
Viewing SARS-CoV-2 Nucleocapsid Protein in Terms of Molecular Flexibility


Abstract
Simple Summary
Abstract
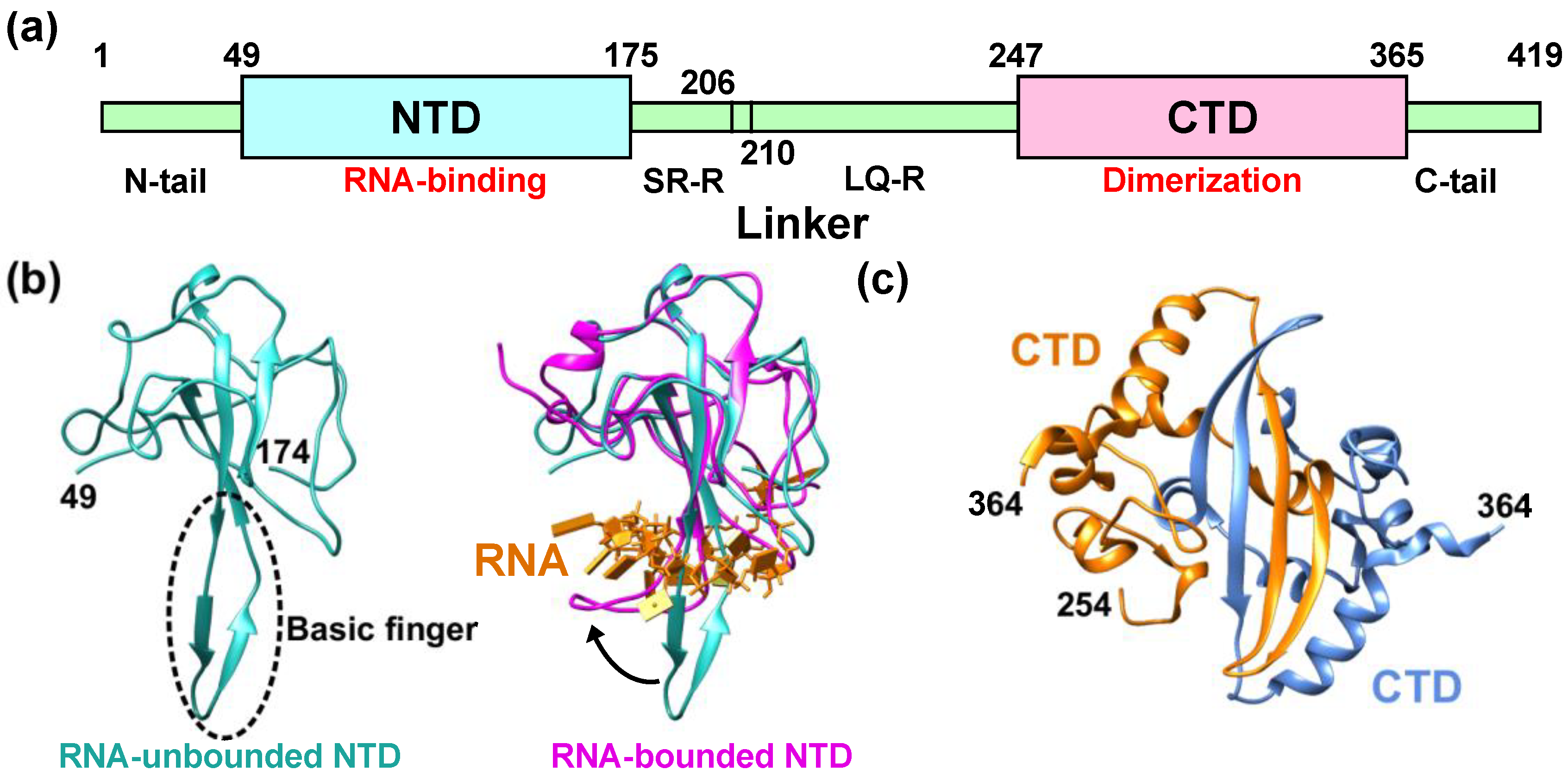
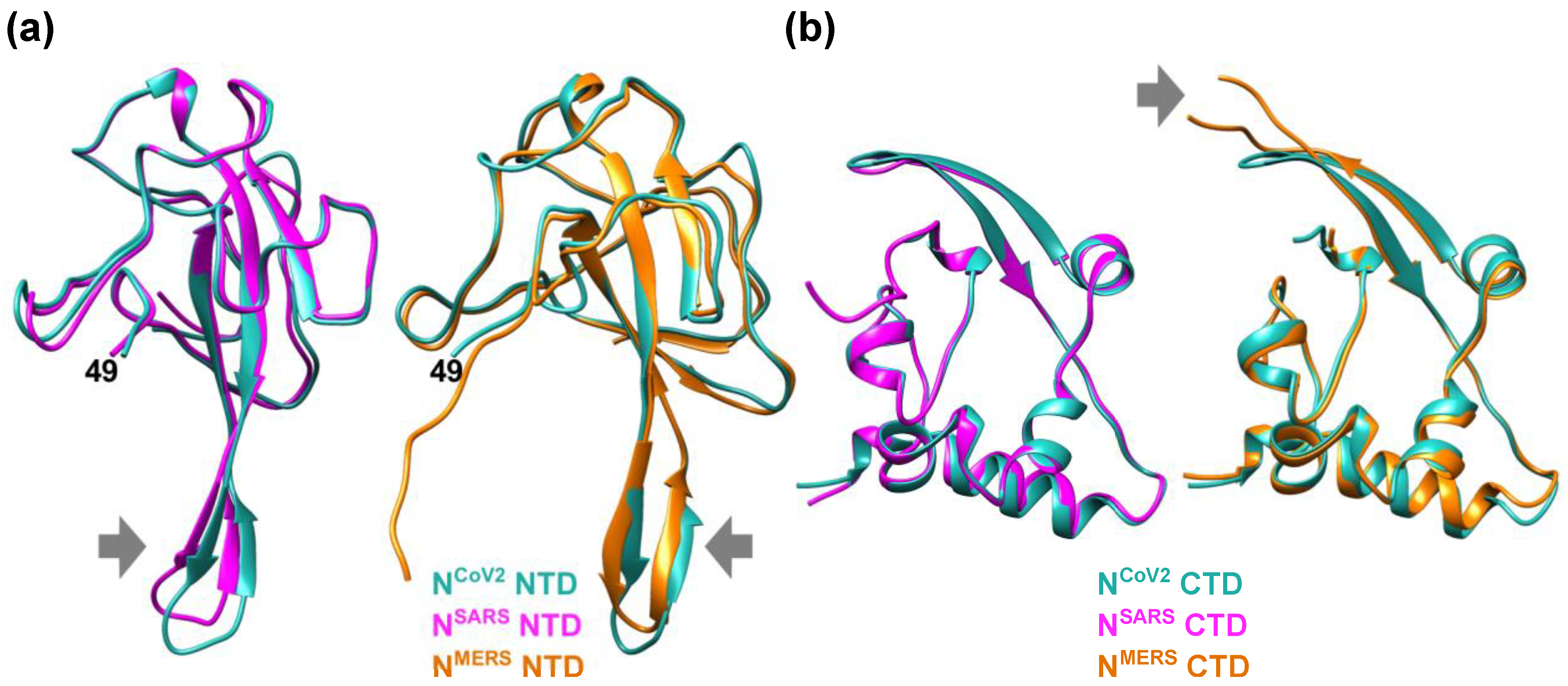
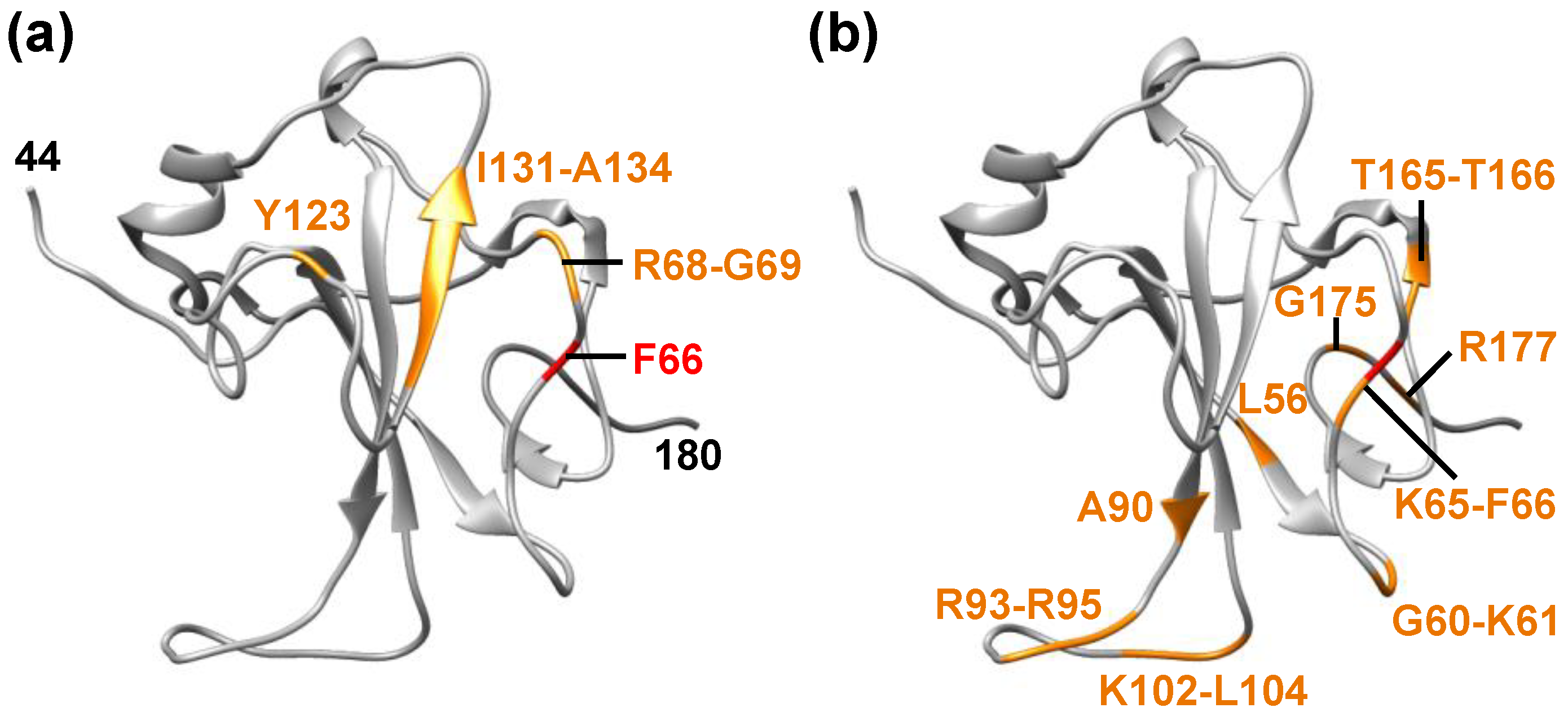
1. SARS-CoV-2 Nucleocapsid Protein
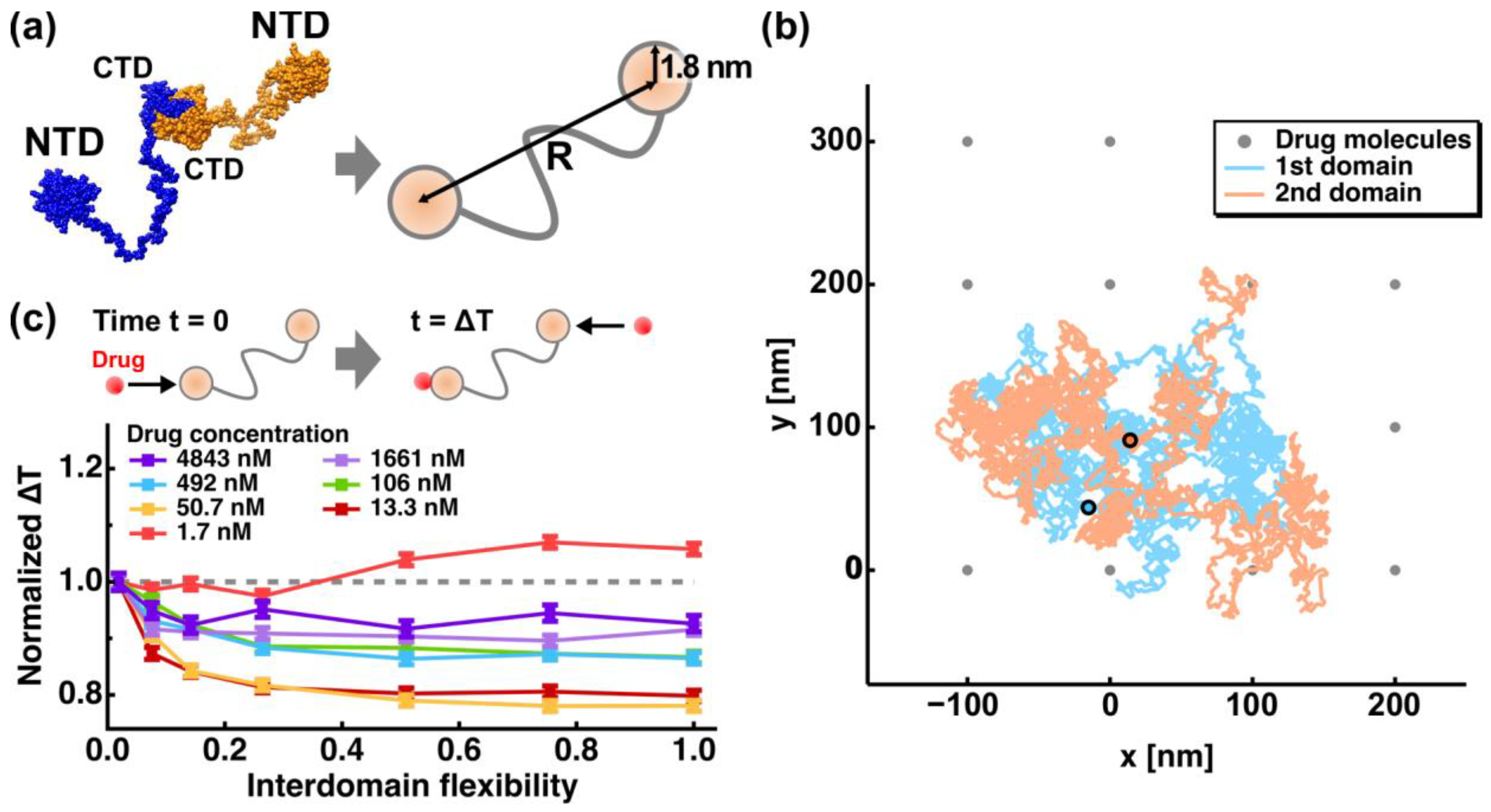
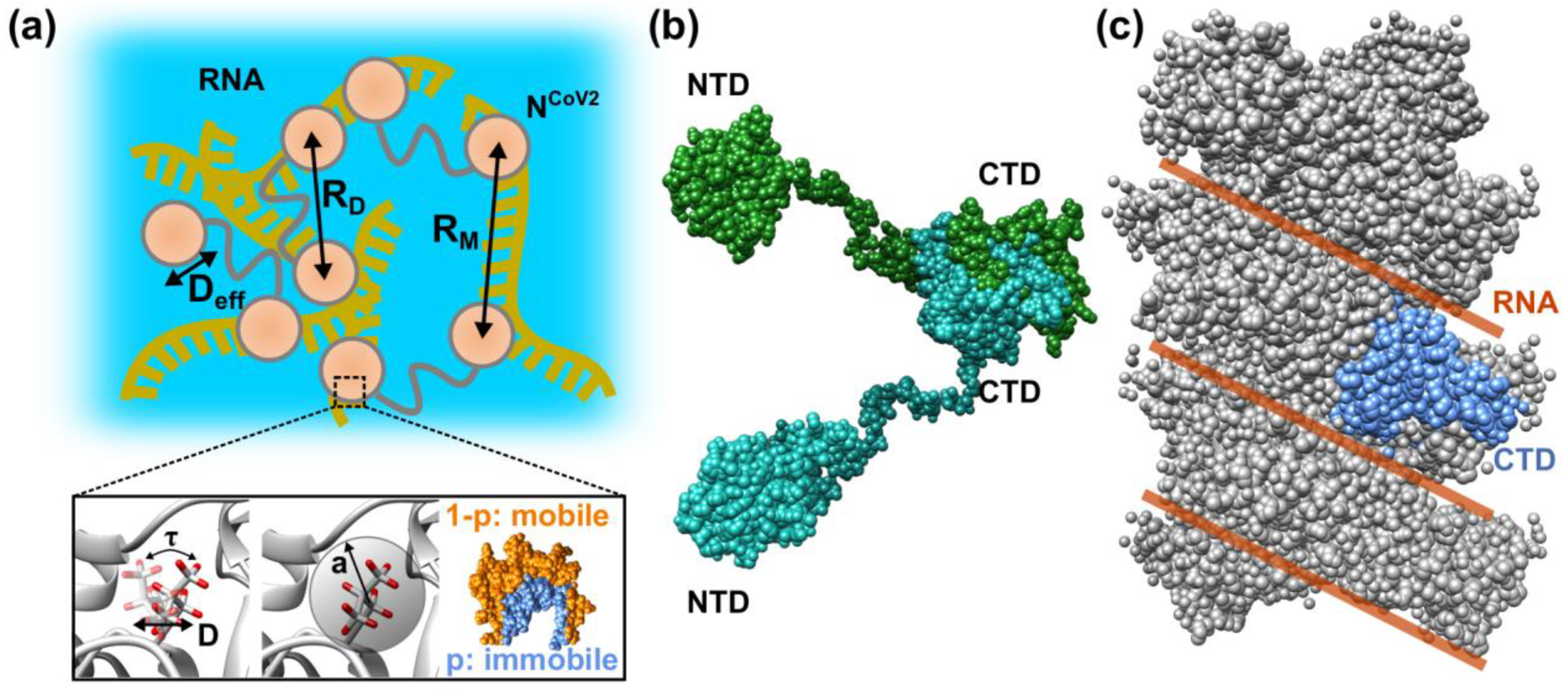
2. Molecular Flexibility and Drug Binding of NCoV2
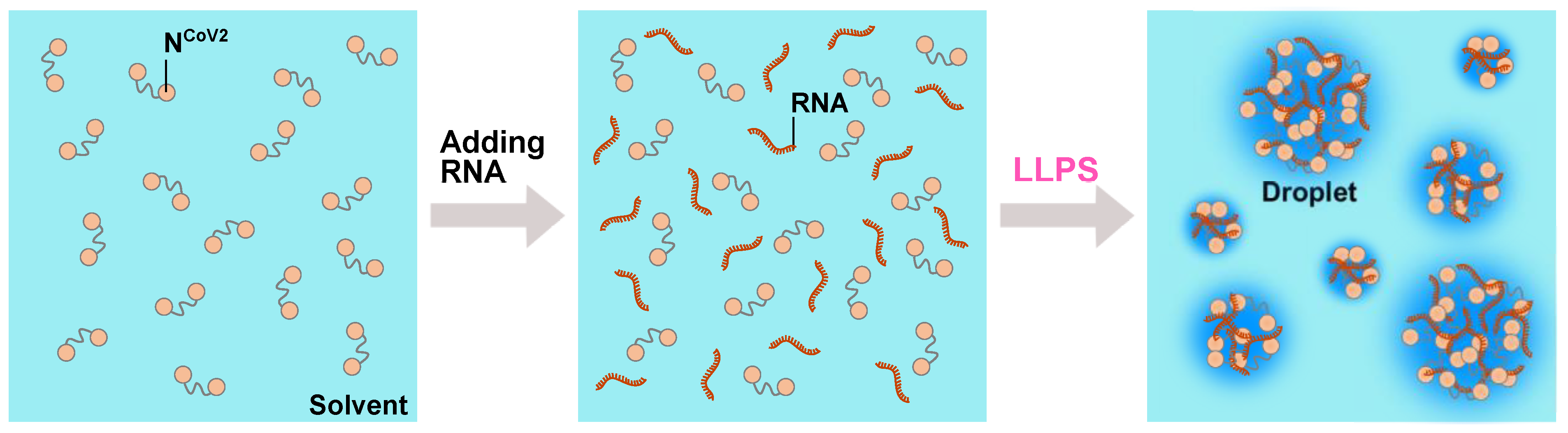
3. Liquid-Liquid Phase Separation (LLPS) of NCoV2
4. Perspective–Toward Structural and Dynamical Characterization of NCoV2 Droplets
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J.; Mayer, C.; Poch, O.; Thompson, J.D. Characterization of accessory genes in coronavirus genomes. Virol. J. 2020, 17, 131. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Astuti, I. Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Lal, S.K. The SARS-CoV nucleocapsid protein: A protein with multifarious activities. Infect. Genet. Evol. 2008, 8, 397–405. [Google Scholar] [CrossRef]
- Chang, C.-K.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T.-H. The SARS coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Zhu, G.; Zhu, C.; Zhu, Y.; Sun, F. Minireview of progress in the structural study of SARS-CoV-2 proteins. Curr. Res. Microb. Sci. 2020, 1, 53–61. [Google Scholar] [CrossRef]
- Chang, C.-K.; Chen, C.-M.M.; Chiang, M.-H.; Hsu, Y.-L.; Huang, T.-H. Transient Oligomerization of the SARS-CoV N Protein—Implication for Virus Ribonucleoprotein Packaging. PLoS ONE 2013, 8, e65045. [Google Scholar] [CrossRef]
- Katsuma, S.; Kokusho, R. A Conserved Glycine Residue Is Required for Proper Functioning of a Baculovirus VP39 Protein. J. Virol. 2017, 91, e02253-16. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.W.; Fang, S.; Fan, H.; Lescar, J.; Liu, D.X. Amino acid residues critical for RNA-binding in the N-terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 2006, 34, 4816–4825. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and self-assembly of the SARS-CoV -2 nucleocapsid protein. Protein Sci. 2020, 29, 1890–1901. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L.; Basquin, J.; Bohn, S.; Beck, F.; Klumpe, S.; Pfeifer, G.; Nagy, I.; Bracher, A.; Hartl, F.U.; Baumeister, W. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the Covid-19 severe acute respiratory syndrome coronavirus. Biochem. Biophys. Res. Commun. 2021, 538, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zeng, R.; Von Brunn, A.; Lei, J. Structural characterization of the C-terminal domain of SARS-CoV-2 nucleocapsid protein. Mol. Biomed. 2020, 1, 1–11. [Google Scholar] [CrossRef]
- Takeda, M.; Chang, C.-K.; Ikeya, T.; Güntert, P.; Chang, Y.-H.; Hsu, Y.-L.; Huang, T.-H.; Kainosho, M. Solution Structure of the C-terminal Dimerization Domain of SARS Coronavirus Nucleocapsid Protein Solved by the SAIL-NMR Method. J. Mol. Biol. 2008, 380, 608–622. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J.; et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C.; Kuhn, P. Ribonucleocapsid Formation of Severe Acute Respiratory Syndrome Coronavirus through Molecular Action of the N-Terminal Domain of N Protein. J. Virol. 2007, 81, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, N.; Lichière, J.; Baklouti, A.; Ferron, F.; Sévajol, M.; Canard, B.; Coutard, B. Structural characterization of the N-terminal part of the MERS-CoV nucleocapsid by X-ray diffraction and small-angle X-ray scattering. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Chang, C.-K.; Chang, Y.-W.; Sue, S.-C.; Bai, H.-I.; Riang, L.; Hsiao, C.-D.; Huang, T.-H. Structure of the SARS Coronavirus Nucleocapsid Protein RNA-binding Dimerization Domain Suggests a Mechanism for Helical Packaging of Viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.H.; Lichière, J.; Canard, B.; Papageorgiou, N.; Attoumani, S.; Ferron, F.; Coutard, B. Structure and oligomerization state of the C-terminal region of the Middle East respiratory syndrome coronavirus nucleoprotein. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takemoto, Y.; Okuno, Y.; Hashimoto, S.; Yoshida, S.; Fukunaga, Y.; Tanaka, T.; Kita, Y.; Kuwayama, S.; Muraki, Y.; et al. The development of vaccines against SARS corona virus in mice and SCID-PBL/hu mice. Vaccine 2005, 23, 2269–2272. [Google Scholar] [CrossRef]
- Gao, W.; Tamin, A.; Soloff, A.; D’Aiuto, L.; Nwanegbo, E.; Robbins, P.D.; Bellini, W.J.; Barratt-Boyes, S.; Gambotto, A. Effects of a SARS-associated coronavirus vaccine in monkeys. Lancet 2003, 362, 1895–1896. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mazumdar, K.; Gordy, J.T. The Nucleocapsid Protein of SARS–CoV-2: A Target for Vaccine Development. J. Virol. 2020, 94, 1–2. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Kumar, S.; Ma, B.; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999, 8, 1181–1190. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLoS Comput. Biol. 2016, 12, e1005067. [Google Scholar] [CrossRef]
- Tatar, G.; Ozyurt, E.; Turhan, K. Computational drug repurposing study of the RNA binding domain of SARS-CoV -2 nucleocapsid protein with antiviral agents. Biotechnol. Prog. 2020, 37, e3110. [Google Scholar] [CrossRef]
- Kaur, H.; Shekhar, N.; Sharma, S.; Sarma, P.; Prakash, A.; Medhi, B. Ivermectin as a potential drug for treatment of COVID-19: An in-sync review with clinical and computational attributes. Pharmacol. Rep. 2021, 1–14. [Google Scholar] [CrossRef]
- Ahamad, S.; Gupta, D.; Kumar, V. Targeting SARS-CoV-2 nucleocapsid oligomerization: Insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Rolta, R.; Yadav, R.; Salaria, D.; Trivedi, S.; Imran, M.; Sourirajan, A.; Baumler, D.J.; Dev, K. In silico screening of hundred phytocompounds of ten medicinal plants as potential inhibitors of nucleocapsid phosphoprotein of COVID-19: An approach to prevent virus assembly. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef]
- Sarma, P.; Shekhar, N.; Prajapat, M.; Avti, P.; Kaur, H.; Kumar, S.; Singh, S.; Kumar, H.; Prakash, A.; Dhibar, D.P.; et al. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). J. Biomol. Struct. Dyn. 2021, 1–9. [Google Scholar] [CrossRef]
- Yadav, R.; Imran, M.; Dhamija, P.; Suchal, K.; Handu, S. Virtual screening and dynamics of potential inhibitors targeting RNA binding domain of nucleocapsid phosphoprotein from SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Abbas, G. Docking study of chloroquine and hydroxychloroquine interaction with RNA binding domain of nucleocapsid phospho-protein—An in silico insight into the comparative efficacy of repurposing antiviral drugs. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Kim, D.-H.; Han, K.-H. Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2018, 19, 3614. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Jensen, M.R.; Ribeiro, E.A., Jr.; Gérard, F.C.A.; Ruigrok, R.W.H.; Blackledge, M.; Jamin, M. The N0-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient α-helices. Protein Sci. 2011, 20, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Houben, K.; Lescop, E.; Blanchard, L.; Ruigrok, R.W.H.; Blackledge, M. Quantitative Conformational Analysis of Partially Folded Proteins from Residual Dipolar Couplings: Application to the Molecular Recognition Element of Sendai Virus Nucleoprotein. J. Am. Chem. Soc. 2008, 130, 8055–8061. [Google Scholar] [CrossRef]
- Feuerstein, S.; Solyom, Z.; Aladag, A.; Favier, A.; Schwarten, M.; Hoffmann, S.; Willbold, D.; Brutscher, B. Transient Structure and SH3 Interaction Sites in an Intrinsically Disordered Fragment of the Hepatitis C Virus Protein NS5A. J. Mol. Biol. 2012, 420, 310–323. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, D.-H.; Han, J.J.; Cha, E.-J.; Lim, J.-E.; Cho, Y.-J.; Lee, C.; Han, K.-H. Understanding Pre-Structured Motifs (PreSMos) in Intrinsically Unfolded Proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Oas, T.G. Osmolyte-Induced Folding of an Intrinsically Disordered Protein: Folding Mechanism in the Absence of Ligand. Biochemistry 2010, 49, 5086–5096. [Google Scholar] [CrossRef]
- Smoyer, C.J.; Jaspersen, S.L. Breaking down the wall: The nuclear envelope during mitosis. Curr. Opin. Cell Biol. 2014, 26, 1–9. [Google Scholar] [CrossRef]
- Stewart, M.P.; Helenius, J.; Toyoda, Y.; Ramanathan, S.P.; Muller, D.J.; Hyman, A.A. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 2011, 469, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Vancraenenbroeck, R.; Harel, Y.S.; Zheng, W.; Hofmann, H. Polymer effects modulate binding affinities in disordered proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 19506–19512. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.M.; Gruebele, M.; Sukenik, S. How does solvation in the cell affect protein folding and binding? Curr. Opin. Struct. Biol. 2018, 48, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Sukenik, S.; Salam, M.; Wang, Y.; Gruebele, M. In-Cell Titration of Small Solutes Controls Protein Stability and Aggregation. J. Am. Chem. Soc. 2018, 140, 10497–10503. [Google Scholar] [CrossRef]
- Moses, D.; Yu, F.; Ginell, G.M.; Shamoon, N.M.; Koenig, P.S.; Holehouse, A.S.; Sukenik, S. Revealing the Hidden Sensitivity of Intrinsically Disordered Proteins to their Chemical Environment. J. Phys. Chem. Lett. 2020, 11, 10131–10136. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T. A theoretical study on the effects of interdomain flexibility on drug encounter rate for coronavirus nucleocapsid-type proteins. Biophys. Chem. 2021, 272, 106574. [Google Scholar] [CrossRef]
- Kuh, H.J.; Jang, S.H.; Wientjes, M.G.; Au, J.L.S. Computational model of intracellular pharmacokinetics of paclitaxel. J. Pharmacol. Exp. Ther. 2000, 293, 761–770. [Google Scholar] [PubMed]
- Steel, R.G.D. A Rank Sum Test for Comparing All Pairs of Treatments. Technometrics 1960, 2, 197–207. [Google Scholar] [CrossRef]
- Dwass, M. Some k-sample rank-order tests. In Contributions to Probability and Statistics; Olkin, I., Hoeffding, W., Madow, W.G., Mann, H.B., Eds.; Stanford University Press: Palo Alto, CA, USA, 1960; pp. 198–202. [Google Scholar]
- Zaccai, G. How Soft Is a Protein? A Protein Dynamics Force Constant Measured by Neutron Scattering. Science 2000, 288, 1604–1607. [Google Scholar] [CrossRef] [PubMed]
- Kovermann, M.; Rogne, P.; Wolf-Watz, M. Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 2016, 49, e6. [Google Scholar] [CrossRef] [PubMed]
- Gabel, F.; Bicout, D.; Lehnert, U.; Tehei, M.; Weik, M.; Zaccai, G. Protein dynamics studied by neutron scattering. Q. Rev. Biophys. 2002, 35, 327–367. [Google Scholar] [CrossRef]
- Bernadó, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. BioSyst. 2012, 8, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T. Usefulness of medium-angle X-ray scattering for structural characterization of flexible proteins studied by computer simulations. Biochem. Biophys. Res. Commun. 2020, 525, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional Characterization of Negri Bodies (NBs) in Rabies Virus-Infected Cells: Evidence that NBs Are Sites of Viral Transcription and Replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. mBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [PubMed]
- Guseva, S.; Milles, S.; Jensen, M.R.; Salvi, N.; Kleman, J.-P.; Maurin, D.; Ruigrok, R.W.H.; Blackledge, M. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 2020, 6, eaaz7095. [Google Scholar] [CrossRef] [PubMed]
- Cascarina, S.M.; Ross, E.D. A proposed role for the SARS-CoV-2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. 2020, 34, 9832–9842. [Google Scholar] [CrossRef]
- Chen, H.; Cui, Y.; Han, X.; Hu, W.; Sun, M.; Zhang, Y.; Wang, P.-H.; Song, G.; Chen, W.; Lou, J. Liquid–liquid phase separation by SARS-CoV-2 nucleocapsid protein and RNA. Cell Res. 2020, 30, 1143–1145. [Google Scholar] [CrossRef]
- Savastano, A.; De Opakua, A.I.; Rankovic, M.; Zweckstetter, M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e4. [Google Scholar] [CrossRef] [PubMed]
- Iserman, C.; Roden, C.A.; Boerneke, M.A.; Sealfon, R.S.; McLaughlin, G.A.; Jungreis, I.; Fritch, E.J.; Hou, Y.J.; Ekena, J.; Weidmann, C.A.; et al. Genomic RNA Elements Drive Phase Separation of the SARS-CoV-2 Nucleocapsid. Mol. Cell 2020, 80, 1078–1091.e6. [Google Scholar] [CrossRef]
- Chen, H.; Gill, A.; Dove, B.K.; Emmett, S.R.; Kemp, C.F.; Ritchie, M.A.; Dee, M.; Hiscox, J.A. Mass Spectroscopic Characterization of the Coronavirus Infectious Bronchitis Virus Nucleoprotein and Elucidation of the Role of Phosphorylation in RNA Binding by Using Surface Plasmon Resonance. J. Virol. 2005, 79, 1164–1179. [Google Scholar] [CrossRef]
- Mohandas, D.V.; Dales, S. Endosomal association of a protein phosphatase with high dephosphorylating activity against a coronavirus nucleocapsid protein. FEBS Lett. 1991, 282, 419–424. [Google Scholar] [CrossRef]
- Banerjee, P.R.; Milin, A.N.; Moosa, M.M.; Onuchic, P.L.; Deniz, A.A. Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew. Chem. Int. Ed. 2017, 56, 11354–11359. [Google Scholar] [CrossRef]
- Onuchic, P.L.; Milin, A.N.; Alshareedah, I.; Deniz, A.A.; Banerjee, P.R. Divalent cations can control a switch-like behavior in heterotypic and homotypic RNA coacervates. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Aumiller, W.M.; Keating, C.D. Phosphorylation-mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat. Chem. 2016, 8, 129–137. [Google Scholar] [CrossRef]
- Jensen, M.R.; Communie, G.; Ribeiro, E.A.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar] [CrossRef]
- Blanchard, L.; Tarbouriech, N.; Blackledge, M.; Timmins, P.; Burmeister, W.P.; Ruigrok, R.W.; Marion, D. Structure and dynamics of the nucleocapsid-binding domain of the Sendai virus phosphoprotein in solution. Virology 2004, 319, 201–211. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Piper, A.; Meilleur, F.; Hernandez, R.; Heller, W.T.; Brown, D.T. Conformational Changes in Sindbis Virus Induced by Decreased pH Are Revealed by Small-Angle Neutron Scattering. J. Virol. 2012, 86, 1982–1987. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Das, N.C.; Warren, G.T.; Kao, C.C.; Dragnea, B.G.; Ni, P.; Sokol, P.E. Small Angle Scattering Study of the Structure and Organization of RNAs and Protein of Brome Mosaic Virus (BMV) Capsid Protein. Phys. Procedia 2014, 60, 101–109. [Google Scholar] [CrossRef][Green Version]
- He, L.; Piper, A.; Meilleur, F.; Myles, D.A.A.; Hernández, R.; Brown, D.T.; Heller, W.T. The Structure of Sindbis Virus Produced from Vertebrate and Invertebrate Hosts as Determined by Small-Angle Neutron Scattering. J. Virol. 2010, 84, 5270–5276. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I.; Koch, M.H.; Timmins, P.A.; May, R.P. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Mahieu, E.; Gabel, F. Biological small-angle neutron scattering: Recent results and development. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Fitter, J.; Gutberlet, T.; Katsaras, J. (Eds.) Neutron Scattering in Biology; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 978-3-540-29108-4. [Google Scholar]
- Heller, W.T. Small-angle neutron scattering and contrast variation: A powerful combination for studying biological structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Gabel, F. Small-Angle Neutron Scattering for Structural Biology of Protein–RNA Complexes. Methods Enzymol. 2015, 558, 391–415. [Google Scholar] [CrossRef]
- Mirandela, G.D.; Tamburrino, G.; Ivanović, M.T.; Strnad, F.M.; Byron, O.; Rasmussen, T.; Hoskisson, P.A.; Hub, J.S.; Zachariae, U.; Gabel, F.; et al. Merging In-Solution X-ray and Neutron Scattering Data Allows Fine Structural Analysis of Membrane–Protein Detergent Complexes. J. Phys. Chem. Lett. 2018, 9, 3910–3914. [Google Scholar] [CrossRef] [PubMed]
- Lapinaite, A.; Carlomagno, T.; Gabel, F. Small-Angle Neutron Scattering of RNA–Protein Complexes. Methods Mol. Biol. 2020, 2113, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Miyazaki, Y.; Summers, M.F. Isotope labeling strategies for NMR studies of RNA. J. Biomol. NMR 2010, 46, 113–125. [Google Scholar] [CrossRef]
- Duss, O.; Lukavsky, P.J.; Allain, F.H.-T. Isotope Labeling and Segmental Labeling of Larger RNAs for NMR Structural Studies BT—Isotope labeling in Biomolecular NMR. In Isotope Labeling in Biomolecular NMR.; Atreya, H.S., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 121–144. ISBN 978-94-007-4954-2. [Google Scholar]
- Lapinaite, A.; Simon, B.; Skjaerven, L.; Rakwalska-Bange, M.; Gabel, F.; Carlomagno, T. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nat. Cell Biol. 2013, 502, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Bée, M. Quasielastic Neutron Scattering; Adam Hilger: Bristol, PA, USA; Philadelphia, PA, USA, 1988; ISBN 9780852743713. [Google Scholar]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | bcoh [10−12 cm] |
|---|---|
| H | −0.37409 |
| D | 0.6674 |
| C | 0.66484 |
| N | 0.936 |
| O | 0.5805 |
| P | 0.513 |
| S | 0.2847 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuo, T. Viewing SARS-CoV-2 Nucleocapsid Protein in Terms of Molecular Flexibility. Biology 2021, 10, 454. https://doi.org/10.3390/biology10060454
Matsuo T. Viewing SARS-CoV-2 Nucleocapsid Protein in Terms of Molecular Flexibility. Biology. 2021; 10(6):454. https://doi.org/10.3390/biology10060454
Chicago/Turabian StyleMatsuo, Tatsuhito. 2021. "Viewing SARS-CoV-2 Nucleocapsid Protein in Terms of Molecular Flexibility" Biology 10, no. 6: 454. https://doi.org/10.3390/biology10060454
APA StyleMatsuo, T. (2021). Viewing SARS-CoV-2 Nucleocapsid Protein in Terms of Molecular Flexibility. Biology, 10(6), 454. https://doi.org/10.3390/biology10060454