
Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy




Abstract
Simple Summary
Abstract
1. Introduction
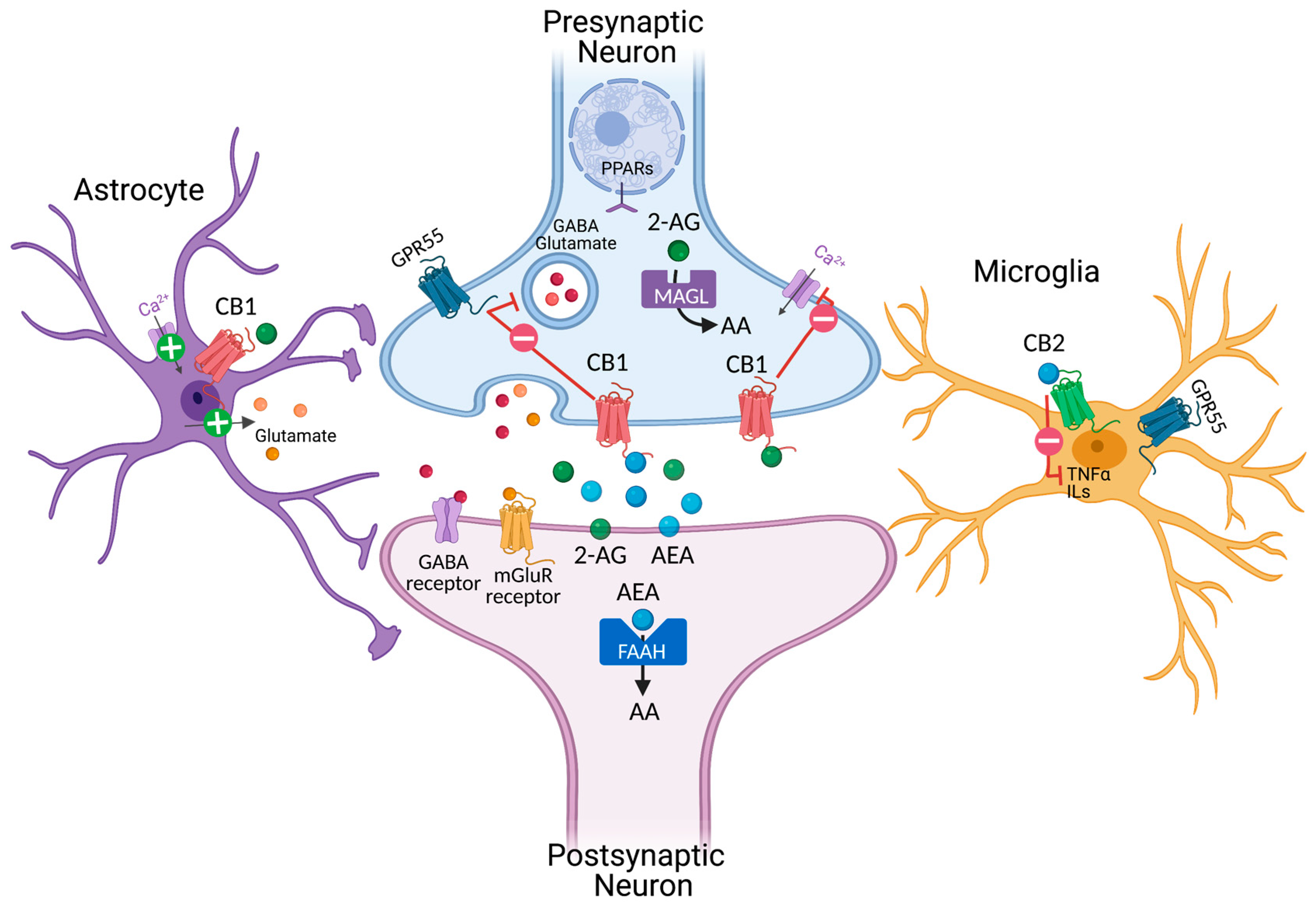
2. Cannabinoids and Endocannabinoid Systems
2.1. Phytocannabinoids and Modulation of Cannabinoid Receptor 1 (CB1)
2.2. THC
2.3. Cannabidiol
2.4. Synthetic CB1 Modulators
2.5. Modulation of Cannabinoid Receptor 2 (CB2)
2.6. Modulation of Endogenous Cannabinoid Anandamide and 2-AG

{kind=link}
{kind=link}
| Compounds | Endocannabinoid System Targets | Beneficial Anti-AD Effects | Adverse/Unwanted Effects |
|---|---|---|---|
| THC | Mixed CB1 and CB2 agonist | Inhibition of achetylcholinesterase [67] Reduce Aβ levels [63] Hippocampal neurogenesis [166] Induce BDNF release [73,74] | Psychotic effects [55] Reduce cognitive functions [54] A deficit in dopamine release [56] |
| CBD | Mixed CB1 and CB2 agonist | No psycoactive effets [80] Neuroprotection [84] Reduce microglia activation [85] Delay cognitive decline [167] | Hypotension at high doses [168] Anxiogenic-like effect [169] |
| WIN 55,212-2 HU 210 CP 55,940 JWH-018 | Mixed CB1 and CB2 agonist | Increase Aβ clearance [116] Promote neurogenesis [111] Prevent cognitive impairment [113,114] | Defect in working memory [95,96,97] Affects long-term potentiation [104,105] Sedation [170] |
| ACEA | Selective CB1 agonist | Anti-inflammatory [117] Prevent spatial memory impairment [118] | N.R. |
| JWH-133 AM-1241 MDA7 | Selective CB2 agonist | Increase Aβ clearance [116] Improve cognitive performance [116] Prevent microglial activation [128] Reduce tau hyper-phosphorylation [132] | Immune suppression [171] |
| URB597 PF-04457845 JZL184 JZL195 | Modulation of endogenous cannabinoid anandamide and 2-AG | Suppress glutamate Aβ42-induced toxicity [147] Reduce proinflammatory interleukin expression [148,156] Restore long-term potentiation [148] Reduce amyloid plaque burden [154] | Cardiac diastolic stiffness [172] |
3. The Orphan G Protein-Coupled Receptors (GPRs)
4. Limits of Cannabinoids in Alzheimer’s Disease Therapy
5. Final Remarks
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385. [Google Scholar] [CrossRef]
- Abate, G.; Memo, M.; Uberti, D. Impact of COVID-19 on Alzheimer’s Disease Risk: Viewpoint for Research Action. Healthcare 2020, 8, 286. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Tonello, S.; Serpelloni, M.; Lopomo, N.F.; Sardini, E.; Abate, G.; Uberti, D.L. Preliminary study of a low-cost point-of-care testing system using screen-printed biosensors: For early biomarkers detection related to Alzheimer Disease. In Proceedings of the 2016 IEEE International Symposium on Medical Measurements and Applications, MeMeA 2016—Proceedings, Benevento, Italy, 15–18 May 2016. [Google Scholar]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.-P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef]
- Marconi, A.; Di Forti, M.; Lewis, C.M.; Murray, R.M.; Vassos, E. Meta-analysis of the Association Between the Level of Cannabis Use and Risk of Psychosis. Schizophr. Bull. 2016, 42, 1262–1269. [Google Scholar] [CrossRef]
- Cohen, K.; Kapitány-Fövény, M.; Mama, Y.; Arieli, M.; Rosca, P.; Demetrovics, Z.; Weinstein, A. The effects of synthetic cannabinoids on executive function. Psychopharmacology 2017, 234, 1121–1134. [Google Scholar] [CrossRef]
- Schuster, R.M.; Gilman, J.; Schoenfeld, D.; Evenden, J.; Hareli, M.; Ulysse, C.; Nip, E.; Hanly, A.; Zhang, H.; Evins, A.E. One Month of Cannabis Abstinence in Adolescents and Young Adults Is Associated With Improved Memory. J. Clin. Psychiatry 2018, 79. [Google Scholar] [CrossRef]
- Levar, N.; Francis, A.N.; Smith, M.J.; Ho, W.C.; Gilman, J.M. Verbal Memory Performance and Reduced Cortical Thickness of Brain Regions Along the Uncinate Fasciculus in Young Adult Cannabis Users. Cannabis Cannabinoid Res. 2018, 3, 56–65. [Google Scholar] [CrossRef]
- Prenderville, J.A.; Kelly, Á.M.; Downer, E.J. The role of cannabinoids in adult neurogenesis. Br. J. Pharmacol. 2015, 172, 3950–3963. [Google Scholar] [CrossRef]
- Volicer, L.; Stelly, M.; Morris, J.; McLaughlin, J.; Volicer, B.J. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 1997, 12, 913–919. [Google Scholar] [CrossRef]
- Palazuelos, J.; Aguado, T.; Egia, A.; Mechoulam, R.; Guzmán, M.; Galve-Roperh, I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006, 20, 2405–2407. [Google Scholar] [CrossRef]
- Oberbarnscheidt, T.; Miller, N.S. The Impact of Cannabidiol on Psychiatric and Medical Conditions. J. Clin. Med. Res. 2020, 12, 393–403. [Google Scholar] [CrossRef]
- Long, J.Z.; Nomura, D.K.; Vann, R.E.; Walentiny, D.M.; Booker, L.; Jin, X.; Burston, J.J.; Sim-Selley, L.J.; Lichtman, A.H.; Wiley, J.L.; et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20270–20275. [Google Scholar] [CrossRef]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef]
- Iversen, L. Cannabis and the brain. Brain 2003, 126, 1252–1270. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Adams, R.; Pease, D.C.; Clark, J.H. Isolation of Cannabinol, Cannabidiol and Quebrachitol from Red Oil of Minnesota Wild Hemp. J. Am. Chem. Soc. 1940, 62, 2194–2196. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- Laun, A.S.; Song, Z.-H. GPR3 and GPR6, novel molecular targets for cannabidiol. Biochem. Biophys. Res. Commun. 2017, 490, 17–21. [Google Scholar] [CrossRef]
- Brown, K.J.; Laun, A.S.; Song, Z.-H. Cannabidiol, a novel inverse agonist for GPR. Biochem. Biophys. Res. Commun. 2017, 493, 451–454. [Google Scholar] [CrossRef]
- Tambaro, S.; Casu, M.A.; Mastinu, A.; Lazzari, P. Evaluation of selective cannabinoid CB(1) and CB(2) receptor agonists in a mouse model of lipopolysaccharide-induced interstitial cystitis. Eur. J. Pharmacol. 2014, 729, 67–74. [Google Scholar] [CrossRef]
- Oliveira da Cruz, J.F.; Robin, L.M.; Drago, F.; Marsicano, G.; Metna-Laurent, M. Astroglial type-1 cannabinoid receptor (CB1): A new player in the tripartite synapse. Neuroscience 2016, 323, 35–42. [Google Scholar] [CrossRef]
- Robin, L.M.; Oliveira da Cruz, J.F.; Langlais, V.C.; Martin-Fernandez, M.; Metna-Laurent, M.; Busquets-Garcia, A.; Bellocchio, L.; Soria-Gomez, E.; Papouin, T.; Varilh, M.; et al. Astroglial CB(1) Receptors Determine Synaptic D-Serine Availability to Enable Recognition Memory. Neuron 2018, 98, 935–944.e5. [Google Scholar] [CrossRef]
- Navarrete, M.; Díez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: An immunohistochemical study. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Basu, S.; Maresz, K.; Bifulco, M.; Dittel, B.N. What we know and do not know about the cannabinoid receptor 2 (CB2). Semin. Immunol. 2014, 26, 369–379. [Google Scholar] [CrossRef]
- Savonenko, A.V.; Melnikova, T.; Wang, Y.; Ravert, H.; Gao, Y.; Koppel, J.; Lee, D.; Pletnikova, O.; Cho, E.; Sayyida, N.; et al. Cannabinoid CB2 Receptors in a Mouse Model of Aβ Amyloidosis: Immunohistochemical Analysis and Suitability as a PET Biomarker of Neuroinflammation. PLoS ONE 2015, 10, e0129618. [Google Scholar] [CrossRef]
- Vogel, Z.; Barg, J.; Levy, R.; Saya, D.; Heldman, E.; Mechoulam, R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J. Neurochem. 1993, 61, 352–355. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M.; et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001, 410, 588–592. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Dinh, T.P.; Freund, T.F.; Piomelli, D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem. Phys. Lipids 2002, 121, 149–158. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Nagre, N.N.; Xie, S.; Subbanna, S. Elevation of endogenous anandamide impairs LTP, learning, and memory through CB1 receptor signaling in mice. Hippocampus 2014, 24, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Dragunow, M.; Faull, R.L. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Bilkei-Gorzo, A.; Racz, I.; Valverde, O.; Otto, M.; Michel, K.; Sastre, M.; Zimmer, A. Early age-related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 15670–15675. [Google Scholar] [CrossRef]
- Lee, J.H.; Agacinski, G.; Williams, J.H.; Wilcock, G.K.; Esiri, M.M.; Francis, P.T.; Wong, P.T.-H.; Chen, C.P.; Lai, M.K.P. Intact cannabinoid CB1 receptors in the Alzheimer’s disease cortex. Neurochem. Int. 2010, 57, 985–989. [Google Scholar] [CrossRef]
- Ramírez, B.G.; Blázquez, C.; Gómez del Pulgar, T.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- Wolff, V.; Rouyer, O.; Geny, B. Adverse health effects of marijuana use. N. Engl. J. Med. 2014, 371, 878. [Google Scholar]
- Gorey, C.; Kuhns, L.; Smaragdi, E.; Kroon, E.; Cousijn, J. Age-related differences in the impact of cannabis use on the brain and cognition: A systematic review. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 37–58. [Google Scholar] [CrossRef]
- Broyd, S.J.; van Hell, H.H.; Beale, C.; Yücel, M.; Solowij, N. Acute and Chronic Effects of Cannabinoids on Human Cognition-A Systematic Review. Biol. Psychiatry 2016, 79, 557–567. [Google Scholar] [CrossRef]
- Kroon, E.; Kuhns, L.; Hoch, E.; Cousijn, J. Heavy cannabis use, dependence and the brain: A clinical perspective. Addiction 2020, 115, 559–572. [Google Scholar] [CrossRef]
- Elsohly, M.A.; Gul, W.; Wanas, A.S.; Radwan, M.M. Synthetic cannabinoids: Analysis and metabolites. Life Sci. 2014, 97, 78–90. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Battisti, R.A.; Roodenrys, S.; Johnstone, S.J.; Respondek, C.; Hermens, D.F.; Solowij, N. Chronic use of cannabis and poor neural efficiency in verbal memory ability. Psychopharmacology 2010, 209, 319–330. [Google Scholar] [CrossRef]
- Murray, R.M.; Englund, A.; Abi-Dargham, A.; Lewis, D.A.; Di Forti, M.; Davies, C.; Sherif, M.; McGuire, P.; D’Souza, D.C. Cannabis-associated psychosis: Neural substrate and clinical impact. Neuropharmacology 2017, 124, 89–104. [Google Scholar] [CrossRef]
- Van de Giessen, E.; Weinstein, J.J.; Cassidy, C.M.; Haney, M.; Dong, Z.; Ghazzaoui, R.; Ojeil, N.; Kegeles, L.S.; Xu, X.; Vadhan, N.P.; et al. Deficits in striatal dopamine release in cannabis dependence. Mol. Psychiatry 2017, 22, 68–75. [Google Scholar] [CrossRef][Green Version]
- Amen, D.G.; Darmal, B.; Raji, C.A.; Bao, W.; Jorandby, L.; Meysami, S.; Raghavendra, C.S. Discriminative Properties of Hippocampal Hypoperfusion in Marijuana Users Compared to Healthy Controls: Implications for Marijuana Administration in Alzheimer’s Dementia. J. Alzheimers Dis. 2017, 56, 261–273. [Google Scholar] [CrossRef]
- Yücel, M.; Solowij, N.; Respondek, C.; Whittle, S.; Fornito, A.; Pantelis, C.; Lubman, D.I. Regional Brain Abnormalities Associated With Long-term Heavy Cannabis Use. Arch. Gen. Psychiatry 2008, 65, 694–701. [Google Scholar] [CrossRef]
- Zalesky, A.; Solowij, N.; Yücel, M.; Lubman, D.I.; Takagi, M.; Harding, I.H.; Lorenzetti, V.; Wang, R.; Searle, K.; Pantelis, C.; et al. Effect of long-term cannabis use on axonal fibre connectivity. Brain 2012, 135, 2245–2255. [Google Scholar] [CrossRef]
- Chandra, S.; Radwan, M.M.; Majumdar, C.G.; Church, J.C.; Freeman, T.P.; ElSohly, M.A. New trends in cannabis potency in USA and Europe during the last decade (2008–2017). Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 5–15. [Google Scholar] [CrossRef]
- Zamengo, L.; Frison, G.; Zwitser, G.; Salomone, A.; Freeman, T.P. Cannabis knowledge and implications for health: Considerations regarding the legalization of non-medical cannabis. Med. Sci. Law 2020, 60, 309–314. [Google Scholar] [CrossRef]
- Schneider, M.; Koch, M. Chronic pubertal, but not adult chronic cannabinoid treatment impairs sensorimotor gating, recognition memory, and the performance in a progressive ratio task in adult rats. Neuropsychopharmacology 2003, 28, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Li, Y.; Liu, H.; Bai, G.; Mayl, J.; Lin, X.; Sutherland, K.; Nabar, N.; Cai, J. The potential therapeutic effects of THC on Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Janefjord, E.; Mååg, J.L.V.; Harvey, B.S.; Smid, S.D. Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell. Mol. Neurobiol. 2014, 34, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Quehenberger, O.M.; Armando, A.; Daugherty, D.; Maher, P.; Schubert, D. Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. NPJ Aging Mech. Dis. 2016, 2, 16012. [Google Scholar] [CrossRef]
- Schubert, D.; Kepchia, D.; Liang, Z.; Dargusch, R.; Goldberg, J.; Maher, P. Efficacy of Cannabinoids in a Pre-Clinical Drug-Screening Platform for Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7719–7730. [Google Scholar] [CrossRef]
- Eubanks, L.M.; Rogers, C.J.; Beuscher, A.E., IV; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K.D. A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef]
- Hampson, A.J.; Grimaldi, M.; Axelrod, J.; Wink, D. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. USA 1998, 95, 8268–8273. [Google Scholar] [CrossRef]
- Fishbein-Kaminietsky, M.; Gafni, M.; Sarne, Y. Ultralow doses of cannabinoid drugs protect the mouse brain from inflammation-induced cognitive damage. J. Neurosci. Res. 2014, 92, 1669–1677. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, J.; Fan, N.; Teng, Z.-Q.; Wu, Y.; Yang, H.; Tang, Y.-P.; Sun, H.; Song, Y.; Chen, C. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell 2013, 155, 1154–1165. [Google Scholar] [CrossRef]
- Aso, E.; Sánchez-Pla, A.; Vegas-Lozano, E.; Maldonado, R.; Ferrer, I. Cannabis-based medicine reduces multiple pathological processes in AβPP/PS1 mice. J. Alzheimers Dis. 2015, 43, 977–991. [Google Scholar] [CrossRef]
- Aso, E.; Andrés-Benito, P.; Ferrer, I. Delineating the Efficacy of a Cannabis-Based Medicine at Advanced Stages of Dementia in a Murine Model. J. Alzheimers Dis. 2016, 54, 903–912. [Google Scholar] [CrossRef]
- Butovsky, E.; Juknat, A.; Goncharov, I.; Elbaz, J.; Eilam, R.; Zangen, A.; Vogel, Z. In vivo up-regulation of brain-derived neurotrophic factor in specific brain areas by chronic exposure to Delta-tetrahydrocannabinol. J. Neurochem. 2005, 93, 802–811. [Google Scholar] [CrossRef]
- Marsicano, G.; Lafenêtre, P. Roles of the endocannabinoid system in learning and memory. Curr. Top. Behav. Neurosci. 2009, 1, 201–230. [Google Scholar] [CrossRef]
- Walther, S.; Mahlberg, R.; Eichmann, U.; Kunz, D. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology 2006, 185, 524–528. [Google Scholar] [CrossRef]
- Van den Elsen, G.A.H.; Ahmed, A.I.A.; Verkes, R.-J.; Kramers, C.; Feuth, T.; Rosenberg, P.B.; van der Marck, M.A.; Olde Rikkert, M.G.M. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: A randomized controlled trial. Neurology 2015, 84, 2338–2346. [Google Scholar] [CrossRef]
- Ahmed, A.I.A.; van den Elsen, G.A.H.; Colbers, A.; Kramers, C.; Burger, D.M.; van der Marck, M.A.; Olde Rikkert, M.G.M. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology 2015, 232, 2587–2595. [Google Scholar] [CrossRef]
- Shelef, A.; Barak, Y.; Berger, U.; Paleacu, D.; Tadger, S.; Plopsky, I.; Baruch, Y. Safety and Efficacy of Medical Cannabis Oil for Behavioral and Psychological Symptoms of Dementia: An-Open Label, Add-On, Pilot Study. J. Alzheimers Dis. 2016, 51, 15–19. [Google Scholar] [CrossRef]
- Herrmann, N.; Ruthirakuhan, M.; Gallagher, D.; Verhoeff, N.P.L.G.; Kiss, A.; Black, S.E.; Lanctôt, K.L. Randomized Placebo-Controlled Trial of Nabilone for Agitation in Alzheimer’s Disease. Am. J. Geriatr. Psychiatry 2019, 27, 1161–1173. [Google Scholar] [CrossRef]
- Woelfl, T.; Rohleder, C.; Mueller, J.K.; Lange, B.; Reuter, A.; Schmidt, A.M.; Koethe, D.; Hellmich, M.; Leweke, F.M. Effects of Cannabidiol and Delta-9-Tetrahydrocannabinol on Emotion, Cognition, and Attention: A Double-Blind, Placebo-Controlled, Randomized Experimental Trial in Healthy Volunteers. Front. Psychiatry 2020, 11, 576877. [Google Scholar] [CrossRef]
- Chung, H.; Fierro, A.; Pessoa-Mahana, C.D. Cannabidiol binding and negative allosteric modulation at the cannabinoid type 1 receptor in the presence of delta-9-tetrahydrocannabinol: An In Silico study. PLoS ONE 2019, 14, e0220025. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; Di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Steardo, L.; Scuderi, C.; Savani, C.; Cuomo, V.; Iuvone, T. CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci. Lett. 2006, 404, 342–346. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef]
- Libro, R.; Diomede, F.; Scionti, D.; Piattelli, A.; Grassi, G.; Pollastro, F.; Bramanti, P.; Mazzon, E.; Trubiani, O. Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. Int. J. Mol. Sci. 2016, 18, 26. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L.J.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef]
- Cheng, D.; Spiro, A.S.; Jenner, A.M.; Garner, B.; Karl, T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2014, 42, 1383–1396. [Google Scholar] [CrossRef]
- Hao, F.; Feng, Y. Cannabidiol (CBD) enhanced the hippocampal immune response and autophagy of APP/PS1 Alzheimer’s mice uncovered by RNA-seq. Life Sci. 2021, 264, 118624. [Google Scholar] [CrossRef] [PubMed]
- Defrancesco, M.; Hofer, A. Cannabinoid as Beneficial Replacement Therapy for Psychotropics to Treat Neuropsychiatric Symptoms in Severe Alzheimer’s Dementia: A Clinical Case Report. Front. Psychiatry 2020, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Todaro, B. Cannabinoids in the treatment of chemotherapy-induced nausea and vomiting. J. Natl. Compr. Cancer Netw. 2012, 10, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Koe, B.K.; Milne, G.M.; Weissman, A.; Johnson, M.R.; Melvin, L.S. Enhancement of brain [3H]flunitrazepam binding and analgesic activity of synthetic cannabimimetics. Eur. J. Pharmacol. 1985, 109, 201–212. [Google Scholar] [CrossRef]
- Lichtman, A.H.; Dimen, K.R.; Martin, B.R. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology 1995, 119, 282–290. [Google Scholar] [CrossRef]
- Clarke, J.R.; Rossato, J.I.; Monteiro, S.; Bevilaqua, L.R.M.; Izquierdo, I.; Cammarota, M. Posttraining activation of CB1 cannabinoid receptors in the CA1 region of the dorsal hippocampus impairs object recognition long-term memory. Neurobiol. Learn. Mem. 2008, 90, 374–381. [Google Scholar] [CrossRef]
- Kosiorek, P.; Hryniewicz, A.; Bialuk, I.; Zawadzka, A.; Winnicka, M.M. Cannabinoids alter recognition memory in rats. Pol. J. Pharmacol. 2003, 55, 903–910. [Google Scholar]
- Avdesh, A.; Hoe, Y.; Martins, R.N.; Martin-Iverson, M.T. Pharmacological effects of cannabinoids on the reference and working memory functions in mice. Psychopharmacology 2013, 225, 483–494. [Google Scholar] [CrossRef]
- Gessa, G.L.; Mascia, M.S.; Casu, M.A.; Carta, G. Inhibition of hippocampal acetylcholine release by cannabinoids: Reversal by SR 141716A. Eur. J. Pharmacol. 1997, 327, R1–R2. [Google Scholar] [CrossRef]
- Braida, D.; Sala, M. Cannabinoid-induced working memory impairment is reversed by a second generation cholinesterase inhibitor in rats. Neuroreport 2000, 11, 2025–2029. [Google Scholar] [CrossRef]
- Auclair, N.; Otani, S.; Soubrie, P.; Crepel, F. Cannabinoids Modulate Synaptic Strength and Plasticity at Glutamatergic Synapses of Rat Prefrontal Cortex Pyramidal Neurons. J. Neurophysiol. 2000, 83, 3287–3293. [Google Scholar] [CrossRef]
- Kucewicz, M.T.; Tricklebank, M.D.; Bogacz, R.; Jones, M.W. Dysfunctional Prefrontal Cortical Network Activity and Interactions following Cannabinoid Receptor Activation. J. Neurosci. 2011, 31, 15560–15568. [Google Scholar] [CrossRef]
- Wise, L.E.; Thorpe, A.J.; Lichtman, A.H. Hippocampal CB1 Receptors Mediate the Memory Impairing Effects of Δ9-Tetrahydrocannabinol. Neuropsychopharmacology 2009, 34, 2072–2080. [Google Scholar] [CrossRef]
- Terranova, J.P.; Michaud, J.C.; Le Fur, G.; Soubrié, P. Inhibition of long-term potentiation in rat hippocampal slices by anandamide and WIN55212-2: Reversal by SR141716 A, a selective antagonist of CB1 cannabinoid receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1995, 352, 576–579. [Google Scholar] [CrossRef]
- Hill, M.N.; Froc, D.J.; Fox, C.J.; Gorzalka, B.B.; Christie, B.R. Prolonged cannabinoid treatment results in spatial working memory deficits and impaired long-term potentiation in the CA1 region of the hippocampus in vivo. Eur. J. Neurosci. 2004, 20, 859–863. [Google Scholar] [CrossRef]
- Robinson, L.; Goonawardena, A.V.; Pertwee, R.G.; Hampson, R.E.; Riedel, G. The synthetic cannabinoid HU210 induces spatial memory deficits and suppresses hippocampal firing rate in rats. Br. J. Pharmacol. 2007, 151, 688–700. [Google Scholar] [CrossRef]
- Goonawardena, A.V.; Riedel, G.; Hampson, R.E. Cannabinoids alter spontaneous firing, bursting, and cell synchrony of hippocampal principal cells. Hippocampus 2011, 21, 520–531. [Google Scholar] [CrossRef]
- Barbieri, M.; Ossato, A.; Canazza, I.; Trapella, C.; Borelli, A.C.; Beggiato, S.; Rimondo, C.; Serpelloni, G.; Ferraro, L.; Marti, M. Synthetic cannabinoid JWH-018 and its halogenated derivatives JWH-018-Cl and JWH-018-Br impair Novel Object Recognition in mice: Behavioral, electrophysiological and neurochemical evidence. Neuropharmacology 2016, 109, 254–269. [Google Scholar] [CrossRef]
- Li, R.-S.; Fukumori, R.; Takeda, T.; Song, Y.; Morimoto, S.; Kikura-Hanajiri, R.; Yamaguchi, T.; Watanabe, K.; Aritake, K.; Tanaka, Y.; et al. Elevation of endocannabinoids in the brain by synthetic cannabinoid JWH-018: Mechanism and effect on learning and memory. Sci. Rep. 2019, 9, 9621. [Google Scholar] [CrossRef]
- Marchalant, Y.; Brothers, H.M.; Norman, G.J.; Karelina, K.; DeVries, A.C.; Wenk, G.L. Cannabinoids attenuate the effects of aging upon neuroinflammation and neurogenesis. Neurobiol. Dis. 2009, 34, 300–307. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Y.; Xiao, L.; Van Cleemput, J.; Ji, S.-P.; Bai, G.; Zhang, X. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J. Clin. Investig. 2005, 115, 3104–3116. [Google Scholar] [CrossRef]
- Jin, K.; Xie, L.; Kim, S.H.; Parmentier-Batteur, S.; Sun, Y.; Mao, X.O.; Childs, J.; Greenberg, D.A. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol. Pharmacol. 2004, 66, 204–208. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Min, X.; Cabral, G.A.; Lokensgard, J.R.; Peterson, P.K. Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1beta-stimulated human astrocytes. Glia 2005, 49, 211–219. [Google Scholar] [CrossRef]
- Velikova, M.; Doncheva, D.; Tashev, R. Subchronic effects of ligands of cannabinoid receptors on learning and memory processes of olfactory bulbectomized rats. Acta Neurobiol. Exp. 2020, 80, 286–296. [Google Scholar] [CrossRef]
- Llorente-Ovejero, A.; Manuel, I.; Lombardero, L.; Giralt, M.T.; Ledent, C.; Giménez-Llort, L.; Rodríguez-Puertas, R. Endocannabinoid and Muscarinic Signaling Crosstalk in the 3xTg-AD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 117–136. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; García-García, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; de Ceballos, M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflamm. 2012, 9, 8. [Google Scholar] [CrossRef]
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB1 agonist ACEA protects neurons and reduces the cognitive impairment of AβPP/PS1 mice. J. Alzheimers Dis. 2012, 30, 439–459. [Google Scholar] [CrossRef]
- Patricio-Martínez, A.; Sánchez-Zavaleta, R.; Angulo-Cruz, I.; Gutierrez-Praxedis, L.; Ramírez, E.; Martínez-García, I.; Limón, I.D. The Acute Activation of the CB1 Receptor in the Hippocampus Decreases Neurotoxicity and Prevents Spatial Memory Impairment in Rats Lesioned with β-Amyloid 25. Neuroscience 2019, 416, 239–254. [Google Scholar] [CrossRef]
- Svízenská, I.; Dubový, P.; Sulcová, A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—A short review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Jhaveri, M.D.; Sagar, D.R.; Elmes, S.J.R.; Kendall, D.A.; Chapman, V. Cannabinoid CB2 receptor-mediated anti-nociception in models of acute and chronic pain. Mol. Neurobiol. 2007, 36, 26–35. [Google Scholar] [CrossRef]
- Whiteside, G.T.; Lee, G.P.; Valenzano, K.J. The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr. Med. Chem. 2007, 14, 917–936. [Google Scholar] [CrossRef]
- Wright, K.L.; Duncan, M.; Sharkey, K.A. Cannabinoid CB2 receptors in the gastrointestinal tract: A regulatory system in states of inflammation. Br. J. Pharmacol. 2008, 153, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Ortega, Z.; Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 2012, 287, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Duff, G.; Argaw, A.; Cecyre, B.; Cherif, H.; Tea, N.; Zabouri, N.; Casanova, C.; Ptito, M.; Bouchard, J.-F. Cannabinoid receptor CB2 modulates axon guidance. PLoS ONE 2013, 8, e70849. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Li, Y. Chronic activation of CB2 cannabinoid receptors in the hippocampus increases excitatory synaptic transmission. J. Physiol. 2015, 593, 871–886. [Google Scholar] [CrossRef]
- Li, Y.; Kim, J. CB2 Cannabinoid Receptor Knockout in Mice Impairs Contextual Long-Term Memory and Enhances Spatial Working Memory. Neural Plast. 2016, 2016, 9817089. [Google Scholar] [CrossRef]
- Grünblatt, E.; Zander, N.; Bartl, J.; Jie, L.; Monoranu, C.-M.; Arzberger, T.; Ravid, R.; Roggendorf, W.; Gerlach, M.; Riederer, P. Comparison analysis of gene expression patterns between sporadic Alzheimer’s and Parkinson’s disease. J. Alzheimers Dis. 2007, 12, 291–311. [Google Scholar] [CrossRef]
- Koppel, J.; Vingtdeux, V.; Marambaud, P.; d’Abramo, C.; Jimenez, H.; Stauber, M.; Friedman, R.; Davies, P. CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer’s disease. Mol. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 791–804. [Google Scholar] [CrossRef]
- Zilka, N.; Kazmerova, Z.; Jadhav, S.; Neradil, P.; Madari, A.; Obetkova, D.; Bugos, O.; Novak, M. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J. Neuroinflamm. 2012, 9, 47. [Google Scholar] [CrossRef]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J. Alzheimers Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef]
- Casarejos, M.J.; Perucho, J.; Gomez, A.; Muñoz, M.P.; Fernandez-Estevez, M.; Sagredo, O.; Fernandez Ruiz, J.; Guzman, M.; de Yebenes, J.G.; Mena, M.A. Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J. Alzheimers Dis. 2013, 35, 525–539. [Google Scholar] [CrossRef]
- Scheiner, M.; Dolles, D.; Gunesch, S.; Hoffmann, M.; Nabissi, M.; Marinelli, O.; Naldi, M.; Bartolini, M.; Petralla, S.; Poeta, E.; et al. Dual-Acting Cholinesterase-Human Cannabinoid Receptor 2 Ligands Show Pronounced Neuroprotection in Vitro and Overadditive and Disease-Modifying Neuroprotective Effects in Vivo. J. Med. Chem. 2019, 62, 9078–9102. [Google Scholar] [CrossRef]
- Montanari, S.; Mahmoud, A.M.; Pruccoli, L.; Rabbito, A.; Naldi, M.; Petralla, S.; Moraleda, I.; Bartolini, M.; Monti, B.; Iriepa, I.; et al. Discovery of novel benzofuran-based compounds with neuroprotective and immunomodulatory properties for Alzheimer’s disease treatment. Eur. J. Med. Chem. 2019, 178, 243–258. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef]
- Panlilio, L.V.; Justinova, Z.; Goldberg, S.R. Inhibition of FAAH and activation of PPAR: New approaches to the treatment of cognitive dysfunction and drug addiction. Pharmacol. Ther. 2013, 138, 84–102. [Google Scholar] [CrossRef]
- Ahn, K.; Smith, S.E.; Liimatta, M.B.; Beidler, D.; Sadagopan, N.; Dudley, D.T.; Young, T.; Wren, P.; Zhang, Y.; Swaney, S.; et al. Mechanistic and pharmacological characterization of PF-04457845: A highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011, 338, 114–124. [Google Scholar] [CrossRef]
- Bilkei-Gorzo, A. The endocannabinoid system in normal and pathological brain ageing. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2012, 367, 3326–3341. [Google Scholar] [CrossRef]
- Piyanova, A.; Lomazzo, E.; Bindila, L.; Lerner, R.; Albayram, O.; Ruhl, T.; Lutz, B.; Zimmer, A.; Bilkei-Gorzo, A. Age-related changes in the endocannabinoid system in the mouse hippocampus. Mech. Ageing Dev. 2015, 150, 55–64. [Google Scholar] [CrossRef]
- Jung, K.-M.; Astarita, G.; Yasar, S.; Vasilevko, V.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. An amyloid β42-dependent deficit in anandamide mobilization is associated with cognitive dysfunction in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1522–1532. [Google Scholar] [CrossRef]
- Pascual, A.C.; Martín-Moreno, A.M.; Giusto, N.M.; de Ceballos, M.L.; Pasquaré, S.J. Normal aging in rats and pathological aging in human Alzheimer’s disease decrease FAAH activity: Modulation by cannabinoid agonists. Exp. Gerontol. 2014, 60, 92–99. [Google Scholar] [CrossRef]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134, 1041–1060. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; De Filippis, D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. 2006, 63, 1410–1424. [Google Scholar] [CrossRef]
- Aguilera-Portillo, G.; Rangel-López, E.; Villeda-Hernández, J.; Chavarría, A.; Castellanos, P.; Elmazoglu, Z.; Karasu, Ç.; Túnez, I.; Pedraza, G.; Königsberg, M.; et al. The Pharmacological Inhibition of Fatty Acid Amide Hydrolase Prevents Excitotoxic Damage in the Rat Striatum: Possible Involvement of CB1 Receptors Regulation. Mol. Neurobiol. 2019, 56, 844–856. [Google Scholar] [CrossRef]
- Maya-López, M.; Ruiz-Contreras, H.A.; de Jesús Negrete-Ruíz, M.; Martínez-Sánchez, J.E.; Benítez-Valenzuela, J.; Colín-González, A.L.; Villeda-Hernández, J.; Sánchez-Chapul, L.; Parra-Cid, C.; Rangel-López, E.; et al. URB597 reduces biochemical, behavioral and morphological alterations in two neurotoxic models in rats. Biomed. Pharmacother. 2017, 88, 745–753. [Google Scholar] [CrossRef]
- Elmazoglu, Z.; Rangel-López, E.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Túnez, I.; Aschner, M.; Santamaría, A.; Karasu, Ç. Cannabinoid-profiled agents improve cell survival via reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Aβ(1-42) peptide in rat hippocampal neurons. Neurochem. Int. 2020, 140, 104817. [Google Scholar] [CrossRef]
- Murphy, N.; Cowley, T.R.; Blau, C.W.; Dempsey, C.N.; Noonan, J.; Gowran, A.; Tanveer, R.; Olango, W.M.; Finn, D.P.; Campbell, V.A.; et al. The fatty acid amide hydrolase inhibitor URB597 exerts anti-inflammatory effects in hippocampus of aged rats and restores an age-related deficit in long-term potentiation. J. Neuroinflamm. 2012, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Scipioni, L.; Arosio, B.; Mari, D.; Oddi, S.; Maccarrone, M. Anti-Inflammatory Effects of Fatty Acid Amide Hydrolase Inhibition in Monocytes/Macrophages from Alzheimer’s Disease Patients. Biomolecules 2021, 11, 502. [Google Scholar] [CrossRef]
- Varvel, S.A.; Wise, L.E.; Niyuhire, F.; Cravatt, B.F.; Lichtman, A.H. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology 2007, 32, 1032–1041. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Liebowitz, M.R.; Stein, M.B.; Grunfeld, J.; Van Hove, I.; Simmons, W.K.; Van Der Ark, P.; Palmer, J.A.; Saad, Z.S.; Pemberton, D.J.; et al. The effects of inhibition of fatty acid amide hydrolase (FAAH) by JNJ-42165279 in social anxiety disorder: A double-blind, randomized, placebo-controlled proof-of-concept study. Neuropsychopharmacology 2020, 46, 1004–1010. [Google Scholar] [CrossRef]
- Johnson, D.S.; Stiff, C.; Lazerwith, S.E.; Kesten, S.R.; Fay, L.K.; Morris, M.; Beidler, D.; Liimatta, M.B.; Smith, S.E.; Dudley, D.T.; et al. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med. Chem. Lett. 2011, 2, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yagyu, K.; Sackett, S.; Zhang, Y. Anti-Inflammatory Effects by Pharmacological Inhibition or Knockdown of Fatty Acid Amide Hydrolase in BV2 Microglial Cells. Cells 2019, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflamm. 2015, 12, 81. [Google Scholar] [CrossRef]
- Anderson, W.B.; Gould, M.J.; Torres, R.D.; Mitchell, V.A.; Vaughan, C.W. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine inflammatory pain model. Neuropharmacology 2014, 81, 224–230. [Google Scholar] [CrossRef]
- Aguado, T.; Monory, K.; Palazuelos, J.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005, 19, 1704–1706. [Google Scholar] [CrossRef]
- Goncalves, M.B.; Suetterlin, P.; Yip, P.; Molina-Holgado, F.; Walker, D.J.; Oudin, M.J.; Zentar, M.P.; Pollard, S.; Yáñez-Muñoz, R.J.; Williams, G.; et al. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol. Cell. Neurosci. 2008, 38, 526–536. [Google Scholar] [CrossRef]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.-Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef]
- Ratano, P.; Petrella, C.; Forti, F.; Passeri, P.P.; Morena, M.; Palmery, M.; Trezza, V.; Severini, C.; Campolongo, P. Pharmacological inhibition of 2-arachidonoilglycerol hydrolysis enhances memory consolidation in rats through CB2 receptor activation and mTOR signaling modulation. Neuropharmacology 2018, 138, 210–218. [Google Scholar] [CrossRef]
- Mazier, W.; Saucisse, N.; Simon, V.; Cannich, A.; Marsicano, G.; Massa, F.; Cota, D. mTORC1 and CB1 receptor signaling regulate excitatory glutamatergic inputs onto the hypothalamic paraventricular nucleus in response to energy availability. Mol. Metab. 2019, 28, 151–159. [Google Scholar] [CrossRef]
- Suliman, N.A.; Taib, C.N.M.; Moklas, M.A.M.; Basir, R. Delta-9-Tetrahydrocannabinol (∆(9)-THC) Induce Neurogenesis and Improve Cognitive Performances of Male Sprague Dawley Rats. Neurotox. Res. 2018, 33, 402–411. [Google Scholar] [CrossRef]
- Khodadadi, H.; Salles, É.L.; Jarrahi, A.; Costigliola, V.; Khan, M.B.; Yu, J.C.; Morgan, J.C.; Hess, D.C.; Vaibhav, K.; Dhandapani, K.M.; et al. Cannabidiol Ameliorates Cognitive Function via Regulation of IL-33 and TREM2 Upregulation in a Murine Model of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 973–977. [Google Scholar] [CrossRef]
- Garberg, H.T.; Solberg, R.; Barlinn, J.; Martinez-Orgado, J.; Løberg, E.-M.; Saugstad, O.D. High-Dose Cannabidiol Induced Hypotension after Global Hypoxia-Ischemia in Piglets. Neonatology 2017, 112, 143–149. [Google Scholar] [CrossRef]
- ElBatsh, M.M.; Assareh, N.; Marsden, C.A.; Kendall, D.A. Anxiogenic-like effects of chronic cannabidiol administration in rats. Psychopharmacolgy 2012, 221, 239–247. [Google Scholar] [CrossRef]
- Jayamanne, A.; Greenwood, R.; Mitchell, V.A.; Aslan, S.; Piomelli, D.; Vaughan, C.W. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br. J. Pharmacol. 2006, 147, 281–288. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacological actions of cannabinoids. Handb. Exp. Pharmacol. 2005, 168, 1–51. [Google Scholar] [CrossRef]
- Mallet, C.; Dubray, C.; Dualé, C. FAAH inhibitors in the limelight, but regrettably. Int. J. Clin. Pharmacol. Ther. 2016, 54, 498–501. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An Update on Non-CB(1), Non-CB(2) Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Ye, L.; Cao, Z.; Wang, W.; Zhou, N. New Insights in Cannabinoid Receptor Structure and Signaling. Curr. Mol. Pharmacol. 2019, 12, 239–248. [Google Scholar] [CrossRef]
- Allende, G.; Chávez-Reyes, J.; Guerrero-Alba, R.; Vázquez-León, P.; Marichal-Cancino, B.A. Advances in Neurobiology and Pharmacology of GPR. Front. Pharmacol. 2020, 11, 628. [Google Scholar] [CrossRef]
- Uhlenbrock, K.; Gassenhuber, H.; Kostenis, E. Sphingosine 1-phosphate is a ligand of the human gpr3, gpr6 and gpr12 family of constitutively active G protein-coupled receptors. Cell. Signal. 2002, 14, 941–953. [Google Scholar] [CrossRef]
- Ignatov, A.; Lintzel, J.; Hermans-Borgmeyer, I.; Kreienkamp, H.-J.; Joost, P.; Thomsen, S.; Methner, A.; Schaller, H.C. Role of the G-protein-coupled receptor GPR12 as high-affinity receptor for sphingosylphosphorylcholine and its expression and function in brain development. J. Neurosci. 2003, 23, 907–914. [Google Scholar] [CrossRef]
- Huang, Y.; Skwarek-Maruszewska, A.; Horré, K.; Vandewyer, E.; Wolfs, L.; Snellinx, A.; Saito, T.; Radaelli, E.; Corthout, N.; Colombelli, J.; et al. Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Sci. Transl. Med. 2015, 7, 309ra164. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.D.; Sheng, M. Gpr3 stimulates Aβ production via interactions with APP and β-Arrestinplos. PLoS ONE 2013, 8, e74680. [Google Scholar] [CrossRef]
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Balenga, N.A.B.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Rempel, V.; Volz, N.; Gläser, F.; Nieger, M.; Bräse, S.; Müller, C.E. Antagonists for the orphan G-protein-coupled receptor GPR55 based on a coumarin scaffold. J. Med. Chem. 2013, 56, 4798–4810. [Google Scholar] [CrossRef]
- Kotsikorou, E.; Lynch, D.L.; Abood, M.E.; Reggio, P.H. Lipid bilayer molecular dynamics study of lipid-derived agonists of the putative cannabinoid receptor, GPR. Chem. Phys. Lipids 2011, 164, 131–143. [Google Scholar] [CrossRef][Green Version]
- Reyes-Resina, I.; Navarro, G.; Aguinaga, D.; Canela, E.I.; Schoeder, C.T.; Załuski, M.; Kieć-Kononowicz, K.; Saura, C.A.; Müller, C.E.; Franco, R. Molecular and functional interaction between GPR18 and cannabinoid CB(2) G-protein-coupled receptors. Relevance in neurodegenerative diseases. Biochem. Pharmacol. 2018, 157, 169–179. [Google Scholar] [CrossRef]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef]
- Musella, A.; Fresegna, D.; Rizzo, F.R.; Gentile, A.; Bullitta, S.; De Vito, F.; Guadalupi, L.; Centonze, D.; Mandolesi, G. A novel crosstalk within the endocannabinoid system controls GABA transmission in the striatum. Sci. Rep. 2017, 7, 7363. [Google Scholar] [CrossRef]
- Hill, J.D.; Zuluaga-Ramirez, V.; Gajghate, S.; Winfield, M.; Sriram, U.; Rom, S.; Persidsky, Y. Activation of GPR55 induces neuroprotection of hippocampal neurogenesis and immune responses of neural stem cells following chronic, systemic inflammation. Brain Behav. Immun. 2019, 76, 165–181. [Google Scholar] [CrossRef]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 2008, 139, 225–236. [Google Scholar] [CrossRef]
- Saliba, S.W.; Jauch, H.; Gargouri, B.; Keil, A.; Hurrle, T.; Volz, N.; Mohr, F.; van der Stelt, M.; Bräse, S.; Fiebich, B.L. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflamm. 2018, 15, 322. [Google Scholar] [CrossRef]
- Wu, C.-S.; Chen, H.; Sun, H.; Zhu, J.; Jew, C.P.; Wager-Miller, J.; Straiker, A.; Spencer, C.; Bradshaw, H.; Mackie, K.; et al. GPR55, a G-Protein Coupled Receptor for Lysophosphatidylinositol, Plays a Role in Motor Coordination. PLoS ONE 2013, 8, e60314. [Google Scholar] [CrossRef]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Méndez-Díaz, M.; Prospéro-García, O. Possible role of hippocampal GPR55 in spatial learning and memory in rats. Acta Neurobiol. Exp. 2018, 78, 41–50. [Google Scholar] [CrossRef]
- Medina-Vera, D.; Rosell-Valle, C.; López-Gambero, A.J.; Navarro, J.A.; Zambrana-Infantes, E.N.; Rivera, P.; Santín, L.J.; Suarez, J.; Rodríguez de Fonseca, F. Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity. Biology 2020, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Croxford, J.L. Therapeutic Potential of Cannabinoids in CNS Disease. CNS Drugs 2003, 17, 179–202. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Kuhns, L.; Cousijn, J. The short-term and long-term effects of cannabis on cognition: Recent advances in the field. Curr. Opin. Psychol. 2021, 38, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Urits, I.; Borchart, M.; Hasegawa, M.; Kochanski, J.; Orhurhu, V.; Viswanath, O. An Update of Current Cannabis-Based Pharmaceuticals in Pain Medicine. Pain Ther. 2019, 8, 41–51. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abate, G.; Uberti, D.; Tambaro, S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology 2021, 10, 542. https://doi.org/10.3390/biology10060542
Abate G, Uberti D, Tambaro S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology. 2021; 10(6):542. https://doi.org/10.3390/biology10060542
Chicago/Turabian StyleAbate, Giulia, Daniela Uberti, and Simone Tambaro. 2021. "Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy" Biology 10, no. 6: 542. https://doi.org/10.3390/biology10060542
APA StyleAbate, G., Uberti, D., & Tambaro, S. (2021). Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology, 10(6), 542. https://doi.org/10.3390/biology10060542