
Exploiting DNA Endonucleases to Advance Mechanisms of DNA Repair


Simple Summary
Abstract
1. Introduction

2. Harnessing CRISPR for Genome Editing
3. Structural Properties of the CRISPR Effector Molecules
4. Incorporating DNA Edits via Cas9 and DSB Repair
5. Cas System Modifications
5.1. Cas Orthologs
5.2. Cas Variants
5.3. Expression Modification Systems
5.4. Base and Prime Editors
6. Mechanistic Advancement of DNA Repair Processes by CRISPR-Controlled Modifications
6.1. Contribution of Gene Editing Technologies to MMR Biology
6.2. Advances in BER Biology by Exploiting Gene Editing Technologies
6.2.1. BER/SSBR Lesion Recognition & Strand Scission
6.2.2. DNA Damage-Induced PARP1 and PARP2 Activation in BER/SSBR, in Response to Replication Stress and Advancing Our Understanding of PARP1/PARP2 Inhibitors
6.2.3. DNA Gap Tailoring in BER/SSBR
6.2.4. PAR Degradation
7. CRISPR Advancements in the Clinical Setting (and Promising Future Advancements)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grissa, I.; Bouchon, P.; Pourcel, C.; Vergnaud, G. On-line resources for bacterial micro-evolution studies using MLVA or CRISPR typing. Biochimie 2008, 90, 660–668. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef]
- Jansen, R.; Embden, J.D.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Kunin, V.; Sorek, R.; Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8, R61. [Google Scholar] [CrossRef]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology (Reading) 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology (Reading) 2005, 151, 653–663. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonte, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Doudna, J.A. Protecting genome integrity during CRISPR immune adaptation. Nat. Struct. Mol. Biol. 2016, 23, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.; Beloglazova, N.; Flick, R.; Graham, C.; Skarina, T.; Nocek, B.; Gagarinova, A.; Pogoutse, O.; Brown, G.; Binkowski, A.; et al. A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 2011, 79, 484–502. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Brown, G.; Zimmerman, M.D.; Proudfoot, M.; Makarova, K.S.; Kudritska, M.; Kochinyan, S.; Wang, S.; Chruszcz, M.; Minor, W.; et al. A novel family of sequence-specific endoribonucleases associated with the clustered regularly interspaced short palindromic repeats. J. Biol. Chem. 2008, 283, 20361–20371. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Zhao, H.; Sheng, G.; Wang, M.; Yin, M.; Wang, Y. Structural and Mechanistic Basis of PAM-Dependent Spacer Acquisition in CRISPR-Cas Systems. Cell 2015, 163, 840–853. [Google Scholar] [CrossRef]
- Rollie, C.; Schneider, S.; Brinkmann, A.S.; Bolt, E.L.; White, M.F. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Westra, E.R.; Semenova, E.; Datsenko, K.A.; Jackson, R.N.; Wiedenheft, B.; Severinov, K.; Brouns, S.J. Type I-E CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013, 9, e1003742. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Pougach, K.; Tikhonov, A.; Wanner, B.L.; Severinov, K.; Semenova, E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012, 3, 945. [Google Scholar] [CrossRef]
- Zhang, Y.; Heidrich, N.; Ampattu, B.J.; Gunderson, C.W.; Seifert, H.S.; Schoen, C.; Vogel, J.; Sontheimer, E.J. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol. Cell 2013, 50, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M.L.; Doudna, J.A. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem. Sci. 2015, 40, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Carte, J.; Wang, R.; Li, H.; Terns, R.M.; Terns, M.P. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008, 22, 3489–3496. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Hochstrasser, M.L.; Taylor, D.W.; Bhat, P.; Guegler, C.K.; Sternberg, S.H.; Nogales, E.; Doudna, J.A. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc. Natl. Acad. Sci. USA 2014, 111, 6618–6623. [Google Scholar] [CrossRef]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef]
- Reeks, J.; Naismith, J.H.; White, M.F. CRISPR interference: A structural perspective. Biochem. J. 2013, 453, 155–166. [Google Scholar] [CrossRef]
- Reeks, J.; Graham, S.; Anderson, L.; Liu, H.; White, M.F.; Naismith, J.H. Structure of the archaeal Cascade subunit Csa5: Relating the small subunits of CRISPR effector complexes. RNA Biol. 2013, 10, 762–769. [Google Scholar] [CrossRef][Green Version]
- Sashital, D.G.; Wiedenheft, B.; Doudna, J.A. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell 2012, 46, 606–615. [Google Scholar] [CrossRef]
- Wright, A.V.; Nunez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Nam, K.H.; Haitjema, C.; Liu, X.; Ding, F.; Wang, H.; DeLisa, M.P.; Ke, A. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 2012, 20, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Garside, E.L.; Schellenberg, M.J.; Gesner, E.M.; Bonanno, J.B.; Sauder, J.M.; Burley, S.K.; Almo, S.C.; Mehta, G.; MacMillan, A.M. Cas5d processes pre-crRNA and is a member of a larger family of CRISPR RNA endonucleases. RNA 2012, 18, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Petit, P.; Flick, R.; Brown, G.; Savchenko, A.; Yakunin, A.F. Structure and activity of the Cas3 HD nuclease MJ0384, an effector enzyme of the CRISPR interference. EMBO J. 2011, 30, 4616–4627. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006, 1, 7. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Szczelkun, M.D.; Tikhomirova, M.S.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Sampson, T.R.; Saroj, S.D.; Llewellyn, A.C.; Tzeng, Y.L.; Weiss, D.S. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 2013, 497, 254–257. [Google Scholar] [CrossRef]
- Anderson, E.M.; Haupt, A.; Schiel, J.A.; Chou, E.; Machado, H.B.; Strezoska, Z.; Lenger, S.; McClelland, S.; Birmingham, A.; Vermeulen, A.; et al. Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. J. Biotechnol. 2015, 211, 56–65. [Google Scholar] [CrossRef]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Van der Oost, J.; Westra, E.R.; Jackson, R.N.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014, 12, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, A.; Yao, M.; Masui, R.; Tanaka, I.; Yokoyama, S.; Kuramitsu, S. Crystal structure of hypothetical protein TTHB192 from Thermus thermophilus HB8 reveals a new protein family with an RNA recognition motif-like domain. Protein Sci. 2006, 15, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Oke, M.; Carter, L.G.; Johnson, K.A.; Liu, H.; McMahon, S.A.; Yan, X.; Kerou, M.; Weikart, N.D.; Kadi, N.; Sheikh, M.A.; et al. The Scottish Structural Proteomics Facility: Targets, methods and outputs. J. Struct. Funct. Genom. 2010, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Lintner, N.G.; Kerou, M.; Brumfield, S.K.; Graham, S.; Liu, H.; Naismith, J.H.; Sdano, M.; Peng, N.; She, Q.; Copie, V.; et al. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE). J. Biol. Chem. 2011, 286, 21643–21656. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H.; Kurinov, I.; Ke, A. Crystal structure of clustered regularly interspaced short palindromic repeats (CRISPR)-associated Csn2 protein revealed Ca2+-dependent double-stranded DNA binding activity. J. Biol. Chem. 2011, 286, 30759–30768. [Google Scholar] [CrossRef]
- Lintner, N.G.; Frankel, K.A.; Tsutakawa, S.E.; Alsbury, D.L.; Copie, V.; Young, M.J.; Tainer, J.A.; Lawrence, C.M. The structure of the CRISPR-associated protein Csa3 provides insight into the regulation of the CRISPR/Cas system. J. Mol. Biol. 2011, 405, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Zhou, K.; Jinek, M.; Coyle, S.M.; Ma, W.; Doudna, J.A. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure 2009, 17, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Koo, Y.; Ka, D.; Kim, E.J.; Suh, N.; Bae, E. Conservation and variability in the structure and function of the Cas5d endoribonuclease in the CRISPR-mediated microbial immune system. J. Mol. Biol. 2013, 425, 3799–3810. [Google Scholar] [CrossRef]
- Cocozaki, A.I.; Ramia, N.F.; Shao, Y.; Hale, C.R.; Terns, R.M.; Terns, M.P.; Li, H. Structure of the Cmr2 subunit of the CRISPR-Cas RNA silencing complex. Structure 2012, 20, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H.; Huang, Q.; Ke, A. Nucleic acid binding surface and dimer interface revealed by CRISPR-associated CasB protein structures. FEBS Lett. 2012, 586, 3956–3961. [Google Scholar] [CrossRef]
- Hrle, A.; Su, A.A.; Ebert, J.; Benda, C.; Randau, L.; Conti, E. Structure and RNA-binding properties of the type III-A CRISPR-associated protein Csm3. RNA Biol. 2013, 10, 1670–1678. [Google Scholar] [CrossRef]
- Lemak, S.; Beloglazova, N.; Nocek, B.; Skarina, T.; Flick, R.; Brown, G.; Popovic, A.; Joachimiak, A.; Savchenko, A.; Yakunin, A.F. Toroidal structure and DNA cleavage by the CRISPR-associated [4Fe-4S] cluster containing Cas4 nuclease SSO0001 from Sulfolobus solfataricus. J. Am. Chem. Soc. 2013, 135, 17476–17487. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef]
- Jackson, R.N.; Golden, S.M.; van Erp, P.B.; Carter, J.; Westra, E.R.; Brouns, S.J.; van der Oost, J.; Terwilliger, T.C.; Read, R.J.; Wiedenheft, B. Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 2014, 345, 1473–1479. [Google Scholar] [CrossRef]
- Huo, Y.; Nam, K.H.; Ding, F.; Lee, H.; Wu, L.; Xiao, Y.; Farchione, M.D., Jr.; Zhou, S.; Rajashankar, K.; Kurinov, I.; et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014, 21, 771–777. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef]
- Yamano, T.; Nishimasu, H.; Zetsche, B.; Hirano, H.; Slaymaker, I.M.; Li, Y.; Fedorova, I.; Nakane, T.; Makarova, K.S.; Koonin, E.V.; et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell 2016, 165, 949–962. [Google Scholar] [CrossRef]
- Gallo, G.; Augusto, G.; Rangel, G.; Zelanis, A.; Mori, M.A.; Campos, C.B.; Wurtele, M. Structural basis for dimer formation of the CRISPR-associated protein Csm2 of Thermotoga maritima. FEBS J. 2016, 283, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yao, D.; Xu, J.G.; Li, A.R.; Xu, J.; Fu, P.; Zhou, Y.; Zhu, Y. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat. Struct. Mol. Biol. 2016, 23, 868–870. [Google Scholar] [CrossRef]
- Yang, H.; Gao, P.; Rajashankar, K.R.; Patel, D.J. PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell 2016, 167, 1814–1828 e1812. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Ma, J.; Li, Z.; You, L.; Wang, J.; Wang, M.; Zhang, X.; Wang, Y. The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 2017, 170, 714–726 e710. [Google Scholar] [CrossRef]
- Zhang, C.; Konermann, S.; Brideau, N.J.; Lotfy, P.; Wu, X.; Novick, S.J.; Strutzenberg, T.; Griffin, P.R.; Hsu, P.D.; Lyumkis, D. Structural Basis for the RNA-Guided Ribonuclease Activity of CRISPR-Cas13d. Cell 2018, 175, 212–223 e217. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Mesa, P.; Kellner, M.J.; Kannan, S.; Brignole, E.; Koob, J.; Feliciano, P.R.; Stella, S.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. High-Resolution Structure of Cas13b and Biochemical Characterization of RNA Targeting and Cleavage. Cell Rep. 2019, 26, 3741–3751 e3745. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.N.; Nakagawa, R.; Okazaki, S.; Hirano, H.; Kobayashi, K.; Kusakizako, T.; Nishizawa, T.; Yamashita, K.; Nishimasu, H.; Nureki, O. Structure of the miniature type V-F CRISPR-Cas effector enzyme. Mol. Cell 2021, 81, 558–570 e553. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Xiao, R.; Chang, L. Mechanisms for target recognition and cleavage by the Cas12i RNA-guided endonuclease. Nat. Struct. Mol. Biol. 2020, 27, 1069–1076. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.; Xiao, R.; Han, R.; Chang, L. Cryo-EM structure of the RNA-guided ribonuclease Cas12g. Nat. Chem. Biol. 2021, 17, 387–393. [Google Scholar] [CrossRef]
- Pyzocha, N.K.; Chen, S. Diverse Class 2 CRISPR-Cas Effector Proteins for Genome Engineering Applications. ACS Chem. Biol. 2018, 13, 347–356. [Google Scholar] [CrossRef]
- Barman, A.; Deb, B.; Chakraborty, S. A glance at genome editing with CRISPR-Cas9 technology. Curr. Genet. 2020, 66, 447–462. [Google Scholar] [CrossRef]
- Gasiunas, G.; Young, J.K.; Karvelis, T.; Kazlauskas, D.; Urbaitis, T.; Jasnauskaite, M.; Grusyte, M.M.; Paulraj, S.; Wang, P.H.; Hou, Z.; et al. A catalogue of biochemically diverse CRISPR-Cas9 orthologs. Nat. Commun. 2020, 11, 5512. [Google Scholar] [CrossRef]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug. Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Lee, C.M.; Cradick, T.J.; Bao, G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. 2016, 24, 645–654. [Google Scholar] [CrossRef]
- Ma, E.; Harrington, L.B.; O’Connell, M.R.; Zhou, K.; Doudna, J.A. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell 2015, 60, 398–407. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [PubMed]
- Hirano, H.; Gootenberg, J.S.; Horii, T.; Abudayyeh, O.O.; Kimura, M.; Hsu, P.D.; Nakane, T.; Ishitani, R.; Hatada, I.; Zhang, F.; et al. Structure and Engineering of Francisella novicida Cas9. Cell 2016, 164, 950–961. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Mali, P.; Braff, J.L.; Moosburner, M.; Yaung, S.J.; Church, G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 2013, 10, 1116–1121. [Google Scholar] [CrossRef]
- Karvelis, T.; Gasiunas, G.; Miksys, A.; Barrangou, R.; Horvath, P.; Siksnys, V. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013, 10, 841–851. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef]
- Liu, L.; Chen, P.; Wang, M.; Li, X.; Wang, J.; Yin, M.; Wang, Y. C2c1-sgRNA Complex Structure Reveals RNA-Guided DNA Cleavage Mechanism. Mol. Cell 2017, 65, 310–322. [Google Scholar] [CrossRef]
- Yan, W.X.; Hunnewell, P.; Alfonse, L.E.; Carte, J.M.; Keston-Smith, E.; Sothiselvam, S.; Garrity, A.J.; Chong, S.; Makarova, K.S.; Koonin, E.V.; et al. Functionally diverse type V CRISPR-Cas systems. Science 2019, 363, 88–91. [Google Scholar] [CrossRef]
- Harrington, L.B.; Ma, E.; Chen, J.S.; Witte, I.P.; Gertz, D.; Paez-Espino, D.; Al-Shayeb, B.; Kyrpides, N.C.; Burstein, D.; Banfield, J.F.; et al. A scoutRNA Is Required for Some Type V CRISPR-Cas Systems. Mol. Cell 2020, 79, 416–424 e415. [Google Scholar] [CrossRef]
- Burstein, D.; Harrington, L.B.; Strutt, S.C.; Probst, A.J.; Anantharaman, K.; Thomas, B.C.; Doudna, J.A.; Banfield, J.F. New CRISPR-Cas systems from uncultivated microbes. Nature 2017, 542, 237–241. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef]
- Karvelis, T.; Bigelyte, G.; Young, J.K.; Hou, Z.; Zedaveinyte, R.; Budre, K.; Paulraj, S.; Djukanovic, V.; Gasior, S.; Silanskas, A.; et al. PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 2020, 48, 5016–5023. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef]
- Strecker, J.; Ladha, A.; Gardner, Z.; Schmid-Burgk, J.L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48–53. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Smargon, A.A.; Cox, D.B.T.; Pyzocha, N.K.; Zheng, K.; Slaymaker, I.M.; Gootenberg, J.S.; Abudayyeh, O.A.; Essletzbichler, P.; Shmakov, S.; Makarova, K.S.; et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol. Cell 2017, 65, 618–630 e617. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676 e614. [Google Scholar] [CrossRef]
- Lewis, K.M.; Ke, A. Building the Class 2 CRISPR-Cas Arsenal. Mol. Cell 2017, 65, 377–379. [Google Scholar] [CrossRef]
- Jiang, F.; Taylor, D.W.; Chen, J.S.; Kornfeld, J.E.; Zhou, K.; Thompson, A.J.; Nogales, E.; Doudna, J.A. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 2016, 351, 867–871. [Google Scholar] [CrossRef]
- Sternberg, S.H.; LaFrance, B.; Kaplan, M.; Doudna, J.A. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 2015, 527, 110–113. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Swarts, D.C.; van der Oost, J.; Jinek, M. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 2017, 66, 221–233 e224. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.N.; van Erp, P.B.; Sternberg, S.H.; Wiedenheft, B. Conformational regulation of CRISPR-associated nucleases. Curr. Opin. Microbiol. 2017, 37, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C.; Jinek, M. Mechanistic Insights into the cis- and trans-Acting DNase Activities of Cas12a. Mol. Cell 2019, 73, 589–600 e584. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Wang, J.; Wang, M.; Chen, P.; Yin, M.; Li, J.; Sheng, G.; Wang, Y. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 2017, 168, 121–134 e112. [Google Scholar] [CrossRef]
- Makarova, K.S.; Anantharaman, V.; Aravind, L.; Koonin, E.V. Live virus-free or die: Coupling of antivirus immunity and programmed suicide or dormancy in prokaryotes. Biol. Direct. 2012, 7, 40. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Tainer, J.A. DNA mismatch repair: The hands of a genome guardian. Structure 2000, 8, R237–R241. [Google Scholar] [CrossRef]
- Boulton, S.J. DNA repair: A heavyweight joins the fray. Nature 2009, 462, 857–858. [Google Scholar] [CrossRef]
- Boulton, S.J. DNA repair: Decision at the break point. Nature 2010, 465, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Parikh, S.S.; Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Envisioning the molecular choreography of DNA base excision repair. Curr. Opin. Struct. Biol. 1999, 9, 37–47. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Corneo, B.; Wendland, R.L.; Deriano, L.; Cui, X.; Klein, I.A.; Wong, S.Y.; Arnal, S.; Holub, A.J.; Weller, G.R.; Pancake, B.A.; et al. Rag mutations reveal robust alternative end joining. Nature 2007, 449, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Hanscom, T.; McVey, M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells 2020, 9, 1657. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, T.A.; Tainer, J.A.; Lees-Miller, S.P. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair 2010, 9, 1307–1314. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Jette, N.; Lees-Miller, S.P. Non-homologous end joining: Emerging themes and unanswered questions. DNA Repair 2014, 17, 2–8. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Song, Y.; Yuan, L.; Wang, Y.; Chen, M.; Deng, J.; Lv, Q.; Sui, T.; Li, Z.; Lai, L. Efficient dual sgRNA-directed large gene deletion in rabbit with CRISPR/Cas9 system. Cell Mol. Life Sci. 2016, 73, 2959–2968. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Chen, L.; Shen, B.; Zhou, J.; Hu, B.; Du, Y.; Tate, P.H.; Huang, X.; Zhang, W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014, 11, 829–835. [Google Scholar] [CrossRef]
- Chen, X.; Xu, F.; Zhu, C.; Ji, J.; Zhou, X.; Feng, X.; Guang, S. Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci. Rep. 2014, 4, 7581. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Suzuki, K.; Izpisua Belmonte, J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018, 63, 157–164. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2018, 9, 691. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, C.; Li, J.; Xing, P.; Li, J.; Zheng, S.; Chen, X. Cell cycle-dependent control of homologous recombination. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 655–668. [Google Scholar] [CrossRef]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef]
- Sung, P.; Krejci, L.; Van Komen, S.; Sehorn, M.G. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003, 278, 42729–42732. [Google Scholar] [CrossRef]
- Liu, J.; Ehmsen, K.T.; Heyer, W.D.; Morrical, S.W. Presynaptic filament dynamics in homologous recombination and DNA repair. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 240–270. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Neuwirth, E.A.; Honma, M.; Grosovsky, A.J. Interchromosomal crossover in human cells is associated with long gene conversion tracts. Mol. Cell Biol. 2007, 27, 5261–5274. [Google Scholar] [CrossRef]
- Aguilera, A.; Klein, H.L. Yeast intrachromosomal recombination: Long gene conversion tracts are preferentially associated with reciprocal exchange and require the RAD1 and RAD3 gene products. Genetics 1989, 123, 683–694. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Kadyk, L.C.; Hartwell, L.H. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992, 132, 387–402. [Google Scholar] [CrossRef]
- Sharma, A.; Toepfer, C.N.; Ward, T.; Wasson, L.; Agarwal, R.; Conner, D.A.; Hu, J.H.; Seidman, C.E. CRISPR/Cas9-Mediated Fluorescent Tagging of Endogenous Proteins in Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2018, 96, 21.11.1–21.11.20. [Google Scholar] [CrossRef]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ’knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Guo, Q.; Mintier, G.; Ma-Edmonds, M.; Storton, D.; Wang, X.; Xiao, X.; Kienzle, B.; Zhao, D.; Feder, J.N. ’Cold shock’ increases the frequency of homology directed repair gene editing in induced pluripotent stem cells. Sci. Rep. 2018, 8, 2080. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Miura, H.; Quadros, R.M.; Gurumurthy, C.B.; Ohtsuka, M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protoc. 2018, 13, 195–215. [Google Scholar] [CrossRef]
- Yoshimi, K.; Kunihiro, Y.; Kaneko, T.; Nagahora, H.; Voigt, B.; Mashimo, T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016, 7, 10431. [Google Scholar] [CrossRef]
- Wu, S.M.; Hochedlinger, K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011, 13, 497–505. [Google Scholar] [CrossRef]
- He, X.; Li, Y.X.; Feng, B. New Turns for High Efficiency Knock-In of Large DNA in Human Pluripotent Stem Cells. Stem Cells Int. 2018, 2018, 9465028. [Google Scholar] [CrossRef]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.T.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [Google Scholar] [CrossRef]
- Kim, S.I.; Matsumoto, T.; Kagawa, H.; Nakamura, M.; Hirohata, R.; Ueno, A.; Ohishi, M.; Sakuma, T.; Soga, T.; Yamamoto, T.; et al. Microhomology-assisted scarless genome editing in human iPSCs. Nat. Commun. 2018, 9, 939. [Google Scholar] [CrossRef]
- Yang, K.; Guo, R.; Xu, D. Non-homologous end joining: Advances and frontiers. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 632–640. [Google Scholar] [CrossRef]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Yamada, M.; Watanabe, Y.; Gootenberg, J.S.; Hirano, H.; Ran, F.A.; Nakane, T.; Ishitani, R.; Zhang, F.; Nishimasu, H.; Nureki, O. Crystal Structure of the Minimal Cas9 from Campylobacter jejuni Reveals the Molecular Diversity in the CRISPR-Cas9 Systems. Mol. Cell 2017, 65, 1109–1121 e1103. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://github.com/mkmellon5/MCI (accessed on 29 March 2021).
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [PubMed]
- Bin Moon, S.; Lee, J.M.; Kang, J.G.; Lee, N.E.; Ha, D.I.; Kim, D.Y.; Kim, S.H.; Yoo, K.; Kim, D.; Ko, J.H.; et al. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3’-overhang. Nat. Commun. 2018, 9, 3651. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ren, S.; Bai, R.; Xiao, P.; Zhou, Q.; Zhou, Y.; Zhou, Z.; Niu, Y.; Ji, W.; Chen, Y. No off-target mutations in functional genome regions of a CRISPR/Cas9-generated monkey model of muscular dystrophy. J. Biol. Chem. 2018, 293, 11654–11658. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- East-Seletsky, A.; O’Connell, M.R.; Burstein, D.; Knott, G.J.; Doudna, J.A. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol. Cell 2017, 66, 373–383 e373. [Google Scholar] [CrossRef]
- Joung, J.; Ladha, A.; Saito, M.; Kim, N.G.; Woolley, A.E.; Segel, M.; Barretto, R.P.J.; Ranu, A.; Macrae, R.K.; Faure, G.; et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. 2020, 383, 1492–1494. [Google Scholar] [CrossRef]
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149. [Google Scholar] [CrossRef]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876 e812. [Google Scholar] [CrossRef]
- Arizti-Sanz, J.; Freije, C.A.; Stanton, A.C.; Petros, B.A.; Boehm, C.K.; Siddiqui, S.; Shaw, B.M.; Adams, G.; Kosoko-Thoroddsen, T.F.; Kemball, M.E.; et al. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat. Commun. 2020, 11, 5921. [Google Scholar] [CrossRef]
- Ikeda, A.; Fujii, W.; Sugiura, K.; Naito, K. High-fidelity endonuclease variant HypaCas9 facilitates accurate allele-specific gene modification in mouse zygotes. Commun. Biol. 2019, 2, 371. [Google Scholar] [CrossRef]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef]
- Saville, K.M.; Clark, J.; Wilk, A.; Rogers, G.D.; Andrews, J.F.; Koczor, C.A.; Sobol, R.W. NAD(+)-mediated regulation of mammalian base excision repair. DNA Repair 2020, 93, 102930. [Google Scholar] [CrossRef]
- Broeders, M.; Herrero-Hernandez, P.; Ernst, M.P.T.; van der Ploeg, A.T.; Pijnappel, W. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience 2020, 23, 100789. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Sternberg, S.H.; Taylor, D.W.; Staahl, B.T.; Bardales, J.A.; Kornfeld, J.E.; Doudna, J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 2015, 112, 2984–2989. [Google Scholar] [CrossRef] [PubMed]
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760. [Google Scholar] [CrossRef]
- Richter, F.; Fonfara, I.; Bouazza, B.; Schumacher, C.H.; Bratovic, M.; Charpentier, E.; Moglich, A. Engineering of temperature- and light-switchable Cas9 variants. Nucleic Acids Res. 2016, 44, 10003–10014. [Google Scholar] [CrossRef]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef]
- Yang, L.Z.; Wang, Y.; Li, S.Q.; Yao, R.W.; Luan, P.F.; Wu, H.; Carmichael, G.G.; Chen, L.L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997 e987. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Limpitikul, W.B.; Dick, I.E.; Tester, D.J.; Boczek, N.J.; Limphong, P.; Yang, W.; Choi, M.H.; Babich, J.; DiSilvestre, D.; Kanter, R.J.; et al. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ. Res. 2017, 120, 39–48. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; E, P.R.I.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.N.; Chang, Y.H.; Truong, V.A.; Lai, P.L.; Nguyen, T.K.N.; Hu, Y.C. CRISPR technologies for stem cell engineering and regenerative medicine. Biotechnol. Adv. 2019, 37, 107447. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993. [Google Scholar] [CrossRef]
- Mitsunobu, H.; Teramoto, J.; Nishida, K.; Kondo, A. Beyond Native Cas9: Manipulating Genomic Information and Function. Trends Biotechnol. 2017, 35, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Schuster, A.; Erasimus, H.; Fritah, S.; Nazarov, P.V.; van Dyck, E.; Niclou, S.P.; Golebiewska, A. RNAi/CRISPR Screens: From a Pool to a Valid Hit. Trends Biotechnol. 2019, 37, 38–55. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- St Martin, A.; Salamango, D.; Serebrenik, A.; Shaban, N.; Brown, W.L.; Donati, F.; Munagala, U.; Conticello, S.G.; Harris, R.S. A fluorescent reporter for quantification and enrichment of DNA editing by APOBEC-Cas9 or cleavage by Cas9 in living cells. Nucleic Acids Res. 2018, 46, e84. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. DNA mismatch repair in eukaryotes and bacteria. J. Nucleic Acids 2010, 2010, 260512. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, M.; Wei, Y.; Sun, Y.; Sun, Y.; Pan, H.; Yao, N.; Zhong, W.; Li, Y.; Li, W.; et al. Highly efficient base editing in human tripronuclear zygotes. Protein Cell 2017, 8, 772–775. [Google Scholar] [CrossRef]
- Li, G.; Liu, Y.; Zeng, Y.; Li, J.; Wang, L.; Yang, G.; Chen, D.; Shang, X.; Chen, J.; Huang, X.; et al. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein Cell 2017, 8, 776–779. [Google Scholar] [CrossRef]
- Liang, P.; Ding, C.; Sun, H.; Xie, X.; Xu, Y.; Zhang, X.; Sun, Y.; Xiong, Y.; Ma, W.; Liu, Y.; et al. Correction of beta-thalassemia mutant by base editor in human embryos. Protein Cell 2017, 8, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, J.; Li, G.; Huang, S.; Yu, W.; Zhang, Y.; Chen, D.; Chen, J.; Liu, J.; Huang, X. Correction of the Marfan Syndrome Pathogenic FBN1 Mutation by Base Editing in Human Cells and Heterozygous Embryos. Mol. Ther. 2018, 26, 2631–2637. [Google Scholar] [CrossRef]
- Sun, H.; Zhi, S.; Wu, G.; Wu, G.; Cao, T.; Hao, H.; Songyang, Z.; Liang, P.; Huang, J. Cost-effective generation of A-to-G mutant mice by zygote electroporation of adenine base editor ribonucleoproteins. J. Genet. Genom. 2020, 47, 337–340. [Google Scholar] [CrossRef]
- Song, C.Q.; Jiang, T.; Richter, M.; Rhym, L.H.; Koblan, L.W.; Zafra, M.P.; Schatoff, E.M.; Doman, J.L.; Cao, Y.; Dow, L.E.; et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020, 4, 125–130. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Eid, A.; Alshareef, S.; Mahfouz, M.M. CRISPR base editors: Genome editing without double-stranded breaks. Biochem. J. 2018, 475, 1955–1964. [Google Scholar] [CrossRef]
- Yang, B.; Yang, L.; Chen, J. Development and Application of Base Editors. CRISPR J. 2019, 2, 91–104. [Google Scholar] [CrossRef]
- Molla, K.A.; Yang, Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019, 37, 1121–1142. [Google Scholar] [CrossRef]
- Matsoukas, I.G. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front. Genet. 2020, 11, 528. [Google Scholar] [CrossRef]
- Marzec, M.; Hensel, G. Prime Editing: Game Changer for Modifying Plant Genomes. Trends Plant Sci. 2020, 25, 722–724. [Google Scholar] [CrossRef]
- Marzec, M.; Braszewska-Zalewska, A.; Hensel, G. Prime Editing: A New Way for Genome Editing. Trends Cell Biol. 2020, 30, 257–259. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in E. coli and Human Mismatch Repair (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 8490–8501. [Google Scholar] [CrossRef]
- Goellner, E.M. Chromatin remodeling and mismatch repair: Access and excision. DNA Repair 2020, 85, 102733. [Google Scholar] [CrossRef]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed]
- Guarne, A. The functions of MutL in mismatch repair: The power of multitasking. Prog. Mol. Biol. Transl. Sci. 2012, 110, 41–70. [Google Scholar] [CrossRef]
- Yang, W. Human MutLalpha: The jack of all trades in MMR is also an endonuclease. DNA Repair 2007, 6, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Liu, L.; Gerson, S.L. Mice defective in the DNA mismatch gene PMS2 are hypersensitive to MNU induced thymic lymphoma and are partially protected by transgenic expression of human MGMT. Oncogene 1999, 18, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, Y.; Tsuzuki, T.; Shiraishi, A.; Kawate, H.; Sekiguchi, M. Alkylation-induced apoptosis of embryonic stem cells in which the gene for DNA-repair, methyltransferase, had been disrupted by gene targeting. Carcinogenesis 1997, 18, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. 1997, 381, 227–241. [Google Scholar] [CrossRef]
- Yan, T.; Berry, S.E.; Desai, A.B.; Kinsella, T.J. DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 2003, 9, 2327–2334. [Google Scholar] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Takeishi, Y.; Fujikane, R.; Rikitake, M.; Obayashi, Y.; Sekiguchi, M.; Hidaka, M. SMARCAD1-mediated recruitment of the DNA mismatch repair protein MutLalpha to MutSalpha on damaged chromatin induces apoptosis in human cells. J. Biol. Chem. 2020, 295, 1056–1065. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef]
- Hung, S.; Saiakhova, A.; Faber, Z.J.; Bartels, C.F.; Neu, D.; Bayles, I.; Ojo, E.; Hong, E.S.; Pontius, W.D.; Morton, A.R.; et al. Mismatch repair-signature mutations activate gene enhancers across human colorectal cancer epigenomes. Elife 2019, 8. [Google Scholar] [CrossRef]
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019, 568, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Lieb, S.; Blaha-Ostermann, S.; Kamper, E.; Rippka, J.; Schwarz, C.; Ehrenhofer-Wolfer, K.; Schlattl, A.; Wernitznig, A.; Lipp, J.J.; Nagasaka, K.; et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895-2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Lynch, H.T.; Lanspa, S.; Shaw, T.; Casey, M.J.; Rendell, M.; Stacey, M.; Townley, T.; Snyder, C.; Hitchins, M.; Bailey-Wilson, J. Phenotypic and genotypic heterogeneity of Lynch syndrome: A complex diagnostic challenge. Fam. Cancer 2018, 17, 403–414. [Google Scholar] [CrossRef]
- Boland, C.R.; Lynch, H.T. The history of Lynch syndrome. Fam. Cancer 2013, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Hampel, H. Recent Advances in Lynch Syndrome: Diagnosis, Treatment, and Cancer Prevention. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 101–109. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Biller, L.H.; Syngal, S.; Yurgelun, M.B. Recent advances in Lynch syndrome. Fam. Cancer 2019, 18, 211–219. [Google Scholar] [CrossRef]
- Golubicki, M.; Bonjoch, L.; Acuna-Ochoa, J.G.; Diaz-Gay, M.; Munoz, J.; Cuatrecasas, M.; Ocana, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Blount, J.; Prakash, A. The changing landscape of Lynch syndrome due to PMS2 mutations. Clin. Genet. 2018, 94, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Heinen, C.D.; Juel Rasmussen, L. Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hered Cancer Clin. Pract. 2012, 10, 9. [Google Scholar] [CrossRef]
- Solomon, I.; Harrington, E.; Hooker, G.; Erby, L.; Axilbund, J.; Hampel, H.; Semotiuk, K.; Blanco, A.; Klein, W.M.P.; Giardiello, F.; et al. Lynch Syndrome Limbo: Patient Understanding of Variants of Uncertain Significance. J. Genet. Couns. 2017, 26, 866–877. [Google Scholar] [CrossRef]
- D’Arcy, B.M.; Blount, J.; Prakash, A. Biochemical and structural characterization of two variants of uncertain significance in the PMS2 gene. Hum. Mutat. 2019, 40, 458–471. [Google Scholar] [CrossRef]
- Drost, M.; Tiersma, Y.; Thompson, B.A.; Frederiksen, J.H.; Keijzers, G.; Glubb, D.; Kathe, S.; Osinga, J.; Westers, H.; Pappas, L.; et al. A functional assay-based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet. Med. 2019, 21, 1486–1496. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.D.; Liberti, S.E.; Lutzen, A.; Drost, M.; Bernstein, I.; Nilbert, M.; Dominguez, M.; Nystrom, M.; Hansen, T.V.; Christoffersen, J.W.; et al. Functional characterization of MLH1 missense variants identified in Lynch syndrome patients. Hum. Mutat. 2012, 33, 1647–1655. [Google Scholar] [CrossRef]
- Drost, M.; Koppejan, H.; de Wind, N. Inactivation of DNA mismatch repair by variants of uncertain significance in the PMS2 gene. Hum. Mutat. 2013, 34, 1477–1480. [Google Scholar] [CrossRef][Green Version]
- Drost, M.; Tiersma, Y.; Glubb, D.; Kathe, S.; van Hees, S.; Calleja, F.; Zonneveld, J.B.M.; Boucher, K.M.; Ramlal, R.P.E.; Thompson, B.A.; et al. Two integrated and highly predictive functional analysis-based procedures for the classification of MSH6 variants in Lynch syndrome. Genet. Med. 2020, 22, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Drost, M.; Zonneveld, J.B.; van Hees, S.; Rasmussen, L.J.; Hofstra, R.M.; de Wind, N. A rapid and cell-free assay to test the activity of lynch syndrome-associated MSH2 and MSH6 missense variants. Hum. Mutat. 2012, 33, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Mishra, A.; Ferreira, V.D.; Hu, C.; Omerza, G.; Kelly, K.; Hesse, A.; Reddi, H.V.; Grady, J.P.; Heinen, C.D. Functional interrogation of Lynch syndrome-associated MSH2 missense variants via CRISPR-Cas9 gene editing in human embryonic stem cells. Hum. Mutat. 2019, 40, 2044–2056. [Google Scholar] [CrossRef]
- Hayashida, G.; Shioi, S.; Hidaka, K.; Fujikane, R.; Hidaka, M.; Tsurimoto, T.; Tsuzuki, T.; Oda, S.; Nakatsu, Y. Differential genomic destabilisation in human cells with pathogenic MSH2 mutations introduced by genome editing. Exp. Cell Res. 2019, 377, 24–35. [Google Scholar] [CrossRef]
- Lau, C.H.; Tin, C.; Suh, Y. CRISPR-based strategies for targeted transgene knock-in and gene correction. Fac. Rev. 2020, 9, 20. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Meira, L.B.; Burgis, N.E.; Samson, L.D. Base excision repair. Adv. Exp. Med. Biol. 2005, 570, 125–173. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef]
- Beard, W.A.; Wilson, S.H. DNA polymerase beta and other gap-filling enzymes in mammalian base excision repair. Enzymes 2019, 45, 1–26. [Google Scholar] [CrossRef]
- Elsakrmy, N.; Zhang-Akiyama, Q.M.; Ramotar, D. The Base Excision Repair Pathway in the Nematode Caenorhabditis elegans. Front. Cell Dev. Biol. 2020, 8, 598860. [Google Scholar] [CrossRef]
- Stratigopoulou, M.; van Dam, T.P.; Guikema, J.E.J. Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences. Front. Immunol. 2020, 11, 1084. [Google Scholar] [CrossRef]
- Thompson, P.S.; Cortez, D. New insights into abasic site repair and tolerance. DNA Repair 2020, 90, 102866. [Google Scholar] [CrossRef]
- Kurthkoti, K.; Kumar, P.; Sang, P.B.; Varshney, U. Base excision repair pathways of bacteria: New promise for an old problem. Future Med. Chem. 2020, 12, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.H.; Kunkel, T.A. Passing the baton in base excision repair. Nat. Struct. Biol. 2000, 7, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.B.; Goellner, E.M.; Wang, X.H.; Trivedi, R.N.; St Croix, C.M.; Jelezcova, E.; Svilar, D.; Brown, A.R.; Sobol, R.W. Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol. Cancer Res. 2010, 8, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ramakodi, M.P.; Devarajan, K.; Blackman, E.; Gibbs, D.; Luce, D.; Deloumeaux, J.; Duflo, S.; Liu, J.C.; Mehra, R.; Kulathinal, R.J.; et al. Integrative genomic analysis identifies ancestry-related expression quantitative trait loci on DNA polymerase beta and supports the association of genetic ancestry with survival disparities in head and neck squamous cell carcinoma. Cancer 2017, 123, 849–860. [Google Scholar] [CrossRef]
- Mahjabeen, I.; Baig, R.M.; Sabir, M.; Kayani, M.A. Genetic and expressional variations of APEX1 are associated with increased risk of head and neck cancer. Mutagenesis 2013, 28, 213–218. [Google Scholar] [CrossRef]
- Gao, D.; Hu, J.; Zhang, X.; Gao, C.; Hong, J. Effect of hOGG1 over-expression on cisplatin resistance in esophageal squamous carcinoma cells. Cancer Biother. Radiopharm. 2013, 28, 433–440. [Google Scholar] [CrossRef]
- Mahjabeen, I.; Ali, K.; Zhou, X.; Kayani, M.A. Deregulation of base excision repair gene expression and enhanced proliferation in head and neck squamous cell carcinoma. Tumour Biol. 2014, 35, 5971–5983. [Google Scholar] [CrossRef]
- Mao, P.; Wyrick, J.J. Organization of DNA damage, excision repair, and mutagenesis in chromatin: A genomic perspective. DNA Repair 2019, 81, 102645. [Google Scholar] [CrossRef]
- Li, C.; Delaney, S. Challenges for base excision repair enzymes: Acquiring access to damaged DNA in chromatin. Enzymes 2019, 45, 27–57. [Google Scholar] [CrossRef]
- Meas, R.; Wyrick, J.J.; Smerdon, M.J. Nucleosomes Regulate Base Excision Repair in Chromatin. Mutat. Res. 2019, 780, 29–36. [Google Scholar] [CrossRef]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Bamps, M.; Glorieux, M.; Zhao, P.; Sablina, A.; Nuyts, S. Radiosensitization approaches for HPV-positive and HPV-negative head and neck squamous carcinomas. Int. J. Cancer 2020, 146, 1075–1085. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. Increased Specificity and Efficiency of Base Excision Repair through Complex Formation. In DNA Damage Recognition; Siede, W., Doetsch, P.W., Kow, Y.W., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2005; pp. 33–64. [Google Scholar]
- Verma, P.; Zhou, Y.; Cao, Z.; Deraska, P.V.; Deb, M.; Arai, E.; Li, W.; Shao, Y.; Puentes, L.; Li, Y.; et al. ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat. Cell Biol. 2021, 23, 160–171. [Google Scholar] [CrossRef]
- Juhasz, S.; Smith, R.; Schauer, T.; Spekhardt, D.; Mamar, H.; Zentout, S.; Chapuis, C.; Huet, S.; Timinszky, G. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Borel, V.; Segura-Bayona, S.; Takaki, T.; Ruis, P.; Bellelli, R.; Lehmann, L.C.; Sommerova, L.; Vancevska, A.; Tomas-Loba, A.; et al. Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol. Cell 2021, 81, 767–783 e711. [Google Scholar] [CrossRef] [PubMed]
- Blessing, C.; Mandemaker, I.K.; Gonzalez-Leal, C.; Preisser, J.; Schomburg, A.; Ladurner, A.G. The Oncogenic Helicase ALC1 Regulates PARP Inhibitor Potency by Trapping PARP2 at DNA Breaks. Mol. Cell 2020, 80, 862–875 e866. [Google Scholar] [CrossRef]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef]
- Gottschalk, A.J.; Trivedi, R.D.; Conaway, J.W.; Conaway, R.C. Activation of the SNF2 family ATPase ALC1 by poly(ADP-ribose) in a stable ALC1.PARP1.nucleosome intermediate. J. Biol. Chem. 2012, 287, 43527–43532. [Google Scholar] [CrossRef]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox. Signal 2011, 14, 2491–2507. [Google Scholar] [CrossRef]
- Fromme, J.C.; Banerjee, A.; Verdine, G.L. DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 2004, 14, 43–49. [Google Scholar] [CrossRef]
- Kunkel, T.A. Celebrating DNA’s Repair Crew. Cell 2015, 163, 1301–1303. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat. Res. 2000, 462, 129–135. [Google Scholar] [CrossRef]
- Roldan-Arjona, T.; Wei, Y.F.; Carter, K.C.; Klungland, A.; Anselmino, C.; Wang, R.P.; Augustus, M.; Lindahl, T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 8016–8020. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef]
- Imai, K.; Sarker, A.H.; Akiyama, K.; Ikeda, S.; Yao, M.; Tsutsui, K.; Shohmori, T.; Seki, S. Genomic structure and sequence of a human homologue (NTHL1/NTH1) of Escherichia coli endonuclease III with those of the adjacent parts of TSC2 and SLC9A3R2 genes. Gene 1998, 222, 287–295. [Google Scholar] [CrossRef]
- Aspinwall, R.; Rothwell, D.G.; Roldan-Arjona, T.; Anselmino, C.; Ward, C.J.; Cheadle, J.P.; Sampson, J.R.; Lindahl, T.; Harris, P.C.; Hickson, I.D. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl. Acad. Sci. USA 1997, 94, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Karahalil, B.; Senturker, S.; Buckley, T.J.; Roldan-Arjona, T. Excision of products of oxidative DNA base damage by human NTH1 protein. Biochemistry 1999, 38, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Zhang, Q.M.; Takao, M.; Yasui, A.; Yonei, S. Escherichia coli Nth and human hNTH1 DNA glycosylases are involved in removal of 8-oxoguanine from 8-oxoguanine/guanine mispairs in DNA. Nucleic Acids Res. 2001, 29, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Hoss, M.; Gunz, D.; Constantinou, A.; Clarkson, S.G.; Doetsch, P.W.; Bolton, P.H.; Wood, R.D.; Lindahl, T. Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell 1999, 3, 33–42. [Google Scholar] [CrossRef]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef]
- Bjoras, M.; Luna, L.; Johnsen, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef]
- Ichinoe, A.; Behmanesh, M.; Tominaga, Y.; Ushijima, Y.; Hirano, S.; Sakai, Y.; Tsuchimoto, D.; Sakumi, K.; Wake, N.; Nakabeppu, Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004, 32, 477–487. [Google Scholar] [CrossRef]
- Hirano, S.; Tominaga, Y.; Ichinoe, A.; Ushijima, Y.; Tsuchimoto, D.; Honda-Ohnishi, Y.; Ohtsubo, T.; Sakumi, K.; Nakabeppu, Y. Mutator phenotype of MUTYH-null mouse embryonic stem cells. J. Biol. Chem. 2003, 278, 38121–38124. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Minko, I.G.; Moellmer, S.A.; Cammann, S.K.; Lloyd, R.S.; McCullough, A.K. Enhanced cytarabine-induced killing in OGG1-deficient acute myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Baquero, J.M.; Benitez-Buelga, C.; Rajagopal, V.; Zhenjun, Z.; Torres-Ruiz, R.; Muller, S.; Hanna, B.M.F.; Loseva, O.; Wallner, O.; Michel, M.; et al. Small molecule inhibitor of OGG1 blocks oxidative DNA damage repair at telomeres and potentiates methotrexate anticancer effects. Sci. Rep. 2021, 11, 3490. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130 e116. [Google Scholar] [CrossRef]
- Donley, N.; Jaruga, P.; Coskun, E.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Small Molecule Inhibitors of 8-Oxoguanine DNA Glycosylase-1 (OGG1). ACS Chem. Biol. 2015, 10, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Visnes, T.; Cazares-Korner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef]
- Spallotta, F.; Cencioni, C.; Atlante, S.; Garella, D.; Cocco, M.; Mori, M.; Mastrocola, R.; Kuenne, C.; Guenther, S.; Nanni, S.; et al. Stable Oxidative Cytosine Modifications Accumulate in Cardiac Mesenchymal Cells From Type2 Diabetes Patients: Rescue by alpha-Ketoglutarate and TET-TDG Functional Reactivation. Circ. Res. 2018, 122, 31–46. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Liu, Y.; Jiang, L.; Yamaguchi, S.; Zhang, Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014, 28, 2103–2119. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.B.; Fu, C.; Prutsch, N.; Wang, M.; He, S.; Look, A.T. Synthetic lethal targeting of TET2-mutant hematopoietic stem and progenitor cells (HSPCs) with TOP1-targeted drugs and PARP1 inhibitors. Leukemia 2020, 34, 2992–3006. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P.; Tricarico, R.; Bhattacharjee, V.; Cosentino, L.; Kadariya, Y.; Jelinek, J.; Nicolas, E.; Einarson, M.; Beeharry, N.; Devarajan, K.; et al. Thymine DNA glycosylase as a novel target for melanoma. Oncogene 2019, 38, 3710–3728. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Miao, G.G.; Curran, T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA 1994, 91, 23–27. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef]
- Lindahl, T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog. Nucleic Acid. Res. Mol. Biol. 1979, 22, 135–192. [Google Scholar] [CrossRef]
- McNeill, D.R.; Whitaker, A.M.; Stark, W.J.; Illuzzi, J.L.; McKinnon, P.J.; Freudenthal, B.D.; Wilson, D.M., 3rd. Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 2020, 35, 27–38. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Freudenthal, B.D. APE1: A skilled nucleic acid surgeon. DNA Repair 2018, 71, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rodriguez, Y.; Ross, R.L.; Zhao, R.; Watts, J.A.; Grunseich, C.; Bruzel, A.; Li, D.; Burdick, J.T.; Prasad, R.; et al. RNA abasic sites in yeast and human cells. Proc. Natl. Acad. Sci. USA 2020, 117, 20689–20695. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Svilar, D.; McClellan, S.; Kim, J.H.; Ahn, E.E.; Vens, C.; Wilson, D.M., 3rd; Sobol, R.W. DNA Repair Molecular Beacon assay: A platform for real-time functional analysis of cellular DNA repair capacity. Oncotarget 2018, 9, 31719–31743. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Song, F.; Gao, Q.; Lu, Z.; Yuan, Y.; Li, X.; Chen, L.; Jia, C.; Yang, R.; Yang, J.; et al. The APEX1/miRNA-27a-5p axis plays key roles in progression, metastasis and targeted chemotherapy of gastric cancer. Int. J. Pharm. 2021, 599, 120446. [Google Scholar] [CrossRef]
- Chen, T.; Liu, C.; Lu, H.; Yin, M.; Shao, C.; Hu, X.; Wu, J.; Wang, Y. The expression of APE1 in triple-negative breast cancer and its effect on drug sensitivity of olaparib. Tumour. Biol. 2017, 39, 1010428317713390. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef]
- Alvarez-Quilon, A.; Wojtaszek, J.L.; Mathieu, M.C.; Patel, T.; Appel, C.D.; Hustedt, N.; Rossi, S.E.; Wallace, B.D.; Setiaputra, D.; Adam, S.; et al. Endogenous DNA 3’ Blocks Are Vulnerabilities for BRCA1 and BRCA2 Deficiency and Are Reversed by the APE2 Nuclease. Mol. Cell 2020, 78, 1152–1165 e1158. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899 e886. [Google Scholar] [CrossRef]
- Wilk, A.; Hayat, F.; Cunningham, R.; Li, J.; Garavaglia, S.; Zamani, L.; Ferraris, D.M.; Sykora, P.; Andrews, J.; Clark, J.; et al. Extracellular NAD(+) enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 2020, 10, 651. [Google Scholar] [CrossRef]
- Aberle, L.; Kruger, A.; Reber, J.M.; Lippmann, M.; Hufnagel, M.; Schmalz, M.; Trussina, I.; Schlesiger, S.; Zubel, T.; Schutz, K.; et al. PARP1 catalytic variants reveal branching and chain length-specific functions of poly(ADP-ribose) in cellular physiology and stress response. Nucleic Acids Res. 2020, 48, 10015–10033. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Koczor, C.A.; Saville, K.M.; Andrews, J.F.; Clark, J.; Fang, Q.; Li, J.; Makarov, M.V.; Migaud, M.; Sobol, R.W. Temporal dynamics of base excision / single-strand break repair protein complex assembly and disassembly are modulated by the PARP1/NAD+/SIRT6 axis. Cell Rep. 2021. submitted, under revision. [Google Scholar]
- Weixler, L.; Scharinger, K.; Momoh, J.; Luscher, B.; Feijs, K.L.H.; Zaja, R. ADP-ribosylation of RNA and DNA: From in vitro characterization to in vivo function. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Liu, F.; Tu, Y.; Cai, Z.; Luo, Y.; Wu, X. PARP1-RNA interaction analysis: PARP1 regulates the expression of extracellular matrix-related genes in HK-2 renal proximal tubular epithelial cells. FEBS Lett. 2021. [Google Scholar] [CrossRef] [PubMed]
- Eleazer, R.; Fondufe-Mittendorf, Y.N. The multifaceted role of PARP1 in RNA biogenesis. Wiley Interdiscip. Rev. RNA 2021, 12, e1617. [Google Scholar] [CrossRef]
- Kim, D.S.; Challa, S.; Jones, A.; Kraus, W.L. PARPs and ADP-ribosylation in RNA biology: From RNA expression and processing to protein translation and proteostasis. Genes Dev. 2020, 34, 302–320. [Google Scholar] [CrossRef]
- Kim, D.S.; Camacho, C.V.; Nagari, A.; Malladi, V.S.; Challa, S.; Kraus, W.L. Activation of PARP-1 by snoRNAs Controls Ribosome Biogenesis and Cell Growth via the RNA Helicase DDX21. Mol. Cell 2019, 75, 1270–1285 e1214. [Google Scholar] [CrossRef]
- Ke, Y.; Zhang, J.; Lv, X.; Zeng, X.; Ba, X. Novel insights into PARPs in gene expression: Regulation of RNA metabolism. Cell Mol. Life Sci. 2019, 76, 3283–3299. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Mathbout, L.F.; Fondufe-Mittendorf, Y.N. PARP1 is a versatile factor in the regulation of mRNA stability and decay. Sci. Rep. 2019, 9, 3722. [Google Scholar] [CrossRef]
- Huang, D.; Kim, D.S.; Kraus, W.L. Specific Binding of snoRNAs to PARP-1 Promotes NAD(+)-Dependent Catalytic Activation. Biochemistry 2020, 59, 1559–1564. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Parsels, L.A.; Engelke, C.G.; Parsels, J.; Flanagan, S.A.; Zhang, Q.; Tanska, D.; Wahl, D.R.; Canman, C.E.; Lawrence, T.S.; Morgan, M.A. Combinatorial Efficacy of Olaparib with Radiation and ATR Inhibitor Requires PARP1 Protein in Homologous Recombination-Proficient Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 263–273. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Gajewski, S.; Hartwig, A. PARP1 Is Required for ATM-Mediated p53 Activation and p53-Mediated Gene Expression after Ionizing Radiation. Chem. Res. Toxicol. 2020, 33, 1933–1940. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef]
- Fang, Q.; Andrews, J.; Sharma, N.; Wilk, A.; Clark, J.; Slyskova, J.; Koczor, C.A.; Lans, H.; Prakash, A.; Sobol, R.W. Stability and sub-cellular localization of DNA polymerase beta is regulated by interactions with NQO1 and XRCC1 in response to oxidative stress. Nucleic Acids Res. 2019, 47, 6269–6286. [Google Scholar] [CrossRef]
- Li, J.; Saville, K.M.; Zeng, X.; McClellan, S.; Angajala, A.; Beiser, A.; Andrews, J.F.; Koczor, C.A.; Clark, J.; Makarov, M.V.; et al. NAD+ bioavailability mediates PARG inhibition-induced replication arrest in glioma stem cells. NAR Cancer 2021. submitted (Under Review). [Google Scholar]
- Ali, R.; Alabdullah, M.; Alblihy, A.; Miligy, I.; Mesquita, K.A.; Chan, S.Y.; Moseley, P.; Rakha, E.A.; Madhusudan, S. PARP1 blockade is synthetically lethal in XRCC1 deficient sporadic epithelial ovarian cancers. Cancer Lett. 2020, 469, 124–133. [Google Scholar] [CrossRef]
- Eckelmann, B.J.; Bacolla, A.; Wang, H.; Ye, Z.; Guerrero, E.N.; Jiang, W.; El-Zein, R.; Hegde, M.L.; Tomkinson, A.E.; Tainer, J.A.; et al. XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer 2020, 2, zcaa013. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331 e313. [Google Scholar] [CrossRef]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Goetze, R.W.; Kim, D.H.; Schinazi, R.F.; Kim, B. A CRISPR/Cas9 approach reveals that the polymerase activity of DNA polymerase beta is dispensable for HIV-1 infection in dividing and nondividing cells. J. Biol. Chem. 2017, 292, 14016–14025. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.W. DNA polymerase beta null mouse embryonic fibroblasts harbor a homozygous null mutation in DNA polymerase iota. DNA Repair 2007, 6, 3–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bebenek, K.; Tissier, A.; Frank, E.G.; McDonald, J.P.; Prasad, R.; Wilson, S.H.; Woodgate, R.; Kunkel, T.A. 5’-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science 2001, 291, 2156–2159. [Google Scholar] [CrossRef] [PubMed]
- Miropolskaya, N.; Petushkov, I.; Kulbachinskiy, A.; Makarova, A.V. Identification of amino acid residues involved in the dRP-lyase activity of human Pol iota. Sci. Rep. 2017, 7, 10194. [Google Scholar] [CrossRef]
- Prasad, R.; Bebenek, K.; Hou, E.; Shock, D.D.; Beard, W.A.; Woodgate, R.; Kunkel, T.A.; Wilson, S.H. Localization of the deoxyribose phosphate lyase active site in human DNA polymerase iota by controlled proteolysis. J. Biol. Chem. 2003, 278, 29649–29654. [Google Scholar] [CrossRef]
- Petta, T.B.; Nakajima, S.; Zlatanou, A.; Despras, E.; Couve-Privat, S.; Ishchenko, A.; Sarasin, A.; Yasui, A.; Kannouche, P. Human DNA polymerase iota protects cells against oxidative stress. EMBO J. 2008, 27, 2883–2895. [Google Scholar] [CrossRef] [PubMed]
- Starostenko, L.V.; Rechkunova, N.I.; Lebedeva, N.A.; Lomzov, A.A.; Koval, V.V.; Lavrik, O.I. Processing of the abasic sites clustered with the benzo[a]pyrene adducts by the base excision repair enzymes. DNA Repair 2017, 50, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Belousova, E.A.; Lavrik, O.I. Repair of Clustered Damage and DNA Polymerase Iota. Biochemistry (Mosc) 2015, 80, 1010–1018. [Google Scholar] [CrossRef]
- Haracska, L.; Prakash, L.; Prakash, S. A mechanism for the exclusion of low-fidelity human Y-family DNA polymerases from base excision repair. Genes Dev. 2003, 17, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Yeom, M.; Hong, J.K.; Kim, J.K.; Guengerich, F.P.; Choi, J.Y. Three Human Pol iota Variants with Impaired Polymerase Activity Fail to Rescue H2O2 Sensitivity in POLI-Deficient Cells. Chem. Res. Toxicol. 2020, 33, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Duan, J.; Shu, S.; Wang, X.; Gao, L.; Guo, J.; Zhang, Y. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining. Proc. Natl. Acad. Sci. USA 2016, 113, 1256–1260. [Google Scholar] [CrossRef]
- Fouquerel, E.; Sobol, R.W. ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair 2014, 23, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.W.; Li, F.; Kruger, A.; Siddeek, B.; Greulich, K.O.; Popp, O.; Meisezahl, C.; et al. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 2013, 4, 2993. [Google Scholar] [CrossRef]
- Bu, X.; Kato, J.; Moss, J. Emerging roles of ADP-ribosyl-acceptor hydrolases (ARHs) in tumorigenesis and cell death pathways. Biochem. Pharmacol. 2019, 167, 44–49. [Google Scholar] [CrossRef]
- Munnur, D.; Ahel, I. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 2017, 284, 4002–4016. [Google Scholar] [CrossRef] [PubMed]
- Perina, D.; Mikoc, A.; Ahel, J.; Cetkovic, H.; Zaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair 2014, 23, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Blessing, C.; Ladurner, A.G. Tickling PARPs into serine action. Nat. Struct. Mol. Biol. 2020, 27, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Prokhorova, E.; Krejcikova, K.; Cihlarova, Z.; Kalasova, I.; Kubovciak, J.; Sachova, J.; Hailstone, R.; Brazina, J.; Ghosh, S.; et al. Pathogenic ARH3 mutations result in ADP-ribose chromatin scars during DNA strand break repair. Nat. Commun. 2020, 11, 3391. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Single Ascending Dose Study in Participants with LCA10. Available online: https://clinicaltrials.gov/ct2/show/NCT03872479 (accessed on 29 May 2021).
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.M.; Sieving, P.A.; Green, D.G. Effect on grating identification of sampling with degenerate arrays. J. Opt. Soc. Am. A 1992, 9, 472–477. [Google Scholar] [CrossRef]
- Geller, A.M.; Sieving, P.A. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vision Res. 1993, 33, 1509–1524. [Google Scholar] [CrossRef]
- Safety and Efficacy of CRISPR/Cas9 mRNA Instantaneous Gene Editing Therapy to Treat Refractory Viral Keratitis. Available online: https://clinicaltrials.gov/ct2/show/NCT04560790 (accessed on 29 May 2021).
- Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NTLA-2001 in Patients With Hereditary Transthyretin Amyloidosis With Polyneuropathy (ATTRv-PN). Available online: https://clinicaltrials.gov/ct2/show/NCT04601051 (accessed on 29 May 2021).
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Plante-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet. J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Kristy Wood, M.P.; Seitzer, J.; Gardner, N.; Alexander, S.; DiMezzo, T.; Amaral, A.; Soukamneuth, S.; Kanjolia, A.; Parmar, R.; Rodhe, E.; et al. Development of NTLA-2001, a CRISPR/Cas9 Genome Editing Therapeutic for the Treatment of ATTR. Available online: https://www.intelliatx.com/wp-content/uploads/2017/05/development-of-ntla-2001.pdf (accessed on 29 May 2021).
- Cherry, J.M. The Saccharomyces Genome Database: Exploring Biochemical Pathways and Mutant Phenotypes. Cold Spring Harb. Protoc. 2015, 2015, pdb.prot088898. [Google Scholar] [CrossRef]
- Carlson, M. The awesome power of yeast biochemical genomics. Trends Genet. 2000, 16, 49–51. [Google Scholar] [CrossRef]
- Mello, C.C.; Conte, D., Jr. Revealing the world of RNA interference. Nature 2004, 431, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Anderson, D.G. CRISPR-Cas: A tool for cancer research and therapeutics. Nat. Rev. Clin. Oncol. 2019, 16, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Cring, M.R.; Sheffield, V.C. Gene therapy and gene correction: Targets, progress, and challenges for treating human diseases. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Chennakesavulu, K.; Singh, H.; Trivedi, P.K.; Jain, M.; Yadav, S.R. State-of-the-Art in CRISPR Technology and Engineering Drought, Salinity, and Thermo-tolerant crop plants. Plant Cell Rep. 2021. [Google Scholar] [CrossRef]
- Khwatenge, C.N.; Nahashon, S.N. Recent Advances in the Application of CRISPR/Cas9 Gene Editing System in Poultry Species. Front. Genet. 2021, 12, 627714. [Google Scholar] [CrossRef]
- Castells-Roca, L.; Tejero, E.; Rodriguez-Santiago, B.; Surralles, J. CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer. Cancers 2021, 13, 1591. [Google Scholar] [CrossRef]




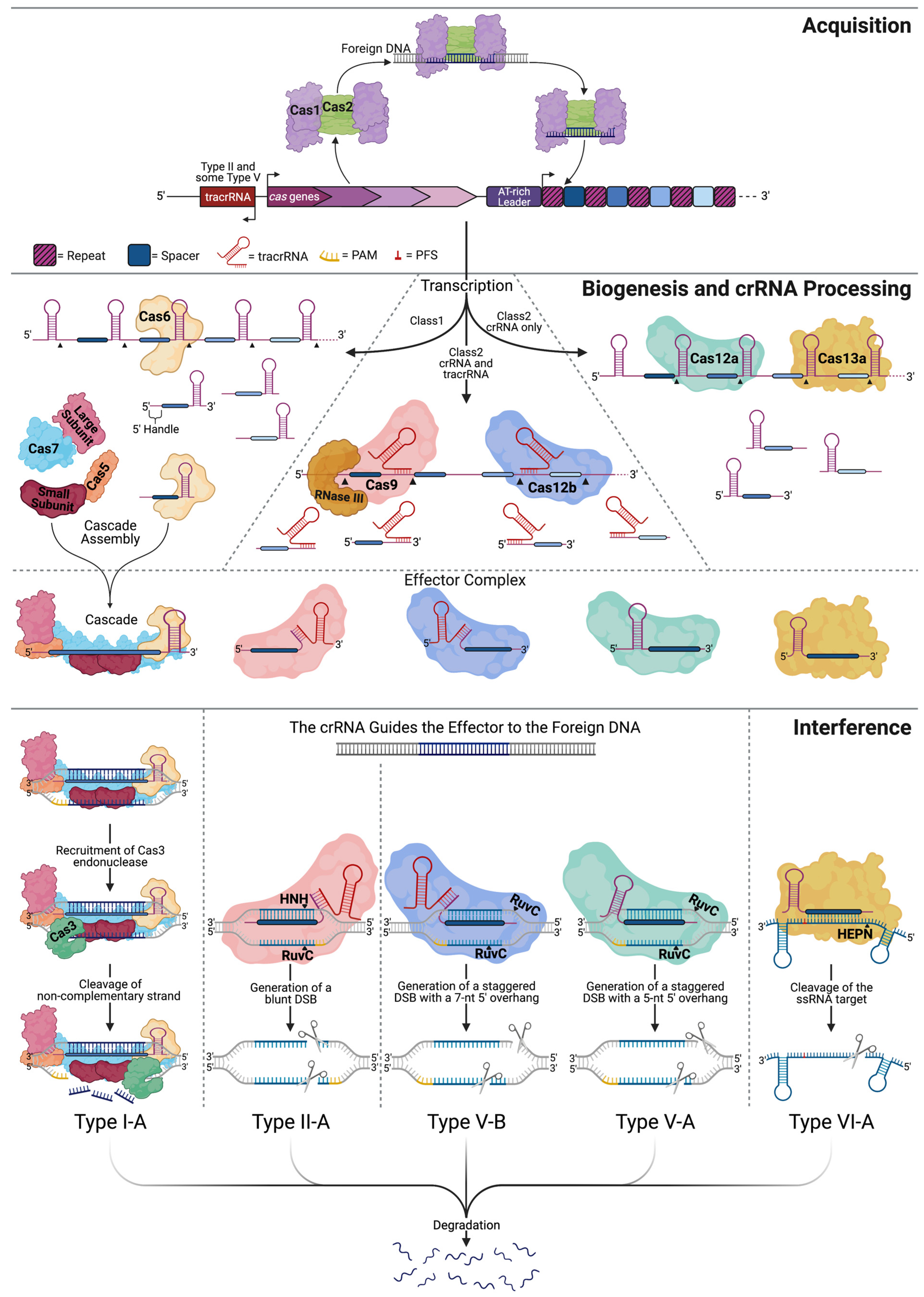{kind=link}
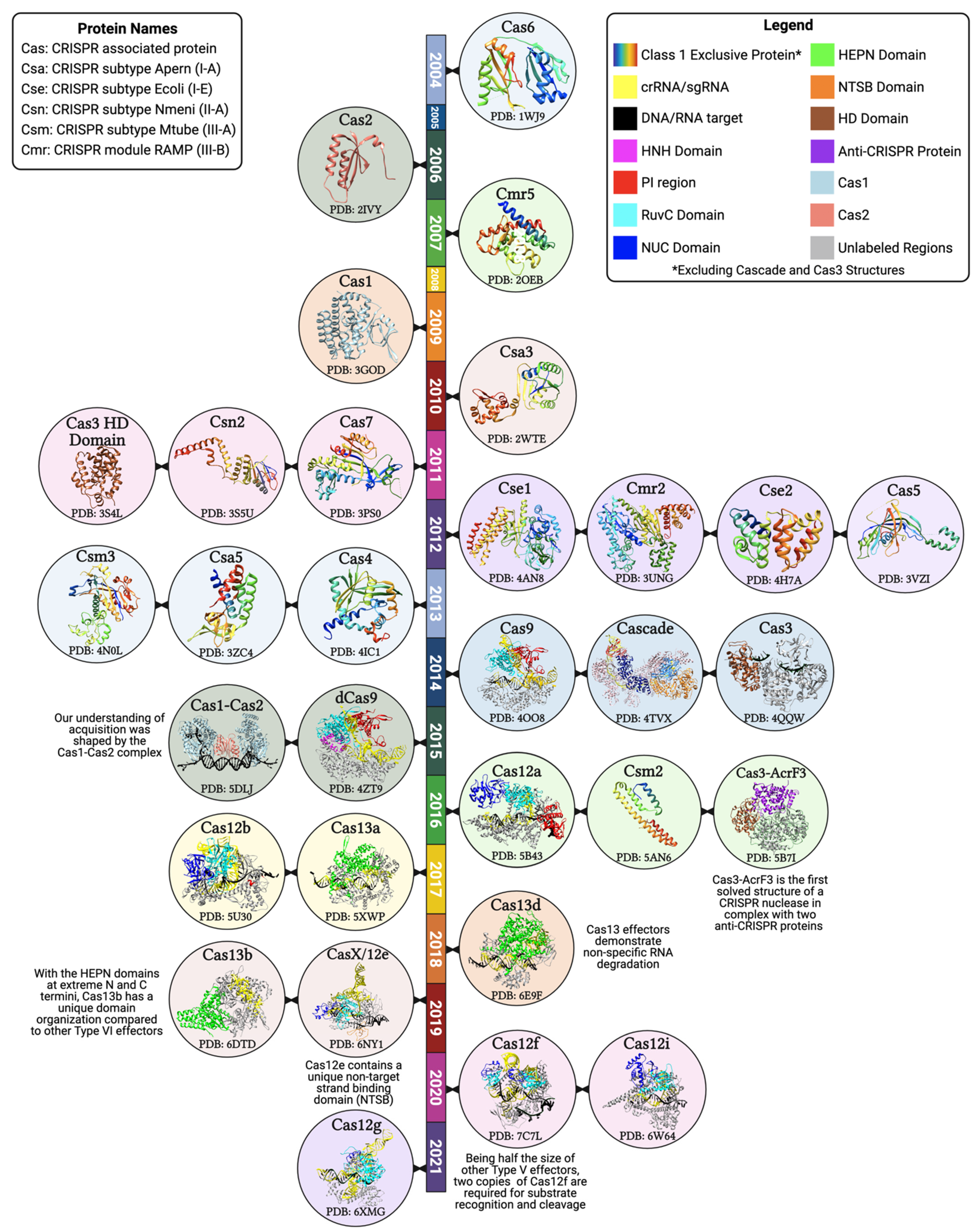{kind=link}
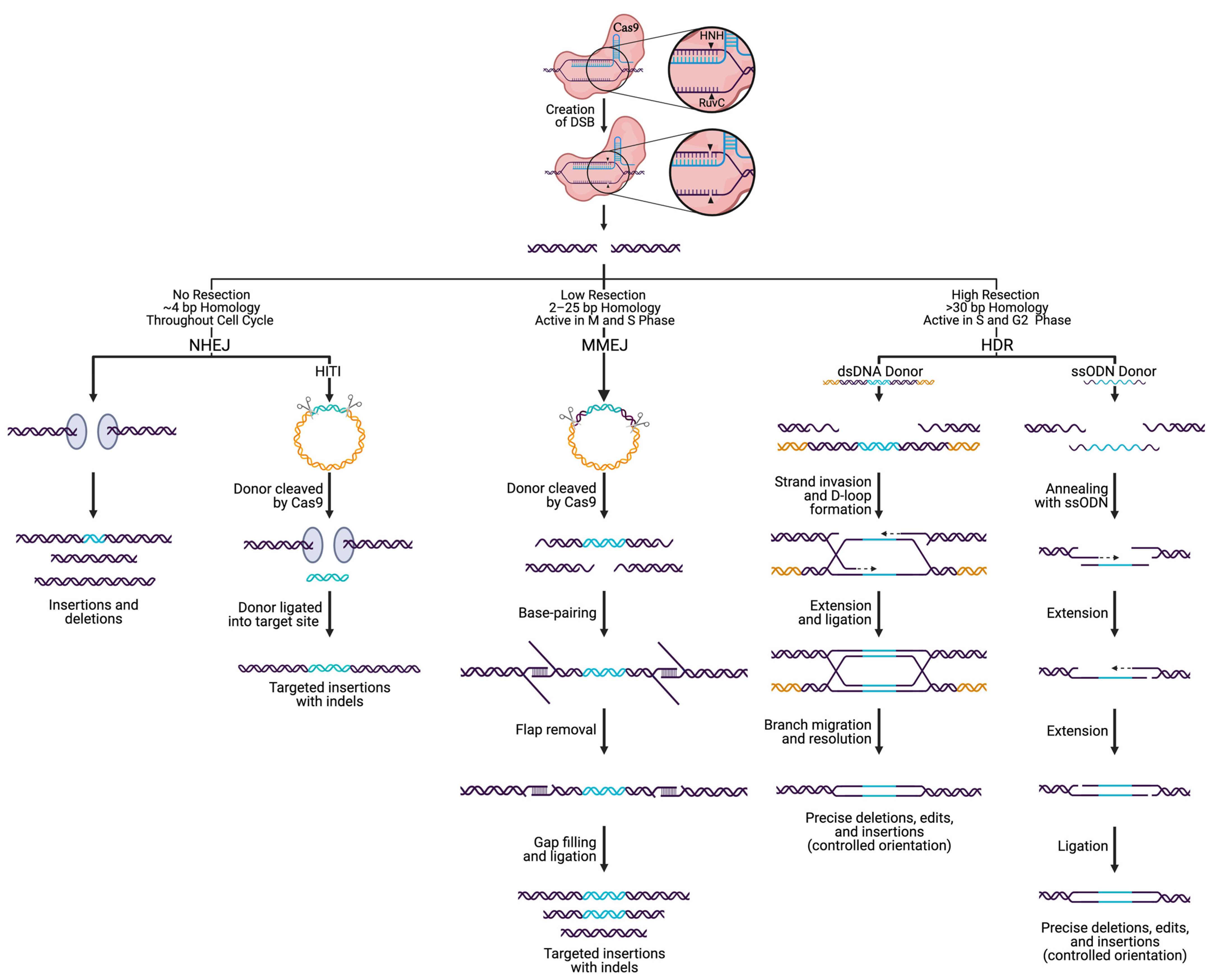{kind=link}
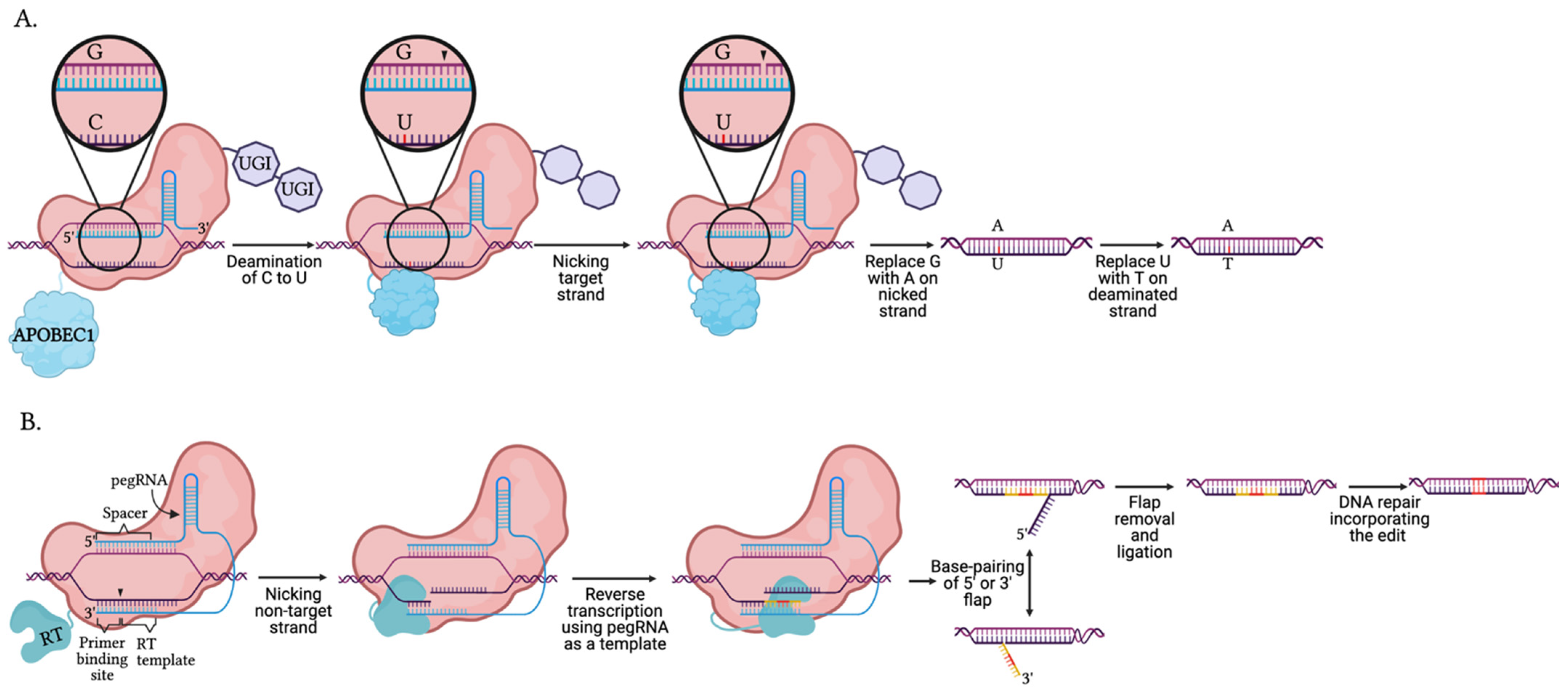{kind=link}
| Cas Ortholog | Size (aa) | Origin | Engineered Variant | Mutation | Target Substrate | Trans-Cleavage | PAM/PFS 5′-3′ | PAM Frequency Compared to spCas9 1 | Feature | Refs |
|---|---|---|---|---|---|---|---|---|---|---|
| SpCas9 | 1368 | Streptococcus pyogenes | - | - | dsDNA | - | NGG | 1.00 | Most commonly used | [79,80] |
| nCas9 | D10A or H840A | dsDNA | - | NGG | 1.00 | Only one active nuclease domain | [26] | |||
| dCas9 | D10A/H840A | dsDNA | - | NGG | 1.00 | Nuclease deficient | [26] | |||
| xCas9 3.7 | A262T/R324L/S409I/E480K/E543D/M694I/E1219V | dsDNA | - | NGN, GAA, GAT | 3.12 | Broadened PAM compatibility | [81,82] | |||
| HypaCas9 | N692A/M694A/Q695A/H698A | dsDNA | - | NGG | 1.00 | Hyper-accurate, increased fidelity | [83] | |||
| Cas9 D1135E | D1135E | dsDNA | - | NGG | 1.00 | Reduced binding to non-canonical PAM (NAG) | [84] | |||
| Cas9 VQR | D1135V/R1335Q/T1337R | dsDNA | - | NGAN, NGNG | 1.59 | Altered PAM | [84] | |||
| Cas9 VRER | D1135V/G1218R/R1335E/T1337R | dsDNA | - | NGCG | 0.04 | Altered PAM | [84] | |||
| Cas9 EQR | D1135E/R1335Q/T1337R | dsDNA | - | NGAG | 0.37 | Altered PAM | [84] | |||
| eCas9 1.1 | K848A/K1003A/R1060A | dsDNA | - | NGG | 1.00 | Refined DNA-NUC lobe contact, enhanced specificity | [85] | |||
| Cas9-HF1 | N497A/R661A/Q695A/Q926A | dsDNA | - | NGG | 1.00 | High fidelity, refined DNA-REC lobe contact, reduce non-specific interactions | [86] | |||
| HifiCas9 | R691A | dsDNA | - | NGG | 1.00 | Designed for RNP delivery | [87] | |||
| NmCas9 | 1082 | Neisseria meningitides | - | - | dsDNA | - | NNNNGATT | 0.10 | Reduced target range and restricted off-target sites | [88] |
| SaCas9 | 1053 | Staphylococcus aureus | - | - | dsDNA | - | NNGRRT | 0.34 | Alternative PAM, smaller Cas protein | [89] |
| Cas9 KKH | E72K/N968K/R1015H | dsDNA | - | NNNRRT | 1.09 | Altered PAM | [90] | |||
| CjCas9 | 984 | Campylobacter jejuni | - | - | dsDNA ssRNA | - | NNNVRYM | 1.11 | Smallest Cas9 | [91] |
| FnCas9 | 1629 | Francisella novicida | - | - | dsDNAssRNA | - | NGG | 1.00 | Can cleave ssRNA | [43,92] |
| FnCas9 RHA | E1369R/E1449H/R1556A | dsDNA | - | YG | 2.00 | Shortest Cas9 PAM | [43,92] | |||
| St1Cas9 | 1121 | Streptococcus thermophilius | - | - | dsDNA | - | NNAGAAW | 0.11 | Alternative PAM | [93] |
| St3Cas9 | 1409 | - | - | dsDNA | - | NGGNG | 0.26 | Alternative PAM | [94] | |
| TdCas9 | 1395 | Treponema denticola | - | - | dsDNA | - | NAAAAC | 0.04 | A-rich PAM | [93] |
| AsCas12a (Cpf1) | 1307 | Acidaminococcus sp. BV3L6 | - | - | DNA | ssDNA | TTTN | 0.50 | Only uses crRNA | [63,95,96] |
| FnCas12a (Cpf1) | 1300 | Francisella novicida | - | - | DNA | ssDNA | TTN | 1.45 | Only uses crRNA | [63,95,96] |
| AacCas12b (C2c1) | 1277 | Alicyclobacillus acidoterrestris | - | - | DNA | ssDNA | TTN | 1.45 | PAM at 5′ end, high specificity, requires tracrRNA | [13,66,97] |
| OspCas12c (C2c3) | 1252 | Oleiphilus sp. | - | - | DNA | ssDNA | TG | 1.88 | Requires scoutRNA for crRNA processing | [98,99] |
| Cas12d (CasY) | 1200 | Uncultivated | - | - | DNA | ssDNA | TA | 1.38 | Requires scoutRNA and crRNA for target cleavage | [99,100] |
| Cas12e (CasX) | 980 | Deltaproteobacteria | - | - | dsDNA | ssDNA | TTCN | 0.49 | Alternative PAM, smaller Cas12, requires scoutRNA for crRNA maturation | [70,99,100] |
| Cas12f (Cas14) | 400–700 | Uncultivated | - | - | ssDNA | ssDNA, dsDNA | TTTR, TTAT | 0.40 | Smallest known Cas, longer sgRNA, non-specific cleavage of ssDNA, requires tracrRNA | [26,101,102,103] |
| Cas12g | 767 | Uncultivated | - | - | ssRNA | ssRNA, ssDNA | No Requirement | - | Does not target dsDNA | [98,100,101] |
| Cas12h | 870 | Uncultivated | - | - | DNA | ssDNA | RTR | 1.17 | Alternative PAM | [98] |
| Cas12i | 1093 | Lachnospiraceae bacterium | - | - | DNA | ssDNA | TTN | 1.45 | Predominantly a nickase cutting the NTS, requires a longer crRNA spacer pairing for TS cleavage | [72,98] |
| Cas12j (Casφ) | ~750 | Biggiephage | - | - | DNA | ssDNA | TBN | 2.93 | Smaller sgRNA, discovered in bacterial phages | [104] |
| Cas12k | ~650 | Scytonema hofmanni | - | - | dsDNA | - | GTN | 1.17 | Requires tracrRNA | [105] |
| LshCas13a (C2c2) | 1389 | Leptotrichia shahii | - | - | ssRNA | ssRNA | H | - | RNA targeting, two HEPN domains, only uses crRNA, cleaves outside the base-pairing region | [13,97,106,107] |
| LwaCas13a (C2c2) | 1152 | Leptotrichia wadei | dCas13a | R474A/R1046A | ssRNA | ssRNA | No requirement | - | Nuclease deficient RNA binding protein | [108] |
| BzCas13b (C2c6) | 1224 | Bergeyella zoohelcum | - | - | ssRNA | ssRNA | D-(PS)-NAN/NNA | - | Recognizes bases on either side of the protospacer | [109] |
| EsCas13d | 954 | Eubacterium siraeum | - | - | ssRNA | ssRNA | No requirement | - | Smaller than other Type VI subtypes | [68,110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, M.K.; Sobol, R.W.; Prakash, A. Exploiting DNA Endonucleases to Advance Mechanisms of DNA Repair. Biology 2021, 10, 530. https://doi.org/10.3390/biology10060530
Thompson MK, Sobol RW, Prakash A. Exploiting DNA Endonucleases to Advance Mechanisms of DNA Repair. Biology. 2021; 10(6):530. https://doi.org/10.3390/biology10060530
Chicago/Turabian StyleThompson, Marlo K., Robert W. Sobol, and Aishwarya Prakash. 2021. "Exploiting DNA Endonucleases to Advance Mechanisms of DNA Repair" Biology 10, no. 6: 530. https://doi.org/10.3390/biology10060530
APA StyleThompson, M. K., Sobol, R. W., & Prakash, A. (2021). Exploiting DNA Endonucleases to Advance Mechanisms of DNA Repair. Biology, 10(6), 530. https://doi.org/10.3390/biology10060530