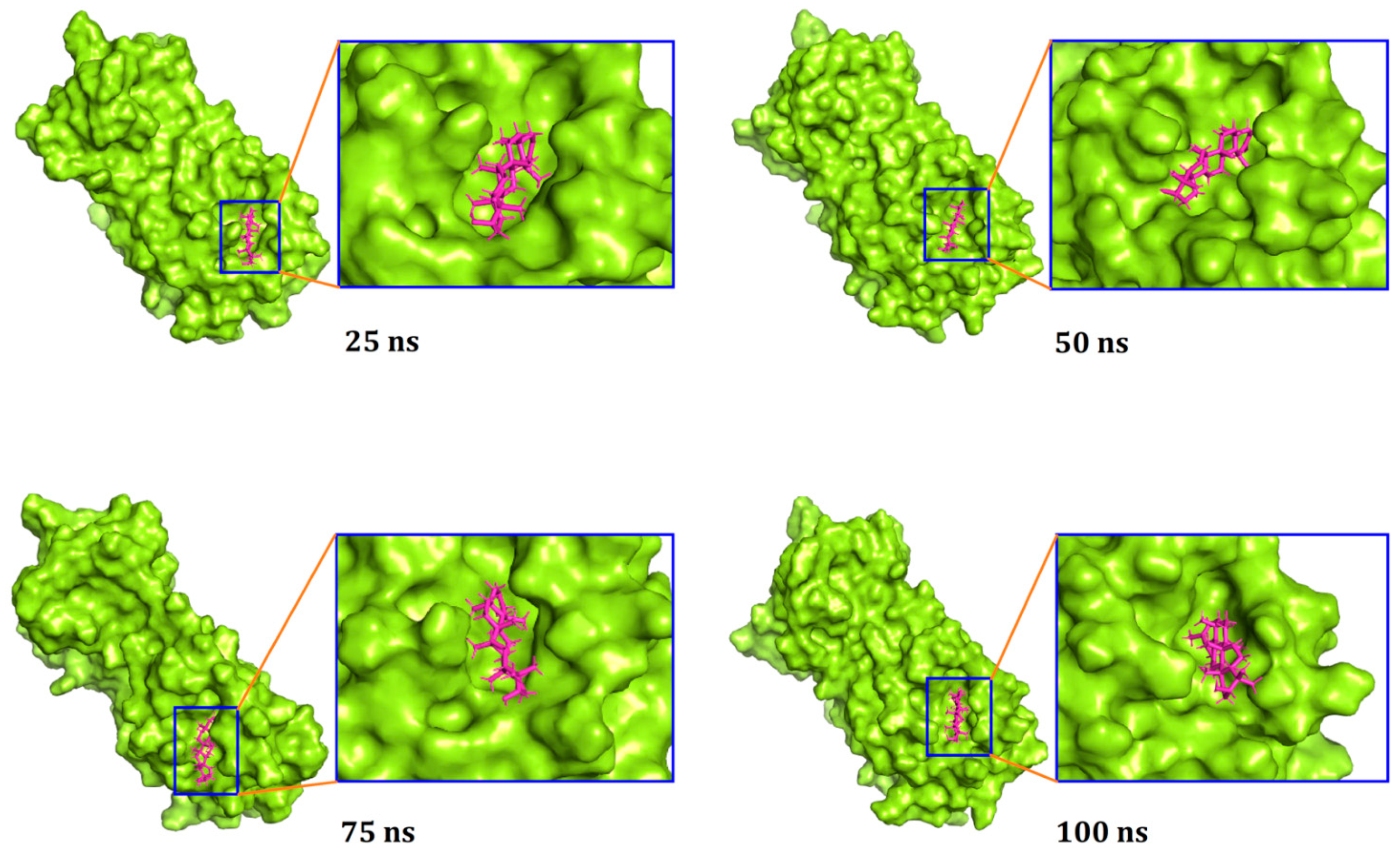
3.3. Molecular Dynamics Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Akhmetzhanov, A.; Mizumoto, K.; Jung, S.; Linton, N.; Omori, R.; Nishiura, H. Estimation of the actual incidence of coronavirus disease (COVID-19) in emergent hotspots: The example of Hokkaido, Japan during February–March 2020. J. Clin. Med. 2020, 10, 2392. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; John, A.; Abburi, C.; Vallamkondu, J.; Reddy, P.H. Current Status of Multiple Drug Molecules, and Vaccines: An Update in SARS-CoV-2 Therapeutics. Mol. Neurobiol. 2020, 57, 4106–4116. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Khullar, N.; Reddy, A.P.; Reddy, P.H. Therapeutic Strategies in the Development of Anti-viral Drugs and Vaccines Against SARS-CoV-2 Infection. Mol. Neurobiol. 2020, 57, 4856–4877. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, P.; Zhang, X.; Yu, Z.; Zhang, W.; Sun, H. SARS-CoV-2 vaccine candidates in rapid development. Hum. Vaccines Immunother. 2020, 17, 1–10. [Google Scholar] [CrossRef]
- Dutta, M.; Nezam, M.; Chowdhury, S.; Rakib, A.; Paul, A.; Sami, S.A.; Uddin, M.Z.; Rana, M.S.; Hossain, S.; Effendi, Y.; et al. Appraisals of the Bangladeshi Medicinal Plant Calotropis gigantea Used by Folk Medicine Practitioners in the Management of COVID-19: A Biochemical and Computational Approach. Front. Mol. Biosci. 2021, 8, 625391. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Li, Z.; Yang, Z.; Shi, M.; Huang, Z.; Song, J.; Song, Z. The possibility of COVID-19 transmission from eye to nose. Acta Ophthalmol. 2020, 98, e388. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.; Wang, H.; Luan, B. In Silico Exploration of the Molecular Mechanism of Clinically Oriented Drugs for Possibly Inhibiting SARS-CoV-2’s Main Protease. J. Phys. Chem. Lett. 2020, 11, 4413–4420. [Google Scholar] [CrossRef] [PubMed]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Shyr, Z.A.; Gorshkov, K.; Chen, C.Z.; Zheng, W. Drug discovery strategies for sars-cov-2. J. Pharmacol. Exp. Ther. 2020, 375, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Yang, C.Y.; Wang, M.; Ou, S.C.; Lo, C.Y.; Tsai, T.L.; Wu, H.Y. Effects of Coronavirus Persistence on the Genome Structure and Subsequent Gene Expression, Pathogenicity and Adaptation Capability. Cells 2020, 9, 2322. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Kuo, R.L.; Shih, S.R. COVID-19: The first documented coronavirus pandemic in history. Biomed. J. 2020, 43, 2–6. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Hadda, T.B.; Berredjem, M.; Almalki, F.A.; Rastija, V.; Bader, A.; Jamalis, J.; Emran, T.B.; Abu-Iznei, T.; Esharkawy, E.; Rodriguez, L.C.; et al. How to face COVID-19: Proposed treatments based on Remdesivir and Hydroxychloroquin in presence of zinc-sulfate and POM theory. J. Biomol. Struct. Dyn. 2021, 2021, 1–14. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Ceraolo, C.; Giorgi, F.M. Genomic variance of the 2019-nCoV coronavirus. J. Med. Virol. 2020, 92, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351.e2. [Google Scholar] [CrossRef]
- Obaidullah, A.J.; Alanazi, M.A.; Alsaif, N.A.; Albassam, H.; Almehizia, A.A.; Alqahtani, A.A.; Mahmud, S.; Sami, S.A.; Emran, T.B. Immunoinformatics-guided design of multi-epitope vaccine from structural proteins of severe acute respiratory syndrome-coronavirus-2. RSC Adv. 2021, 11, 18103–18121. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 Coronavirus Structure, Mechanism of Action, Antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 39, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genomics 2020, 47, 119–121. [Google Scholar] [CrossRef]
- Ngo, S.T.; Quynh Anh Pham, N.; Thi Le, L.; Pham, D.H.; Vu, V.V. Computational Determination of Potential Inhibitors of SARS-CoV-2 Main Protease. J. Chem. Inf. Model. 2020, 60, 5771–5780. [Google Scholar] [CrossRef]
- Fauquet, C.M.; Fargette, D. International Committee on Taxonomy of Viruses and the 3,142 unassigned species. Virol. J. 2005, 2, 1–10. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Havranek, B.; Islam, S.M. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J. Biomol. Struct. Dyn. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Mahmud, S.; Paul, G.K.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Hossain, A.; Promi, M.; Ema, F.K.; et al. Prospective role of peptide-based antiviral therapy against the main protease of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 628585. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. (3CL pro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Sarmah, S.; Lyndem, S.; Singha Roy, A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020, 39, 1–11. [Google Scholar] [CrossRef]
- Mahmud, S.; Paul, G.K.; Afroze, M.; Islam, S.; Gupt, S.B.R.; Razu, M.H.; Biswas, S.; Zaman, S.; Uddin, M.S.; Khan, M.; et al. Efficacy of Phytochemicals Derived from Avicennia officinalis for the Management of COVID-19: A Combined In Silico and Biochemical Study. Molecules 2021, 28, 2210. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Yarmolinsky, L.; Porat, D.; Dahan, A. Antiviral effect of phytochemicals from medicinal plants: Applications and drug delivery strategies. Drug Deliv. Transl. Res. 2020, 10, 354–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attia, Y.A.; Alagawany, M.M.; Farag, M.R.; Alkhatib, F.M.; Khafaga, A.F.; Abdel-Moneim, A.M.E.; Asiry, K.A.; Mesalam, N.M.; Shafi, M.E.; Al-Harthi, M.A.; et al. Phytogenic Products and Phytochemicals as a Candidate Strategy to Improve Tolerance to Coronavirus. Front. Vet. Sci. 2020, 7, 1–18. [Google Scholar] [CrossRef]
- Kapoor, R.; Sharma, B.; Kanwar, S.S. Antiviral Phytochemicals: An Overview. Biochem. Physiol. 2017, 6. [Google Scholar] [CrossRef]
- Del Mar, C.; Doshi, P.; Hama, R.; Jones, M.; Jefferson, T.; Heneghan, C.; Onakpoya, I.; Howick, J. Neuraminidase inhibitors for influenza complications. Lancet 2014, 384, 1260–1261. [Google Scholar] [CrossRef]
- Rakib, A.; Sami, S.A.; Islam, M.A.; Ahmed, S.; Faiz, F.B.; Khanam, B.H.; Marma, K.K.S.; Rahman, M.; Uddin, M.M.N.; Nainu, F.; et al. Epitope-based immunoinformatics approach on nucleocapsid protein of severe acute respiratory syndrome-coronavirus-2. Molecules 2020, 25, 5088. [Google Scholar] [CrossRef] [PubMed]
- Paintaud, G.; Alván, G.; Berninger, E.; Gustafsson, L.L.; Idrizbegovic, E.; Karlsson, K.K.; Wakelkamp, M. The concentration-effect relationship of quinine-induced hearing impairment. Clin. Pharmacol. Ther. 1994, 55, 317–323. [Google Scholar] [CrossRef]
- Rakib, A.; Paul, A.; Ahmed, S.; Chy, M.N.U.; Sami, S.A.; Baral, S.K.; Majumder, M.; Tareq, A.T.; Amin, M.N.; Shahriar, A.; et al. Biochemical and computational approach of phytocompounds from Tinospora crispa in the management of COVID-19. Molecules 2020, 25, 3936. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Cao, H.Y.; Liu, P.; Cheng, G.H.; Sun, M.Y. Glycyrrhizic acid in the treatment of liver diseases: Literature review. Biomed. Res. Int. 2014, 2014, 872139. [Google Scholar] [CrossRef]
- Heo, Y.A. Baloxavir: First Global Approval. Drugs 2018, 78, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Melendez, D.P.; Razonable, R.R. Letermovir and inhibitors of the terminase complex: A promising new class of investigational antiviral drugs against human cytomegalovirus. Infect. Drug Resist. 2015, 8, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F.; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Myers, R.P.; Shah, H.; Burak, K.W.; Cooper, C.; Feld, J.J. An update on the management of chronic hepatitis C: 2015 consensus guidelines from the Canadian Association for the Study of the Liver. Can. J. Gastroenterol. Hepatol. 2015, 29, 19–34. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.1; Schrödinger LLC: New York, NY, USA, 2002. [Google Scholar] [CrossRef] [Green Version]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Softw. Inc.: San Diego, CA, USA, 2015. [Google Scholar]
- Kaplan, W.; Littlejohn, T.G. Software review Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Halgren, T.A. Performance of MMFF94*. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Swargiary, A.; Mahmud, S.; Saleh, M.A. Screening of phytochemicals as potent inhibitor of 3-chymotrypsin and papain-like proteases of SARS-CoV2: An in silico approach to combat COVID-19. J. Biomol. Struct. Dyn. 2020, 2020, 1–15. [Google Scholar] [CrossRef]
- Mahmud, S.; Parves, M.R.; Riza, Y.M.; Sujon, K.M.; Ray, S.; Tithi, F.A.; Zaoti, Z.F.; Alam, S.; Absar, N. Exploring the potent inhibitors and binding modes of phospholipase A2 through in silico investigation. J. Biomol. Struct. Dyn. 2020, 38, 4221–4231. [Google Scholar] [CrossRef] [PubMed]
- Bappy, S.S.; Sultana, S.; Adhikari, J.; Mahmud, S.; Khan, M.A.; Kibria, K.M.K.; Rahman, M.M.; Shibly, A.Z. Extensive Immunoinformatics study for the prediction of novel peptide-based epitope vaccine with docking confirmation against Envelope protein of Chikungunya virus: A Computational Biology Approach. J. Biomol. Struct. Dyn. 2020, 39, 1139–1154. [Google Scholar] [CrossRef]
- Chowdhury, K.H.; Chowdhury, M.R.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.S.M.A.; Emran, T.B.; Simal-Gandara, J. Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 2020, 10, 2. [Google Scholar] [CrossRef]
- Khan, M.A.; Mahmud, S.; Alam, A.S.M.R.U.; Rahman, M.E.; Ahmed, F.; Rahmatullah, M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: A molecular docking study. J. Biomol. Struct. Dyn. 2020, 2020, 1–7. [Google Scholar] [CrossRef]
- Mahmud, S.; Uddin, M.A.R.; Paul, G.K.; Shimu, M.S.S.; Islam, S.; Rahman, E.; Islam, A.; Islam, M.S.; Promi, M.M.; Emran, T.B.; et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief. Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Islam, M.J.; Parves, M.R.; Mahmud, S.; Tithi, F.A.; Reza, M.A. Assessment of structurally and functionally high-risk nsSNPs impacts on human bone morphogenetic protein receptor type IA (BMPR1A) by computational approach. Comput. Biol. Chem. 2019, 80, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Samiul Islam, M.; Mahmud, S.; Sultana, R.; Dong, W. Identification and in silico molecular modelling study of newly isolated Bacillus subtilis SI-18 strain against S9 protein of Rhizoctonia solani. Arab. J. Chem. 2020, 13, 8600–8612. [Google Scholar] [CrossRef]
- Mahmud, S.; Uddin, M.A.R.; Zaman, M.; Sujon, K.M.; Rahman, M.E.; Shehab, M.N.; Islam, A.; Alom, M.W.; Amin, A.; Akash, A.S.; et al. Molecular docking and dynamics study of natural compound for potential inhibition of main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Pramanik, S.K.; Mahmud, S.; Paul, G.K.; Jabin, T.; Naher, K.; Uddin, M.S.; Zaman, S.; Saleh, M.A. Fermentation optimization of cellulase production from sugarcane bagasse by Bacillus pseudomycoides and molecular modeling study of cellulase. Curr. Res. Microb. Sci. 2021, 2, 100013. [Google Scholar] [CrossRef]
- Rakib, A.; Nain, Z.; Sami, S.A.; Mahmud, S.; Islam, A.; Ahmed, S.; Siddiqui, A.B.F.; Babu, S.M.O.F.; Hossain, P.; Shahriar, A.; et al. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Brief. Bioinform. 2021, 22, 1476–1498. [Google Scholar] [CrossRef]
- Tsaioun, K.; Bottlaender, M.; Mabondzo, A. ADDME-Avoiding Drug Development Mistakes Early: Central nervous system drug discovery perspective. BMC Neurol. 2009, 9, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) Through Computational Drug Repurposing Study. J. Chem. Inf. Modeling 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Rakib, A.; Sami, S.A.; Mimi, N.J.; Chowdhury, M.M.; Eva, T.A.; Nainu, F.; Paul, A.; Shahriar, A.; Tareq, A.M.; Emon, N.U.; et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Computers Biol. Med. 2020, 124, 103967. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of Chymotrypsin-like Protease Inhibitors of SARS-CoV-2 Via Integrated Computational Approach. J. Biomol. Struct. Dyn. 2021, 39, 2607–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harapan, H.; Ryan, M.; Yohan, B.; Abidin, R.S.; Nainu, F.; Rakib, A.; Jahan, I.; Emran, T.B.; Ullah, I.; Panta, K.; et al. COVID-19 and dengue: Double punches for dengue-endemic countries in Asia. Rev. Med. Virol. 2021, 31, e2161. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Lu, I.L.; Mahindroo, N.; Liang, P.H.; Peng, Y.H.; Kuo, C.J.; Tsai, K.C.; Hsieh, H.P.; Chao, Y.S.; Wu, S.Y. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006, 49, 5154–5161. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Hengphasatporn, K.; Garon, A.; Wolschann, P.; Langer, T.; Yasuteru, S.; Huynh, T.N.T.; Chavasiri, W.; Saelee, T.; Boonyasuppayakorn, S.; Rungrotmongkol, T. Multiple virtual screening strategies for the discovery of novel compounds active against dengue virus: A hit identification study. Sci. Pharm. 2020, 88, 2. [Google Scholar] [CrossRef] [Green Version]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; MFSA Cerqueira, N.; Fernandes, P.A.; Joao Ramos, M. Virtual Screening in Drug Design and Development. Comb. Chem. High Throughput Screen. 2010, 13, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Schichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Li, Y.; Liu, Z.; Lin, Y.; Huang, J. An Anti-melanogenic effects of epigallocatechin-3-gallate (EGCG), epicatechin-3-gallate (ECG) and gallocatechin-3-gallate (GCG) via down-regulation of cAMP/CREB/MITF signaling pathway in B16F10 melanoma cells. Fitoterapia 2020, 145, 104634. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Chiou, Y.S.; Wang, Y.J.; Ho, C.T.; Lin, J.K. Multistage carcinogenesis process as molecular targets in cancer chemoprevention by epicatechin-3-gallate. Food Funct. 2011, 2, 101–110. [Google Scholar] [CrossRef]
- Chen, T.; Yang, Y.; Zhu, S.; Lu, Y.; Zhu, L.; Wang, Y.; Wang, X. Inhibition of Aβ aggregates in Alzheimer’s disease by epigallocatechin and epicatechin-3-gallate from green tea. Bioorg. Chem. 2020, 105, 104382. [Google Scholar] [CrossRef]
- Huang, S.F.; Horng, C.T.; Hsieh, Y.S.; Hsieh, Y.H.; Chu, S.C.; Chen, P.N. Epicatechin-3-gallate reverses TGF-β1-induced epithelial-to-mesenchymal transition and inhibits cell invasion and protease activities in human lung cancer cells. Food Chem. Toxicol. 2016, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 3, 1210–1213. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Muhammad, J.; Khan, A.; Sule, A.A.; Tirupathi, R.; Mutair, A.A.; Alhumaid, S.; Al-Omari, A.; Dhawan, M.; et al. Role of Inflammatory Cytokines in COVID-19 Patients: A Review on Molecular Mechanisms, Immune Functions, Immunopathology and Immunomodulatory Drugs to Counter Cytokine Storm. Vaccine 2021, 9, 436. [Google Scholar] [CrossRef]
- Akihisa, T.; Yasukawa, K.; Oinuma, H.; Kasahara, Y.; Yamanouchi, S.; Takido, M.; Kumaki, K.; Tamura, T. Triterpene alcohols from the flowers of compositae and their anti- inflammatory effects. Phytochemistry 1996, 43, 1255–1260. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chemie Int. Ed. 2003, 42, 4120. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Garout, M.A.; Al-Qaaneh, A.M.; Sule, A.A.; Tirupathi, R.; Mutair, A.A.; Alhumaid, S.; Al-Omari, A.; Hasan, A.; et al. Diverse Immunological Factors Influencing Pathogenesis in Patients with COVID-19: A Review on Viral Dissemination, Immunotherapeutic Options to Counter Cytokine Storm and Inflammatory Responses. Pathogens 2021, 10, 565. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. The lipogenesis pathway as a cancer target. J. Med. Chem. 2011, 54, 5615–5638. [Google Scholar] [CrossRef]
- Kürbitz, C.; Heise, D.; Redmer, T.; Goumas, F.; Arlt, A.; Lemke, J.; Rimbach, G.; Kalthoff, H.; Trauzold, A. Epicatechin gallate and catechin gallate are superior to epigallocatechin gallate in growth suppression and anti-inflammatory activities in pancreatic tumor cells. Cancer Sci. 2011, 102, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Chen, W.; Yi, K.; Wu, Z.; Si, Y.; Han, W.; Zhao, Y. The evaluation of catechins that contain a galloyl moiety as potential HIV-1 integrase inhibitors. Clin. Immunol. 2010, 137, 347–356. [Google Scholar] [CrossRef]
- Strobel, P.; Allard, C.; Perez-Acle, T.; Calderon, R.; Aldunate, R.; Leighton, F. Myricetin, quercetin and catechin-gallate inhibit glucose uptake in isolated rat adipocytes. Biochem. J. 2005, 386, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Rabaan, A.A.; Tirupathi, R.; Sule, A.A.; Aldali, J.; Al Mutair, A.; Alhumaid, S.; Muzaheed; Gupta, N.; Koritala, T.; Adhikari, R.; et al. Viral dynamics and real-time RT-PCR Ct values correlation with disease severity in COVID-19. Diagnostics 2021, 11, 1091. [Google Scholar] [CrossRef]
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica 2020, 2020, 6307457. [Google Scholar] [CrossRef]
- Bhowmick, S.; Saha, A.; Osman, S.M.; Alasmary, F.A.; Almutairi, T.M.; Islam, M.A. Structure-based identification of SARS-CoV-2 main protease inhibitors from anti-viral specific chemical libraries: An exhaustive computational screening approach. Mol. Divers. 2021, 2021, 1–19. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Marinho, E.M.; Batista de Andrade Neto, J.; Silva, J.; Rocha da Silva, C.; Cavalcanti, B.C.; Marinho, E.S.; Nobre Júnior, H.V. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microb. Pathog. 2020, 148, 104365. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | PubChem CID | Binding Affinity (kcal/mol) | Residues in Contact | Interaction Type | Distance in Å |
|---|---|---|---|---|---|
| Epicatechin-3-O-gallate | 65056 | −8.4 | HIS164 | Conventional hydrogen bond | 2.51938 |
| HIS163 | Conventional hydrogen bond | 2.28965 | |||
| ASN142 | Conventional hydrogen bond | 2.57732 | |||
| GLN189 | Carbon hydrogen bond | 2.19623 | |||
| MET165 | Pi-alkyl | 5.3972 | |||
| PRO168 | Pi-alkyl | 5.49119 | |||
| Psi-taraxasterol | 5270605 | −8.5 | MET165 | Alkyl | 5.04103 |
| MET49 | Alkyl | 3.86071 | |||
| CYS145 | Alkyl | 5.38342 | |||
| HIS41 | Pi-alkyl | 5.35384 | |||
| Catechin gallate | 6419835 | −8.8 | LEU141 | Conventional hydrogen bond | 2.05522 |
| HIS163 | Conventional hydrogen bond | 1.92422 | |||
| ARG188 | Conventional hydrogen bond | 2.46163 | |||
| THR190 | Conventional hydrogen bond | 2.05509 | |||
| GLN189 | Carbon hydrogen bond | 2.3535 | |||
| HIS41 | Pi-Pi T-shaped | 5.22838 | |||
| MET165 | Pi-alkyl | 4.91554 | |||
| CYS145 | Pi-alkyl | 5.4117 | |||
| MET49 | Pi-alkyl | 5.21898 |
| Parameters | Epicatechin-3-O-gallate | Psi-taraxasterol | Catechin gallate |
|---|---|---|---|
| Molecular weight | 442.4 g/moL | 426.7 g/moL | 442.4 g/moL |
| H-bond acceptor | 10 | 1 | 10 |
| H-bond donor | 7 | 1 | 7 |
| CNS | −3.743 | −1.992 | −3.743 |
| CYP2D6 substrate | No | No | No |
| CYP3A4 substrate | No | Yes | No |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | No | No | No |
| CYP2C9 inhibitor | No | No | No |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | No | No | No |
| Carcinogenicity | Non-carcinogenic | Non-carcinogenic | Non-carcinogenic |
| Hepatotoxicity | No | No | No |
| P-glycoprotein inhibitor | No | No | No |
| Human intestinal absorption | +0.9942 | +0.9919 | +0.9942 |
| Ames mutagenesis | −0.5000 | −0.8500 | −0.5000 |
| Acute oral toxicity | No | No | No |
| Lipinski rule of five | Yes | Yes | Yes |
| Complex | Residues | Interactions | Distance |
|---|---|---|---|
| Epicatechin-3-O-gallate | Ser144 His163 Phe140 Cys145 His41 Leu141 Met49 Cys165 | H H H H H PA PA PA | 1.78 2.73 1.96 2.50 2.89 4.21 5.18 5.23 |
| Psi-taraxasterol | Cys145 Met165 Met49 His41 | H H H PA | 1.23 2.21 2.73 4.33 |
| Catechin gallate | Asn142 Leu141 Glu166 Met49 Val186 Arg188 Met165 Cys145 His41 | H H H H H H H PA PA | 1.81 2.24 1.50 3.03 2.92 1.83 2.39 5.21 5.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmud, S.; Biswas, S.; Paul, G.K.; Mita, M.A.; Promi, M.M.; Afrose, S.; Hasan, M.R.; Zaman, S.; Uddin, M.S.; Dhama, K.; et al. Plant-Based Phytochemical Screening by Targeting Main Protease of SARS-CoV-2 to Design Effective Potent Inhibitors. Biology 2021, 10, 589. https://doi.org/10.3390/biology10070589
Mahmud S, Biswas S, Paul GK, Mita MA, Promi MM, Afrose S, Hasan MR, Zaman S, Uddin MS, Dhama K, et al. Plant-Based Phytochemical Screening by Targeting Main Protease of SARS-CoV-2 to Design Effective Potent Inhibitors. Biology. 2021; 10(7):589. https://doi.org/10.3390/biology10070589
Chicago/Turabian StyleMahmud, Shafi, Suvro Biswas, Gobindo Kumar Paul, Mohasana Akter Mita, Maria Meha Promi, Shamima Afrose, Md. Robiul Hasan, Shahriar Zaman, Md. Salah Uddin, Kuldeep Dhama, and et al. 2021. "Plant-Based Phytochemical Screening by Targeting Main Protease of SARS-CoV-2 to Design Effective Potent Inhibitors" Biology 10, no. 7: 589. https://doi.org/10.3390/biology10070589
APA StyleMahmud, S., Biswas, S., Paul, G. K., Mita, M. A., Promi, M. M., Afrose, S., Hasan, M. R., Zaman, S., Uddin, M. S., Dhama, K., Emran, T. B., Saleh, M. A., & Simal-Gandara, J. (2021). Plant-Based Phytochemical Screening by Targeting Main Protease of SARS-CoV-2 to Design Effective Potent Inhibitors. Biology, 10(7), 589. https://doi.org/10.3390/biology10070589