
17O NMR Spectroscopy: A Novel Probe for Characterizing Protein Structure and Folding



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
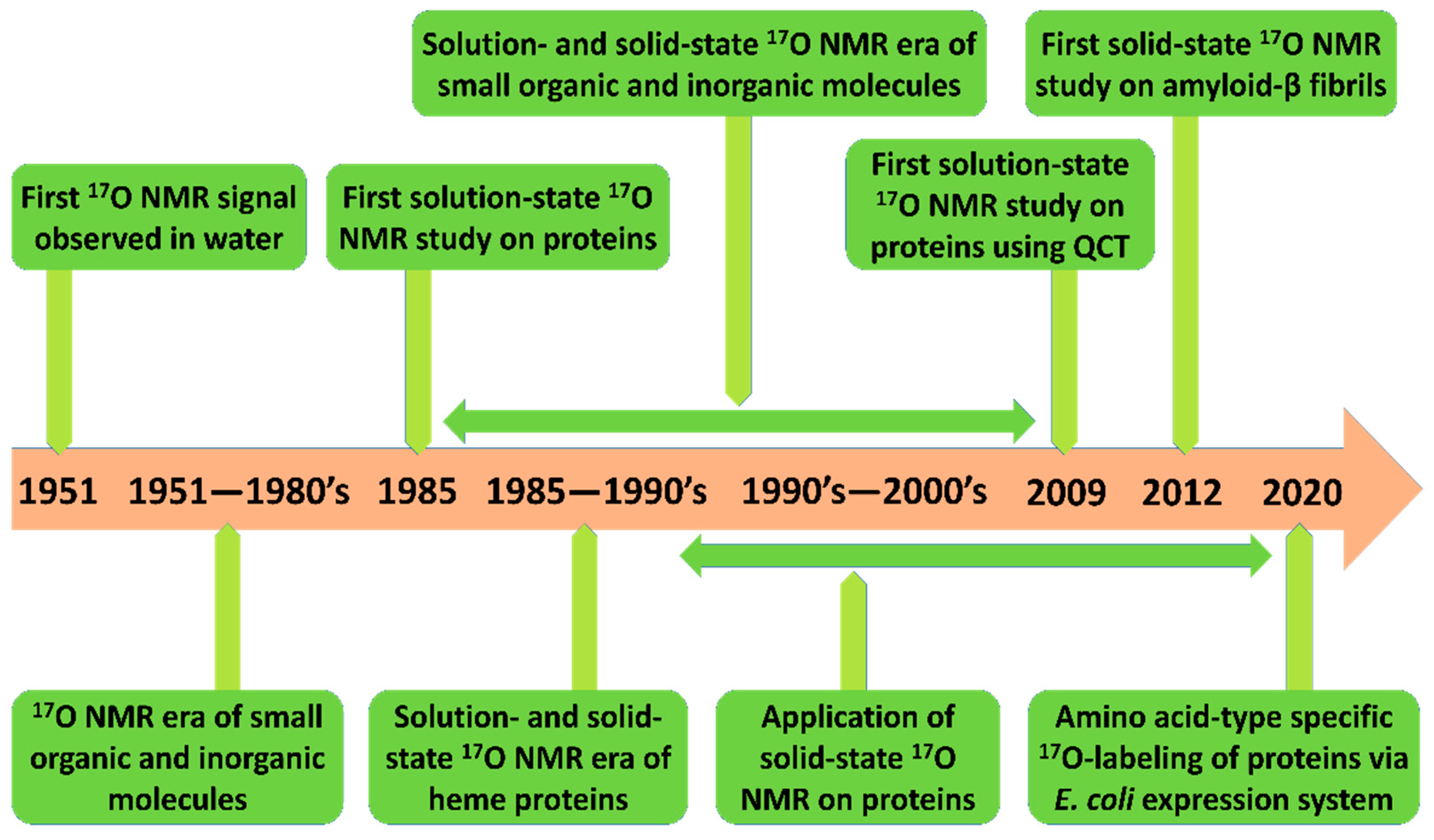
1. Introduction
2. Solution- and Solid-State 17O NMR as a Probe for Studying Protein Structure
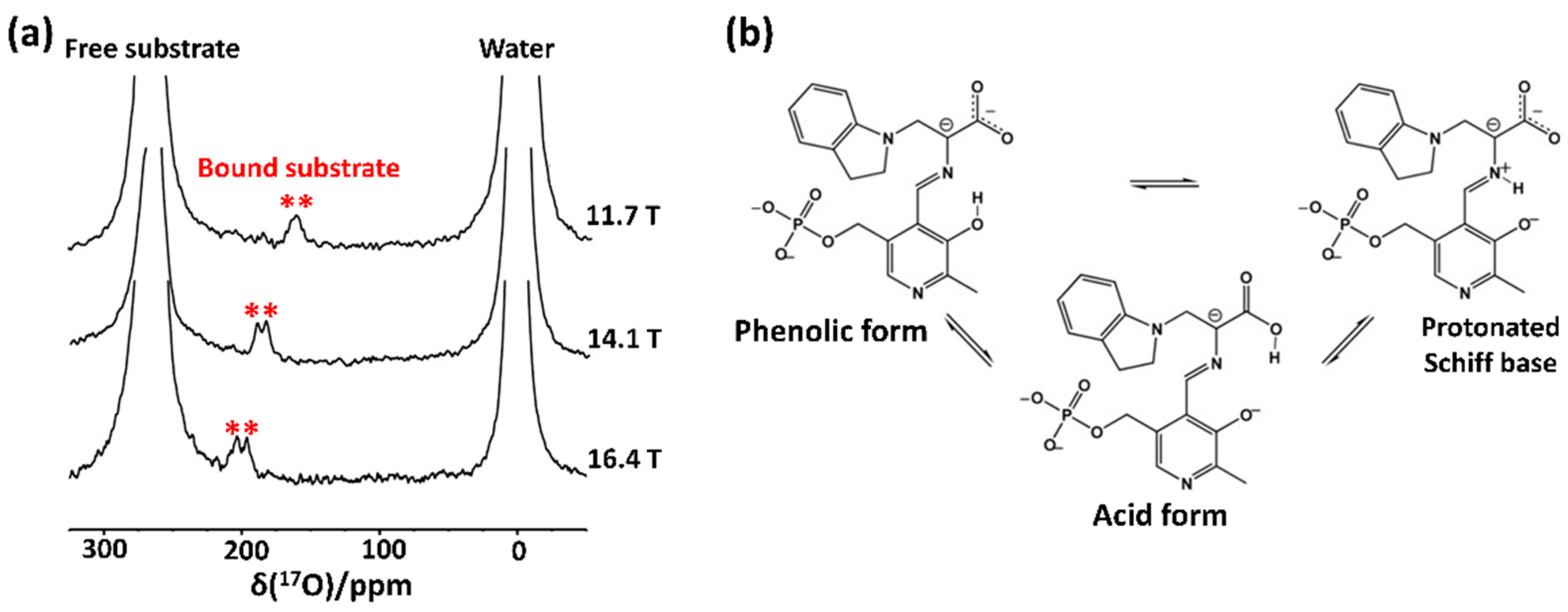
2.1. Early Solution-State 17O NMR-Based Studies for Biomolecules
2.2. Solid-state 17O NMR-Based Approaches
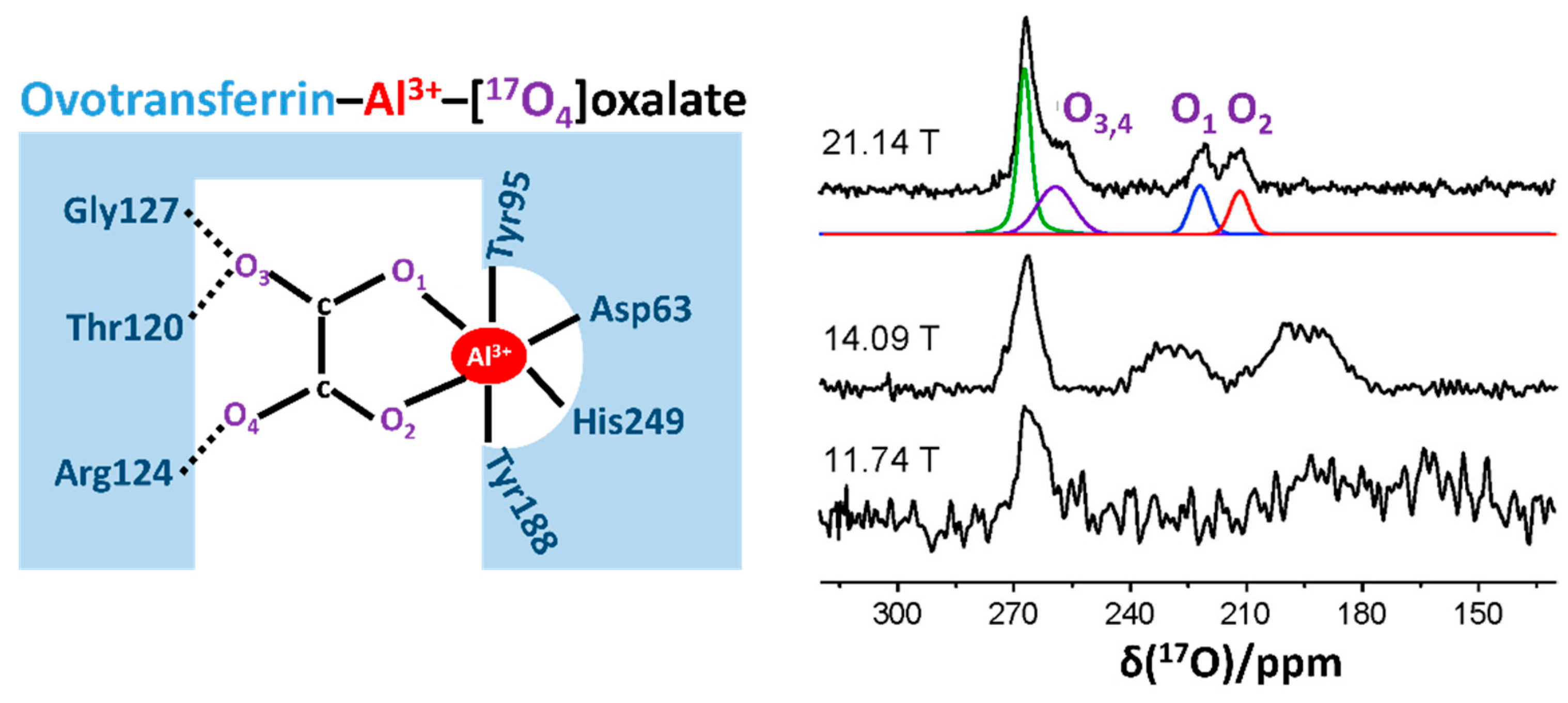
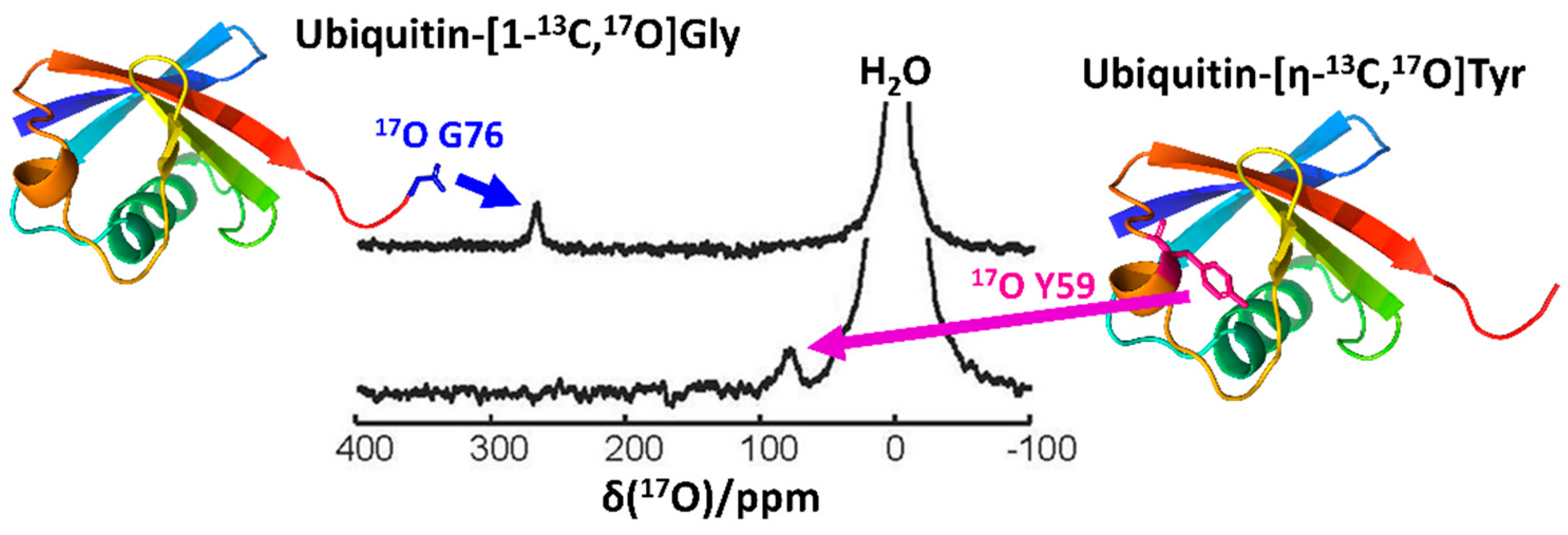
2.3. Recent Developments in Solution-State 17O NMR Spectroscopy for Large Proteins
3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cooper, G.M. The Molecular Composition of Cells. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Cary, NC, USA, 2000. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Protein Function. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Protein Structure and Function. In Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Wang, H.W.; Wang, J.W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef]
- Shoemaker, S.C.; Ando, N. X-rays in the cryo-electron microscopy era: Structural biology’s dynamic future. Biochemistry 2018, 57, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Gauto, D.F.; Estrozi, L.F.; Schwieters, C.D.; Effantin, G.; Macek, P.; Sounier, R.; Sivertsen, A.C.; Schmidt, E.; Kerfah, R.; Mas, G.; et al. Integrated NMR and cryo-EM atomic-resolution structure determination of a half-megadalton enzyme complex. Nat. Commun. 2019, 10, 2697. [Google Scholar] [CrossRef] [PubMed]
- Bax, A.; Clore, G.M. Protein NMR: Boundless opportunities. J. Magn. Reson. 2019, 306, 187–191. [Google Scholar] [CrossRef]
- Geraets, J.A.; Pothula, K.R.; Schröder, G.F. Integrating cryo-EM and NMR data. Curr. Opin. Struct. Biol. 2020, 61, 173–181. [Google Scholar] [CrossRef]
- Muchmore, D.C.; McIntosh, L.P.; Russell, C.B.; Anderson, D.E.; Dahlquist, F.W. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol. 1989, 177, 44–73. [Google Scholar] [CrossRef]
- Gardner, K.H.; Kay, L.E. The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 357–406. [Google Scholar] [CrossRef]
- Kay, L.E.; Ikura, M.; Tschudin, R.; Bax, A. Three-dimensional triple-resonance NMR Spectroscopy of isotopically enriched proteins. J. Magn. Reson. 2011, 213, 423–441. [Google Scholar] [CrossRef]
- Chang, R. Chemistry. In Chemistry, 10th ed.; McGraw-Hill Education: New York, NY, USA, 2010; p. 52. ISBN 007766695X. [Google Scholar]
- Westhof, E.; Fritsch, V. RNA folding: Beyond Watson-Crick pairs. Structure 2000, 8, R55–R65. [Google Scholar] [CrossRef]
- Kool, E.T. Hydrogen bonding, base stacking, and steric effects in DNA replication. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 1–22. [Google Scholar] [CrossRef]
- Wu, G. Solid-state 17O NMR studies of organic and biological molecules. Prog. Nucl. Magn. Reson. Spectrosc. 2008, 52, 118–169. [Google Scholar] [CrossRef]
- Wu, G. Solid-State O-17 NMR studies of organic and biological molecules: Recent advances and future directions. Solid State Nucl. Magn. Reson. 2016, 73, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. O-17 NMR studies of organic and biological molecules in aqueous solution and in the solid state. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114, 135–191. [Google Scholar] [CrossRef]
- Zhu, J.F.; Kwan, I.C.M.; Wu, G. Quadrupole-central-transition O-17 NMR spectroscopy of protein-ligand complexes in solution. J. Am. Chem. Soc. 2009, 131, 14206–14207. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, G. Quadrupole central transition 17O NMR spectroscopy of biological macromolecules in aqueous solution. J. Am. Chem. Soc. 2011, 133, 920–932. [Google Scholar] [CrossRef]
- Lin, B.; Hung, I.; Gan, Z.; Chien, P.H.; Spencer, H.L.; Smith, S.P.; Wu, G. 17O NMR studies of yeast ubiquitin in aqueous solution and in the solid state. ChemBioChem 2021, 22, 826–829. [Google Scholar] [CrossRef]
- Alder, F.; Yu, F.C. On the spin and magnetic moment of O-17. Phys. Rev. 1951, 81, 1067–1068. [Google Scholar] [CrossRef]
- Yamada, K. Recent Applications of Solid-State 17O NMR. In Annual Reports on NMR Spectroscopy; Academic Press: Cambridge, MA, USA, 2010; Volume 70, pp. 115–158. [Google Scholar]
- Wisner, D.A.; Steginsky, C.A.; Shyy, Y.J.; Tsai, M.D. Mechanism of adenylate kinase. 1. Use of 17O NMR to study the binding properties of substrates. J. Am. Chem. Soc. 1985, 107, 2814–2815. [Google Scholar] [CrossRef]
- Lee, H.C.; Cummings, K.; Hall, K.; Hager, L.P.; Oldfield, E. Oxygen-17 nuclear magnetic resonance spectroscopic studies of carbonmonoxyperoxidases. J. Biol. Chem. 1988, 263, 16118–16124. [Google Scholar] [CrossRef]
- Lee, H.C.; Oldfield, E. Oxygen-17 nuclear magnetic resonance spectroscopic studies of carbonmonoxy Hemoproteins. J. Am. Chem. Soc. 1989, 111, 1584–1590. [Google Scholar] [CrossRef]
- Gerothanassis, I.P.; Momenteau, M.; Loock, B. Hydrogen-bond stabilization of dioxygen: Conformation excitation and autoxidation mechanism in hemoprotein models as revealed by oxygen-17 NMR spectroscopy. J. Am. Chem. Soc. 1989, 111, 7006–7012. [Google Scholar] [CrossRef]
- Oldfield, E.; Lee, H.C.; Coretsopoulos, C.; Adebodun, F.; Park, K.D.; Yang, S.T.; Chung, J.; Phillips, B. Solid-state O-17 nuclear-magnetic-resonance spectroscopic studies of [(O2)-O-17] picket fence porphyrin, myoglobin, and hemoglobin. J. Am. Chem. Soc. 1991, 113, 8680–8685. [Google Scholar] [CrossRef]
- Andrew, E.R.; Bradbury, A.; Eades, R.G. Nuclear magnetic resonance spectra from a crystal rotated at high speed. Nature 1958, 182, 1659. [Google Scholar] [CrossRef]
- Lowe, I.J. Free induction decays of rotating solids. Phys. Rev. Lett. 1959, 2, 285–287. [Google Scholar] [CrossRef]
- Wong, A.; Howes, A.P.; Pike, K.J.; Lemaitre, V.; Watts, A.; Anupold, T.; Past, J.; Samoson, A.; Dupree, R.; Smith, M.E. New limits for solid-state O-17 NMR spectroscopy: Complete resolution of multiple oxygen sites in a simple biomolecule. J. Am. Chem. Soc. 2006, 128, 7744–7745. [Google Scholar] [CrossRef]
- Zhu, J.F.; Lau, J.Y.C.; Wu, G. A Solid-state O-17 NMR study of L-Tyrosine in different ionization states: Implications for probing tyrosine side chains in proteins. J. Phys. Chem. B 2010, 114, 11681–11688. [Google Scholar] [CrossRef]
- Wong, A.; Howes, A.P.; Yates, J.R.; Watts, A.; Anupold, T.; Past, J.; Samoson, A.; Dupree, R.; Smith, M.E. Ultra-high resolution O-17 solid-state NMR spectroscopy of biomolecules: A comprehensive spectral analysis of monosodium L-glutamate center dot monohydrate. Phys. Chem. Chem. Phys. 2011, 13, 12213–12224. [Google Scholar] [CrossRef]
- Llor, A.; Virlet, J. Towards high-resolution NMR of more nuclei in solids-sample spinning with time-dependent spinner axis angle. Chem. Phys. Lett. 1988, 152, 248–253. [Google Scholar] [CrossRef]
- Samoson, A.; Lippmaa, E.; Pines, A. High-resolution solid-state NMR averaging of 2nd-order effects by means of a double-rotor. Mol. Phys. 1988, 65, 1013–1018. [Google Scholar] [CrossRef]
- Chmelka, B.F.; Mueller, K.T.; Pines, A.; Stebbins, J.; Wu, Y.; Zwanziger, J.W. O-17 NMR in solids by dynamic-angle spinning and double rotation. Nature 1989, 339, 42–43. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, B.Q.; Pines, A.; Samoson, A.; Lippmaa, E. NMR experiments with a new double rotor. J. Magn. Reson. 1990, 89, 297–309. [Google Scholar] [CrossRef]
- Mueller, K.T.; Sun, B.Q.; Chingas, G.C.; Zwanziger, J.W.; Terao, T.; Pines, A. Dynamic-angle spinning of quadrupolar nuclei. J. Magn. Reson. 1990, 86, 470–487. [Google Scholar] [CrossRef]
- Frydman, L.; Harwood, J.S. Isotropic Spectra of Half-Integer Quadrupolar Spins from Bidimensional Magic-Angle-Spinning NMR. J. Am. Chem. Soc. 1995, 117, 5367–5368. [Google Scholar] [CrossRef]
- Medek, A.; Harwood, J.S.; Frydman, L. Multiple-quantum magic-angle spinning NMR: A new method for the study of quadrupolar nuclei in solids. J. Am. Chem. Soc. 1995, 117, 12779–12787. [Google Scholar] [CrossRef]
- Gan, Z.H. Isotropic NMR spectra of half-integer quadrupolar nuclei using satellite transitions and magic-angle spinning. J. Am. Chem. Soc. 2000, 122, 3242–3243. [Google Scholar] [CrossRef]
- Yamauchi, K.; Kuroki, S.; Ando, I.; Ozaki, T.; Shoji, A. O-17 NMR chemical shifts and quadrupole coupling constants in solid poly(L-alanine)s determined using a high-speed MAS technique. Chem. Phys. Lett. 1999, 302, 331–336. [Google Scholar] [CrossRef]
- Wu, G.; Dong, S.; Ida, R. Solid-state O-17 NMR of thymine: A potential new probe to nucleic acid base pairing. Chem. Commun. 2001, 891–892. [Google Scholar] [CrossRef]
- Wu, G.; Dong, S.; Ida, R.; Reen, N. A solid-state O-17 nuclear magnetic resonance study of nucleic acid bases. J. Am. Chem. Soc. 2002, 124, 1768–1777. [Google Scholar] [CrossRef]
- Lemaître, V.; De Planque, M.R.R.R.; Howes, A.P.; Smith, M.E.; Dupree, R.; Watts, A. Solid-state 17 O NMR as a probe for structural studies of proteins in biomembranes. J. Am. Chem. Soc. 2004, 126, 15320–15321. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chekmenev, E.Y.; Gan, Z.; Gor’kov, P.L.; Saha, S.; Brey, W.W.; Cross, T.A. Ion solvation by channel carbonyls characterized by 17 O solid-state NMR at 21 T. J. Am. Chem. Soc. 2005, 127, 11922–11923. [Google Scholar] [CrossRef]
- Wong, A.; Beevers, A.J.; Kukol, A.; Dupree, R.; Smith, M.E. Solid-state 17O NMR spectroscopy of a phospholemman transmembrane domain protein: Implications for the limits of detecting dilute 17O sites in biomaterials. Solid State Nucl. Magn. Reson. 2008, 33, 72–75. [Google Scholar] [CrossRef]
- Zhu, J.; Ye, E.; Terskikh, V.; Wu, G. Solid-State 17O NMR Spectroscopy of large protein-ligand complexes. Angew. Chem. 2010, 122, 8577–8580. [Google Scholar] [CrossRef][Green Version]
- Tang, A.W.; Kong, X.Q.; Terskikh, V.; Wu, G. Solid-state O-17 NMR of unstable acyl-enzyme intermediates: A direct probe of hydrogen bonding interactions in the oxyanion hole of serine proteases. J. Phys. Chem. B 2016, 120, 11142–11150. [Google Scholar] [CrossRef] [PubMed]
- Gullion, T.; Yamauchi, K.; Okonogi, M.; Asakura, T. 13 C− 17 O REAPDOR NMR as a tool for determining secondary structure in polyamides. Macromolecules 2007, 40, 1363–1365. [Google Scholar] [CrossRef]
- Hung, I.; Uldry, A.C.; Becker-Baldus, J.; Webber, A.L.; Wong, A.; Smith, M.E.; Joyce, S.A.; Yates, J.R.; Pickard, C.J.; Dupree, R.; et al. Probing heteronuclear 15N-17O and13C- 17O connectivities and proximities by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2009, 131, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Antzutkin, O.N.; Iuga, D.; Filippov, A.V.; Kelly, R.T.; Becker-Baldus, J.; Brown, S.P.; Dupree, R. Hydrogen bonding in Alzheimer’s amyloid-β fibrils probed by 15 N{ 17 O} REAPDOR solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2012, 51, 10289–10292. [Google Scholar] [CrossRef]
- Wei, J.; Antzutkin, O.N.; Filippov, A.V.; Iuga, D.; Lam, P.Y.; Barrow, M.P.; Dupree, R.; Brown, S.P.; O’Connor, P.B. Amyloid hydrogen bonding polymorphism evaluated by N-15{O-17}REAPDOR solid-state NMR and ultra-high resolution fourier transform ion cyclotron resonance mass spectrometry. Biochemistry 2016, 55, 2065–2068. [Google Scholar] [CrossRef] [PubMed]
- Hanashima, S.; Fujiwara, N.; Matsumoto, K.; Iwasaki, N.; Zheng, G.Q.; Torigoe, H.; Suzuki, K.; Taniguchi, N.; Yamaguchi, Y.; Taniguchibgh, N.; et al. A solution 17O-NMR approach for observing an oxidized cysteine residue in Cu, Zn-superoxide dismutase. Chem. Commun. 2013, 49, 1449–1451. [Google Scholar] [CrossRef]
- Young, R.P.; Caulkins, B.G.; Borchardt, D.; Bulloch, D.N.; Larive, C.K.; Dunn, M.F.; Mueller, L.J. Solution-state O-17 quadrupole central-transition NMR spectroscopy in the active site of tryptophan Synthase. Angew. Chem.-Int. Ed. 2016, 55, 1350–1354. [Google Scholar] [CrossRef]
- Ohki, S.Y.; Kainosho, M. Stable isotope labeling methods for protein NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2008, 53, 208–226. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen bonds: Simple after all? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, M.P.; Hasnain, S.S.; Antonyuk, S.V. Sub-atomic resolution X-ray crystallography and neutron crystallography: Promise, challenges and potential. IUCrJ 2015, 2, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, E.; Chen, J.C.H.; Fisher, S.Z. Neutron crystallography for the study of hydrogen bonds in macromolecules. Molecules 2017, 22, 596. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Otting, G.; Liepinsh, E.; Wüthrich, K. Protein hydration in aqueous solution. Science 1991, 254, 974–980. [Google Scholar] [CrossRef]
- Ernst, J.A.; Clubb, R.T.; Zhou, H.X.; Gronenborn, A.M.; Clore, G.M. Demonstration of positionally disordered water within a protein hydrophobic cavity by NMR. Science 1995, 67, 1813–1817. [Google Scholar] [CrossRef]
- Royer, W.E.; Pardananii, A.; Gibson, Q.H.; Peterson, E.S.; Friedman, J.M. Ordered water molecules as key allosteric mediators in a cooperative dimeric hemoglobin. Proc. Natl. Acad. Sci. USA 1996, 93, 14526–14531. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N. Water and proteins: A love-hate relationship. Proc. Natl. Acad. Sci. USA 2004, 101, 3325–3326. [Google Scholar] [CrossRef]
- Schiebel, J.; Gaspari, R.; Wulsdorf, T.; Ngo, K.; Sohn, C.; Schrader, T.E.; Cavalli, A.; Ostermann, A.; Heine, A.; Klebe, G. Intriguing role of water in protein-ligand binding studied by neutron crystallography on trypsin complexes. Nat. Commun. 2018, 9, 3559. [Google Scholar] [CrossRef]
- Maurer, M.; Oostenbrink, C. Water in protein hydration and ligand recognition. J. Mol. Recognit. 2019, 32. [Google Scholar] [CrossRef]
- Matricon, P.; Suresh, R.R.; Gao, Z.G.; Panel, N.; Jacobson, K.A.; Carlsson, J. Ligand design by targeting a binding site water. Chem. Sci. 2021, 12, 960–968. [Google Scholar] [CrossRef]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.F.; Rau, D.C.; Parsegian, V.A. Protein solvation in allosteric regulation: A water effect on hemoglobin. Science 1992, 256, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, N. Allosteric transitions in hemoglobin revisited. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129335. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muniyappan, S.; Lin, Y.; Lee, Y.-H.; Kim, J.H. 17O NMR Spectroscopy: A Novel Probe for Characterizing Protein Structure and Folding. Biology 2021, 10, 453. https://doi.org/10.3390/biology10060453
Muniyappan S, Lin Y, Lee Y-H, Kim JH. 17O NMR Spectroscopy: A Novel Probe for Characterizing Protein Structure and Folding. Biology. 2021; 10(6):453. https://doi.org/10.3390/biology10060453
Chicago/Turabian StyleMuniyappan, Srinivasan, Yuxi Lin, Young-Ho Lee, and Jin Hae Kim. 2021. "17O NMR Spectroscopy: A Novel Probe for Characterizing Protein Structure and Folding" Biology 10, no. 6: 453. https://doi.org/10.3390/biology10060453
APA StyleMuniyappan, S., Lin, Y., Lee, Y.-H., & Kim, J. H. (2021). 17O NMR Spectroscopy: A Novel Probe for Characterizing Protein Structure and Folding. Biology, 10(6), 453. https://doi.org/10.3390/biology10060453