
Macronutrient Determinants of Obesity, Insulin Resistance and Metabolic Health


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Carbohydrate
2.1. Types of Carbohydrates and Their Metabolic Effects
2.2. Glycaemic Index and Metabolic Effects of Carbohydrates
2.3. Molecular Mechanisms of Metabolic Benefits of Fibre Intake
2.4. Molecular Mechanisms of Adverse Effects of Carbohydrate Intake
3. Fat
3.1. Types of Fatty Acids and Their Metabolic Effect
3.2. Molecular Mechanisms Involved in HFD-Induced Insulin Resistance
4. Protein
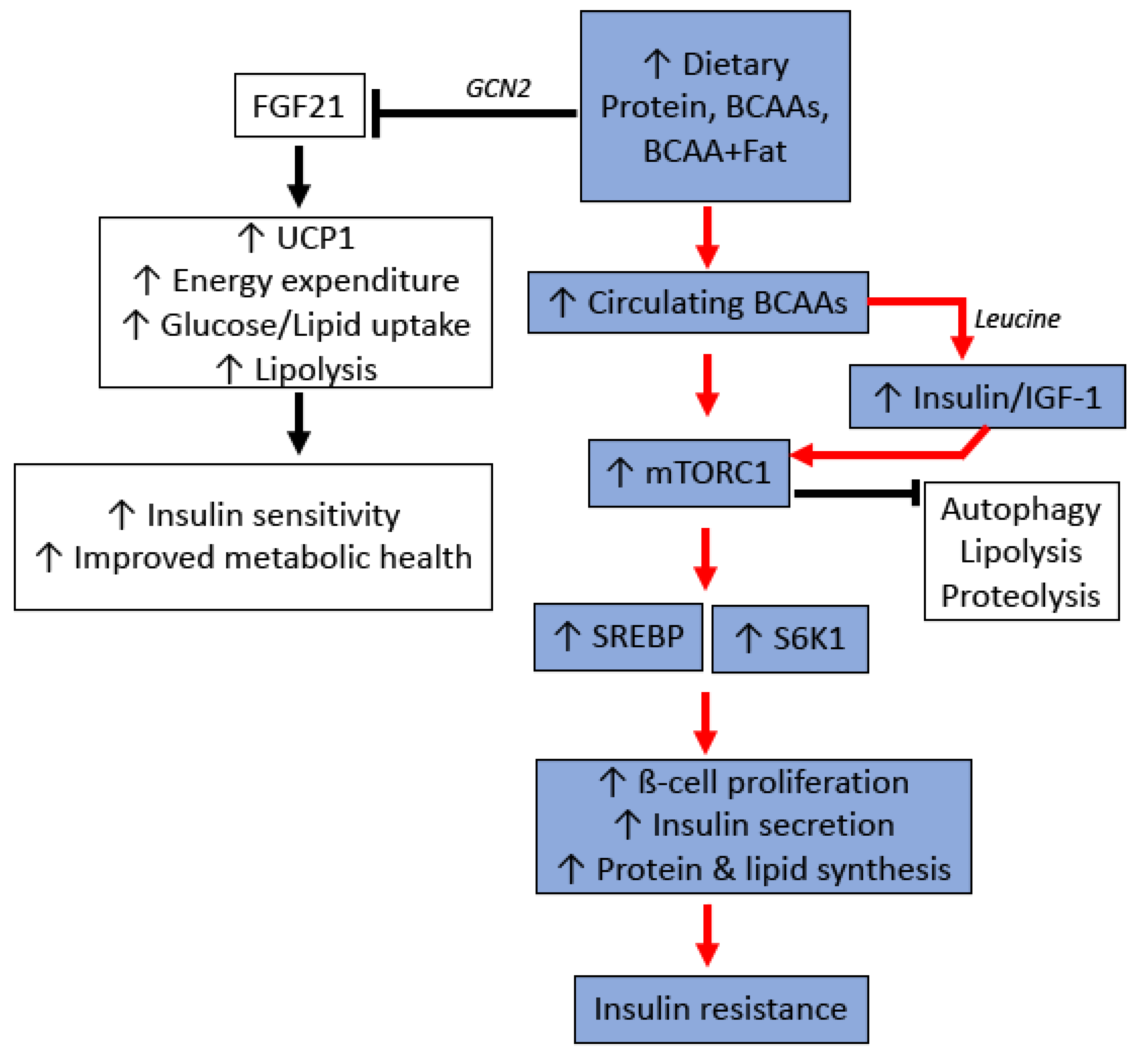
4.1. Protein, Branch-Chained Amino Acids, and Their Metabolic Effects
4.2. Molecular Mechanisms of Protein/BCAA Induced Insulin Resistance
5. Limitations of the Single Nutrient Approach
6. Importance of Dietary Protein Content and the Value of a Multi-Nutrient Mixture Approach to Research in Understanding the Nutritional Basis of Metabolic Physiology
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collaboration, N.R.F. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary fat: From foe to friend? Science 2018, 362, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Elia, M.; Cummings, J.H. Physiological aspects of energy metabolism and gastrointestinal effects of carbohydrates. Eur. J. Clin. Nutr. 2007, 61 (Suppl. 1), S40–S74. [Google Scholar] [CrossRef] [PubMed]
- Raubenheimer, D.; Gosby, A.K.; Simpson, S.J. Integrating nutrients, foods, diets, and appetites with obesity and cardiometabolic health. Obesity 2015, 23, 1741–1742. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Lopez-Otin, C.; Madeo, F.; de Cabo, R. Carbotoxicity-Noxious Effects of Carbohydrates. Cell 2018, 175, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, M.; Mente, A.; Zhang, X.; Swaminathan, S.; Li, W.; Mohan, V.; Iqbal, R.; Kumar, R.; Wentzel-Viljoen, E.; Rosengren, A.; et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): A prospective cohort study. Lancet 2017, 390, 2050–2062. [Google Scholar] [CrossRef]
- Willett, W.C.; Stampfer, M.J. Current evidence on healthy eating. Annu. Rev. Public Health 2013, 34, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.Y.; Micha, R.; Mozaffarian, D. Dietary fats and cardiometabolic disease: Mechanisms and effects on risk factors and outcomes. Nat. Rev. Cardiol. 2019, 16, 581–601. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Wahl, D.; Clark, X.; Bagley, E.E.; Gregoriou, G.C.; Senior, A.M.; Wang, Q.-P.; Brandon, A.E.; et al. Branched-chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat. Metab. 2019, 1, 532–545. [Google Scholar] [CrossRef]
- Yap, Y.W.; Rusu, P.M.; Chan, A.Y.; Fam, B.C.; Jungmann, A.; Solon-Biet, S.M.; Barlow, C.K.; Creek, D.J.; Huang, C.; Schittenhelm, R.B.; et al. Restriction of essential amino acids dictates the systemic metabolic response to dietary protein dilution. Nat. Commun. 2020, 11, 2894. [Google Scholar] [CrossRef]
- Mair, W.; Piper, M.D.W.; Partridge, L. Calories Do Not Explain Extension of Life Span by Dietary Restriction in Drosophila. PLoS Biol. 2005, 3, e223. [Google Scholar] [CrossRef]
- Cummings, N.E.; Williams, E.M.; Kasza, I.; Konon, E.N.; Schaid, M.D.; Schmidt, B.A.; Poudel, C.; Sherman, D.S.; Yu, D.; Arriola Apelo, S.I.; et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J. Physiol. 2017, 596, 623–645. [Google Scholar] [CrossRef]
- Fontana, L.; Cummings, N.E.; Arriola Apelo, S.I.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Piper, M.D.W.; Soultoukis, G.A.; Blanc, E.; Mesaros, A.; Herbert, S.L.; Juricic, P.; He, X.; Atanassov, I.; Salmonowicz, H.; Yang, M.; et al. Matching Dietary Amino Acid Balance to the In Silico-Translated Exome Optimizes Growth and Reproduction without Cost to Lifespan. Cell Metab. 2017, 25, 1206. [Google Scholar] [CrossRef] [PubMed]
- Wali, J.A.; Jarzebska, N.; Raubenheimer, D.; Simpson, S.J.; Rodionov, R.N.; O’Sullivan, J.F. Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms-A Narrative Review. Nutrients 2020, 12, 1505. [Google Scholar] [CrossRef]
- Westman, E.C. Is dietary carbohydrate essential for human nutrition? Am. J. Clin. Nutr. 2002, 75, 951–953. [Google Scholar] [CrossRef]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef]
- Mente, A.; Dehghan, M.; Rangarajan, S.; McQueen, M.; Dagenais, G.; Wielgosz, A.; Lear, S.; Li, W.; Chen, H.; Yi, S. Association of dietary nutrients with blood lipids and blood pressure in 18 countries: A cross-sectional analysis from the PURE study. Lancet Diabetes Endocrinol. 2017, 5, 774–787. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Hu, F.B.; Tappy, L.; Brand-Miller, J. Dietary carbohydrates: Role of quality and quantity in chronic disease. BMJ 2018, 361, k2340. [Google Scholar] [CrossRef]
- Keenan, M.J.; Zhou, J.; Hegsted, M.; Pelkman, C.; Durham, H.A.; Coulon, D.B.; Martin, R.J. Role of resistant starch in improving gut health, adiposity, and insulin resistance. Adv. Nutr. 2015, 6, 198–205. [Google Scholar] [CrossRef]
- Reynolds, A.; Mann, J.; Cummings, J.; Winter, N.; Mete, E.; Te Morenga, L. Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 2019, 393, 434–445. [Google Scholar] [CrossRef]
- Choo, V.L.; Viguiliouk, E.; Blanco Mejia, S.; Cozma, A.I.; Khan, T.A.; Ha, V.; Wolever, T.M.S.; Leiter, L.A.; Vuksan, V.; Kendall, C.W.C.; et al. Food sources of fructose-containing sugars and glycaemic control: Systematic review and meta-analysis of controlled intervention studies. BMJ 2018, 363. [Google Scholar] [CrossRef]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Blaut, M. Gut microbiota and energy balance: Role in obesity. Proc. Nutr. Soc. 2015, 74, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I. Dietary Fibre and Incidence of Type 2 Diabetes in Eight European Countries: The EPIC-InterAct Study and a Meta-analysis of Prospective Studies; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- King, D.E.; Mainous III, A.G.; Lambourne, C.A. Trends in dietary fiber intake in the United States, 1999–2008. J. Acad. Nutr. Diet. 2012, 112, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.X.; Zhou, J.W.; Liu, Y.L.; Lu, X.B.; Han, C.X.; Zhang, W.Y.; Xu, Y.H.; Yan, Y.M. Biosynthesis and Regulation of Wheat Amylose and Amylopectin from Proteomic and Phosphoproteomic Characterization of Granule-binding Proteins. Sci. Rep. 2016, 6, 33111. [Google Scholar] [CrossRef]
- Aller, E.E.; Abete, I.; Astrup, A.; Martinez, J.A.; Baak, M.A.v. Starches, sugars and obesity. Nutrients 2011, 3, 341–369. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, S.H.; Rao, M.B.; Deshpande, V.V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 1996, 60, 280–300. [Google Scholar] [CrossRef]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The Third National Health and Nutrition Examination Survey. Medscape J. Med. 2008, 10, 160. [Google Scholar]
- Stanhope, K.L.; Griffen, S.C.; Bair, B.R.; Swarbrick, M.M.; Keim, N.L.; Havel, P.J. Twenty-four-hour endocrine and metabolic profiles following consumption of high-fructose corn syrup-, sucrose-, fructose-, and glucose-sweetened beverages with meals. Am. J. Clin. Nutr. 2008, 87, 1194–1203. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef]
- Carden, T.J.; Carr, T.P. Food availability of glucose and fat, but not fructose, increased in the U.S. between 1970 and 2009: Analysis of the USDA food availability data system. Nutr. J. 2013, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar] [CrossRef] [PubMed]
- De Koning, L.; Malik, V.S.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Sugar-sweetened and artificially sweetened beverage consumption and risk of type 2 diabetes in men. Am. J. Clin. Nutr. 2011, 93, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.; Imamura, F.; Lentjes, M.A.; Khaw, K.-T.; Wareham, N.J.; Forouhi, N.G. Prospective associations and population impact of sweet beverage intake and type 2 diabetes, and effects of substitutions with alternative beverages. Diabetologia 2015, 58, 1474–1483. [Google Scholar] [CrossRef]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.-P.; Willett, W.C.; Hu, F.B. Sugar sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483. [Google Scholar] [CrossRef]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef]
- Stanhope, K.L. More pieces of the fructose puzzle. J. Intern. Med. 2017, 282, 202–204. [Google Scholar] [CrossRef]
- Rippe, J.M.; Angelopoulos, T.J. Fructose-containing sugars and cardiovascular disease. Adv. Nutr. 2015, 6, 430–439. [Google Scholar] [CrossRef]
- Lustig, R.H. Fructose: It’s “Alcohol Without the Buzz”; Oxford University Press: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- Lustig, R.H. Sickeningly Sweet: Does Sugar Cause Type 2 Diabetes? Yes. Can J. Diabetes 2016, 40, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Te Morenga, L.; Mallard, S.; Mann, J. Dietary sugars and body weight: Systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ 2013, 346, e7492. [Google Scholar] [CrossRef]
- Lean, M.E.; Te Morenga, L. Sugar and Type 2 diabetes. Br. Med. Bull. 2016, 120, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Rosset, R.; Surowska, A.; Tappy, L. Pathogenesis of Cardiovascular and Metabolic Diseases: Are Fructose-Containing Sugars More Involved Than Other Dietary Calories? Curr. Hypertens Rep. 2016, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.J.; Heitmann, B.L. Intake of calorically sweetened beverages and obesity. Obes. Rev. 2009, 10, 68–75. [Google Scholar] [CrossRef]
- Reid, M.; Hammersley, R.; Hill, A.J.; Skidmore, P. Long-term dietary compensation for added sugar: Effects of supplementary sucrose drinks over a 4-week period. Br. J. Nutr. 2007, 97, 193–203. [Google Scholar] [CrossRef]
- Mattes, R.D. Dietary compensation by humans for supplemental energy provided as ethanol or carbohydrate in fluids. Physiol. Behav. 1996, 59, 179–187. [Google Scholar] [CrossRef]
- Stice, E.; Burger, K.S.; Yokum, S. Relative ability of fat and sugar tastes to activate reward, gustatory, and somatosensory regions. Am. J. Clin. Nutr. 2013, 98, 1377–1384. [Google Scholar] [CrossRef]
- Luo, S.; Monterosso, J.R.; Sarpelleh, K.; Page, K.A. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc. Natl. Acad. Sci. USA 2015, 112, 6509–6514. [Google Scholar] [CrossRef]
- Avena, N.M.; Rada, P.; Hoebel, B.G. Evidence for sugar addiction: Behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci. Biobehav. Rev. 2008, 32, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Sievenpiper, J.L. Controversies about sugars: Results from systematic reviews and meta-analyses on obesity, cardiometabolic disease and diabetes. Eur. J. Nutr. 2016, 55, 25–43. [Google Scholar] [CrossRef]
- Rippe, J.M.; Kris Etherton, P.M. Fructose, sucrose, and high fructose corn syrup: Modern scientific findings and health implications. Adv. Nutr. 2012, 3, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Vega-López, S.; Venn, B.J.; Slavin, J.L. Relevance of the Glycemic Index and Glycemic Load for Body Weight, Diabetes, and Cardiovascular Disease. Nutrients 2018, 10, 1361. [Google Scholar] [CrossRef] [PubMed]
- Radulian, G.; Rusu, E.; Dragomir, A.; Posea, M. Metabolic effects of low glycaemic index diets. Nutr. J. 2009, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Brand-Miller, J.; Buyken, A.E. The Relationship between Glycemic Index and Health. Nutrients 2020, 12, 536. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, A.A.; Sexton-Oates, A.; O’Brien, E.C.; Alberdi, G.; Fransquet, P.; Saffery, R.; McAuliffe, F.M. A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial. Nutrients 2018, 10, 455. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, Y.; Wang, L.; Yang, W.; Li, C.; Wang, L.; Gu, P.; Xia, Y.; Yan, J.; Shen, Y.; et al. Maternal dietary glycaemic change during gestation influences insulin-related gene methylation in the placental tissue: A genome-wide methylation analysis. Genes Nutr. 2019, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Lim, S.; Egger, G. The effect of a low glycaemic index breakfast on blood glucose, insulin, lipid profiles, blood pressure, body weight, body composition and satiety in obese and overweight individuals: A pilot study. J. Am. Coll. Nutr. 2008, 27, 387–393. [Google Scholar] [CrossRef]
- Krog-Mikkelsen, I.; Sloth, B.; Dimitrov, D.; Tetens, I.; Björck, I.; Flint, A.; Holst, J.J.; Astrup, A.; Elmståhl, H.; Raben, A. A low glycemic index diet does not affect postprandial energy metabolism but decreases postprandial insulinemia and increases fullness ratings in healthy women. J. Nutr. 2011, 141, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Aston, L.M.; Stokes, C.S.; Jebb, S.A. No effect of a diet with a reduced glycaemic index on satiety, energy intake and body weight in overweight and obese women. Int. J. Obes. 2008, 32, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Gilhooly, C.H.; Golden, J.K.; Pittas, A.G.; Fuss, P.J.; Cheatham, R.A.; Tyler, S.; Tsay, M.; McCrory, M.A.; Lichtenstein, A.H.; et al. Long-term effects of 2 energy-restricted diets differing in glycemic load on dietary adherence, body composition, and metabolism in CALERIE: A 1-y randomized controlled trial. Am. J. Clin. Nutr. 2007, 85, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Juanola-Falgarona, M.; Salas-Salvadó, J.; Ibarrola-Jurado, N.; Rabassa-Soler, A.; Díaz-López, A.; Guasch-Ferré, M.; Hernández-Alonso, P.; Balanza, R.; Bulló, M. Effect of the glycemic index of the diet on weight loss, modulation of satiety, inflammation, and other metabolic risk factors: A randomized controlled trial. Am. J. Clin. Nutr. 2014, 100, 27–35. [Google Scholar] [CrossRef]
- Murakami, K.; Sasaki, S.; Okubo, H.; Takahashi, Y.; Hosoi, Y.; Itabashi, M. Dietary fiber intake, dietary glycemic index and load, and body mass index: A cross-sectional study of 3931 Japanese women aged 18-20 years. Eur. J. Clin. Nutr. 2007, 61, 986–995. [Google Scholar] [CrossRef]
- Silva, F.M.; Steemburgo, T.; de Mello, V.D.; Tonding, S.F.; Gross, J.L.; Azevedo, M.J. High dietary glycemic index and low fiber content are associated with metabolic syndrome in patients with type 2 diabetes. J. Am. Coll Nutr. 2011, 30, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Abete, I.; Parra, D.; Martinez, J.A. Energy-restricted diets based on a distinct food selection affecting the glycemic index induce different weight loss and oxidative response. Clin. Nutr. 2008, 27, 545–551. [Google Scholar] [CrossRef]
- Cheng, I.S.; Liao, S.F.; Liu, K.L.; Liu, H.Y.; Wu, C.L.; Huang, C.Y.; Mallikarjuna, K.; Smith, R.W.; Kuo, C.H. Effect of dietary glycemic index on substrate transporter gene expression in human skeletal muscle after exercise. Eur. J. Clin. Nutr. 2009, 63, 1404–1410. [Google Scholar] [CrossRef][Green Version]
- Mendez, M.A.; Covas, M.I.; Marrugat, J.; Vila, J.; Schröder, H. Glycemic load, glycemic index, and body mass index in Spanish adults. Am. J. Clin. Nutr. 2009, 89, 316–322. [Google Scholar] [CrossRef]
- Milton, J.E.; Briche, B.; Brown, I.J.; Hickson, M.; Robertson, C.E.; Frost, G.S. Relationship of glycaemic index with cardiovascular risk factors: Analysis of the National Diet and Nutrition Survey for people aged 65 and older. Public Health Nutr. 2007, 10, 1321–1335. [Google Scholar] [CrossRef]
- Castro-Quezada, I.; Artacho, R.; Molina-Montes, E.; Serrano, F.A.; Ruiz-López, M.D. Dietary glycaemic index and glycaemic load in a rural elderly population (60-74 years of age) and their relationship with cardiovascular risk factors. Eur. J. Nutr. 2015, 54, 523–534. [Google Scholar] [CrossRef]
- Karl, J.P.; Roberts, S.B.; Schaefer, E.J.; Gleason, J.A.; Fuss, P.; Rasmussen, H.; Saltzman, E.; Das, S.K. Effects of carbohydrate quantity and glycemic index on resting metabolic rate and body composition during weight loss. Obesity 2015, 23, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, S.; Cosentino, L.; Rosafio, G.; Morgana, M.; Mattina, A.; Sprini, D.; Verga, S.; Rini, G.B. Effects of hypocaloric diets with different glycemic indexes on endothelial function and glycemic variability in overweight and in obese adult patients at increased cardiovascular risk. Clin. Nutr. 2013, 32, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Philippou, E.; McGowan, B.M.; Brynes, A.E.; Dornhorst, A.; Leeds, A.R.; Frost, G.S. The effect of a 12-week low glycaemic index diet on heart disease risk factors and 24 h glycaemic response in healthy middle-aged volunteers at risk of heart disease: A pilot study. Eur. J. Clin. Nutr. 2008, 62, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Sichieri, R.; Moura, A.S.; Genelhu, V.; Hu, F.; Willett, W.C. An 18-mo randomized trial of a low-glycemic-index diet and weight change in Brazilian women. Am. J. Clin. Nutr. 2007, 86, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Villegas, R.; Liu, S.; Gao, Y.T.; Yang, G.; Li, H.; Zheng, W.; Shu, X.O. Prospective study of dietary carbohydrates, glycemic index, glycemic load, and incidence of type 2 diabetes mellitus in middle-aged Chinese women. Arch. Intern. Med. 2007, 167, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Nakamura, K.; Miura, K.; Takamura, T.; Yoshita, K.; Morikawa, Y.; Ishizaki, M.; Kido, T.; Naruse, Y.; Suwazono, Y.; et al. Dietary glycemic index and risk of type 2 diabetes mellitus in middle-aged Japanese men. Metabolism 2012, 61, 47–55. [Google Scholar] [CrossRef]
- Barclay, A.W.; Flood, V.M.; Rochtchina, E.; Mitchell, P.; Brand-Miller, J.C. Glycemic index, dietary fiber, and risk of type 2 diabetes in a cohort of older Australians. Diabetes Care 2007, 30, 2811–2813. [Google Scholar] [CrossRef]
- Mosdøl, A.; Witte, D.R.; Frost, G.; Marmot, M.G.; Brunner, E.J. Dietary glycemic index and glycemic load are associated with high-density-lipoprotein cholesterol at baseline but not with increased risk of diabetes in the Whitehall II study. Am. J. Clin. Nutr. 2007, 86, 988–994. [Google Scholar] [CrossRef]
- Jenkins, D.J.; Kendall, C.W.; McKeown-Eyssen, G.; Josse, R.G.; Silverberg, J.; Booth, G.L.; Vidgen, E.; Josse, A.R.; Nguyen, T.H.; Corrigan, S.; et al. Effect of a low-glycemic index or a high-cereal fiber diet on type 2 diabetes: A randomized trial. JAMA 2008, 300, 2742–2753. [Google Scholar] [CrossRef]
- Wolever, T.M.; Gibbs, A.L.; Mehling, C.; Chiasson, J.L.; Connelly, P.W.; Josse, R.G.; Leiter, L.A.; Maheux, P.; Rabasa-Lhoret, R.; Rodger, N.W.; et al. The Canadian Trial of Carbohydrates in Diabetes (CCD), a 1-y controlled trial of low-glycemic-index dietary carbohydrate in type 2 diabetes: No effect on glycated hemoglobin but reduction in C-reactive protein. Am. J. Clin. Nutr. 2008, 87, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Bantle, J.P. Is fructose the optimal low glycemic index sweetener? Nestle Nutr. Workshop Ser. Clin. Perform Programme 2006, 11, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Livesey, G.; Taylor, R.; Livesey, H.F.; Buyken, A.E.; Jenkins, D.J.A.; Augustin, L.S.A.; Sievenpiper, J.L.; Barclay, A.W.; Liu, S.; Wolever, T.M.S.; et al. Dietary Glycemic Index and Load and the Risk of Type 2 Diabetes: A Systematic Review and Updated Meta-Analyses of Prospective Cohort Studies. Nutrients 2019, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.J.A.; Dehghan, M.; Mente, A.; Bangdiwala, S.I.; Rangarajan, S.; Srichaikul, K.; Mohan, V.; Avezum, A.; Díaz, R.; Rosengren, A.; et al. Glycemic Index, Glycemic Load, and Cardiovascular Disease and Mortality. N. Engl. J. Med. 2021, 384, 1312–1322. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef]
- Bindels, L.B.; Walter, J.; Ramer-Tait, A.E. Resistant starches for the management of metabolic diseases. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 559–565. [Google Scholar] [CrossRef]
- Kieffer, D.A.; Piccolo, B.D.; Marco, M.L.; Kim, E.B.; Goodson, M.L.; Keenan, M.J.; Dunn, T.N.; Knudsen, K.E.; Martin, R.J.; Adams, S.H. Mice Fed a High-Fat Diet Supplemented with Resistant Starch Display Marked Shifts in the Liver Metabolome Concurrent with Altered Gut Bacteria. J. Nutr. 2016, 146, 2476–2490. [Google Scholar] [CrossRef]
- Koay, Y.C.; Wali, J.A.; Luk, A.W.S.; Macia, L.; Cogger, V.C.; Pulpitel, T.J.; Wahl, D.; Solon-Biet, S.M.; Holmes, A.; Simpson, S.J.; et al. Ingestion of resistant starch by mice markedly increases microbiome-derived metabolites. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 8033–8042. [Google Scholar] [CrossRef]
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. The microbial metabolite indole-3-propionic acid improves glucose metabolism in rats, but does not affect behaviour. Arch. Physiol. Biochem. 2018, 124, 306–312. [Google Scholar] [CrossRef]
- Robertson, M.D.; Bickerton, A.S.; Dennis, A.L.; Vidal, H.; Frayn, K.N. Insulin-sensitizing effects of dietary resistant starch and effects on skeletal muscle and adipose tissue metabolism. Am. J. Clin. Nutr. 2005, 82, 559–567. [Google Scholar] [CrossRef]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef]
- Li, H.; Gao, Z.; Zhang, J.; Ye, X.; Xu, A.; Ye, J.; Jia, W. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes 2012, 61, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kishi, M.; Fushimi, T.; Ugajin, S.; Kaga, T. Vinegar intake reduces body weight, body fat mass, and serum triglyceride levels in obese Japanese subjects. Biosci. Biotechnol. Biochem. 2009, 73, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Graff, E.C.; Judd, R.L. Effects of high fat diet on GPR109A and GPR81 gene expression. Biochem. Biophys. Res. Commun. 2012, 425, 278–283. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Jones, M.A.; Fromal, O.; Powell, F.L.; Khurana, S.; Singh, N.; Martin, P.M. Loss of GPR109A/HCAR2 induces aging-associated hepatic steatosis. Aging 2019, 11, 386–400. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Marek, G.; Pannu, V.; Shanmugham, P.; Pancione, B.; Mascia, D.; Crosson, S.; Ishimoto, T.; Sautin, Y.Y. Adiponectin resistance and proinflammatory changes in the visceral adipose tissue induced by fructose consumption via ketohexokinase-dependent pathway. Diabetes 2015, 64, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, H.; Haass, W.; Castaneda, T.R.; Schurmann, A.; Koebnick, C.; Dombrowski, F.; Otto, B.; Nawrocki, A.R.; Scherer, P.E.; Spranger, J.; et al. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes. Res. 2005, 13, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Faeh, D.; Minehira, K.; Schwarz, J.-M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef]
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001, 15, 1206–1216. [Google Scholar] [CrossRef]
- Yang, J.; Goldstein, J.L.; Hammer, R.E.; Moon, Y.A.; Brown, M.S.; Horton, J.D. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc. Natl. Acad. Sci. USA 2001, 98, 13607–13612. [Google Scholar] [CrossRef]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e734. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Lu, C.; Tutnauer, J.; Hartman, T.E.; Hwang, S.-K.; Murphy, C.J.; Pauli, C.; Morris, R.; Taylor, S.; Bosch, K. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019, 363, 1345–1349. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and-independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Bortolin, R.C.; Vargas, A.R.; Gasparotto, J.; Chaves, P.R.; Schnorr, C.E.; Martinello, K.B.; Silveira, A.K.; Rabelo, T.K.; Gelain, D.P.; Moreira, J.C.F. A new animal diet based on human Western diet is a robust diet-induced obesity model: Comparison to high-fat and cafeteria diets in term of metabolic and gut microbiota disruption. Int. J. Obes. 2018, 42, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.; Feinglos, M.; Rodin, J.; Sutherland, A.; Petro, A.; Opara, E.; Kuhn, C.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and AJ mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Kraegen, E.W.; Cooney, G.J. Free fatty acids and skeletal muscle insulin resistance. Curr. Opin. Lipidol. 2008, 19, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Yanagita, T. Medium-chain fatty acids: Functional lipids for the prevention and treatment of the metabolic syndrome. Pharm. Res. 2010, 61, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Aidaros, A.A.; Sharma, C.; Langhans, C.D.; J, G.O.; Hoffmann, G.F.; Dasouki, M.; Chakraborty, P.; Aljasmi, F.; O, Y.A.-D. Targeted Metabolomic Profiling of Total Fatty Acids in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry. Metabolites 2020, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Kish-Trier, E.; Schwarz, E.L.; Pasquali, M.; Yuzyuk, T. Quantitation of total fatty acids in plasma and serum by GC-NCI-MS. Clin. Mass Spectrom. 2016, 2, 11–17. [Google Scholar] [CrossRef]
- 2015–2020 Dietary Guidelines for Americans; U.S. Department of Health and Human Services (HHS); U.S. Department of Agriculture: Washington, DC, USA, 2015.
- Small, L.; Brandon, A.E.; Turner, N.; Cooney, G.J. Modeling insulin resistance in rodents by alterations in diet: What have high-fat and high-calorie diets revealed? Am. J. Physiol. Endocrinol. Metab. 2018, 314, E251–E265. [Google Scholar] [CrossRef] [PubMed]
- Fiamoncini, J.; Turner, N.; Hirabara, S.M.; Salgado, T.M.; Marcal, A.C.; Leslie, S.; da Silva, S.M.; Deschamps, F.C.; Luz, J.; Cooney, G.J.; et al. Enhanced peroxisomal beta-oxidation is associated with prevention of obesity and glucose intolerance by fish oil-enriched diets. Obesity 2013, 21, 1200–1207. [Google Scholar] [CrossRef]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Dube, J.J.; Coen, P.M.; DiStefano, G.; Chacon, A.C.; Helbling, N.L.; Desimone, M.E.; Stafanovic-Racic, M.; Hames, K.C.; Despines, A.A.; Toledo, F.G.; et al. Effects of acute lipid overload on skeletal muscle insulin resistance, metabolic flexibility, and mitochondrial performance. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1117–E1124. [Google Scholar] [CrossRef]
- Hoeg, L.D.; Sjoberg, K.A.; Jeppesen, J.; Jensen, T.E.; Frosig, C.; Birk, J.B.; Bisiani, B.; Hiscock, N.; Pilegaard, H.; Wojtaszewski, J.F.; et al. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes 2011, 60, 64–73. [Google Scholar] [CrossRef]
- De Vogel-van den Bosch, J.; van den Berg, S.A.; Bijland, S.; Voshol, P.J.; Havekes, L.M.; Romijn, H.A.; Hoeks, J.; van Beurden, D.; Hesselink, M.K.; Schrauwen, P.; et al. High-fat diets rich in medium- versus long-chain fatty acids induce distinct patterns of tissue specific insulin resistance. J. Nutr. Biochem. 2011, 22, 366–371. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Osborne, B.; Brown, S.H.; Small, L.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J. Lipid Res. 2013, 54, 3322–3333. [Google Scholar] [CrossRef]
- Turner, N.; Hariharan, K.; TidAng, J.; Frangioudakis, G.; Beale, S.M.; Wright, L.E.; Zeng, X.Y.; Leslie, S.J.; Li, J.Y.; Kraegen, E.W.; et al. Enhancement of muscle mitochondrial oxidative capacity and alterations in insulin action are lipid species dependent: Potent tissue-specific effects of medium-chain fatty acids. Diabetes 2009, 58, 2547–2554. [Google Scholar] [CrossRef]
- Liu, M.; Montgomery, M.K.; Fiveash, C.E.; Osborne, B.; Cooney, G.J.; Bell-Anderson, K.; Turner, N. PPARalpha-independent actions of omega-3 PUFAs contribute to their beneficial effects on adiposity and glucose homeostasis. Sci. Rep. 2014, 4, 5538. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, L.; Wang, Z.; Zheng, X.; Hou, Z.; Zhao, D.; Long, J.; Liu, J. Omega-3 polyunsaturated fatty acids prevent obesity by improving tricarboxylic acid cycle homeostasis. J. Nutr. Biochem. 2020, 88, 108503. [Google Scholar] [CrossRef] [PubMed]
- Rudy, L.; Carmen, R.; Daniel, R.; Artemio, R.; Moises, R.O. Anticonvulsant mechanisms of the ketogenic diet and caloric restriction. Epilepsy Res. 2020, 168, 106499. [Google Scholar] [CrossRef] [PubMed]
- Deemer, S.E.; Plaisance, E.P.; Martins, C. Impact of ketosis on appetite regulation-a review. Nutr. Res. 2020, 77, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lundsgaard, A.M.; Holm, J.B.; Sjoberg, K.A.; Bojsen-Moller, K.N.; Myrmel, L.S.; Fjaere, E.; Jensen, B.A.H.; Nicolaisen, T.S.; Hingst, J.R.; Hansen, S.L.; et al. Mechanisms Preserving Insulin Action during High Dietary Fat Intake. Cell Metab. 2019, 29, 229. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef]
- Brandon, A.E.; Liao, B.M.; Diakanastasis, B.; Parker, B.L.; Raddatz, K.; McManus, S.A.; O’Reilly, L.; Kimber, E.; van der Kraan, A.G.; Hancock, D.; et al. Protein Kinase C Epsilon Deletion in Adipose Tissue, but Not in Liver, Improves Glucose Tolerance. Cell Metab. 2019, 29, 183–191.e187. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C. Deconstructing the Role of PKC Epsilon in Glucose Homeostasis. Trends Endocrinol. Metab. 2020, 31, 344–356. [Google Scholar] [CrossRef]
- Straczkowski, M.; Kowalska, I.; Nikolajuk, A.; Dzienis-Straczkowska, S.; Kinalska, I.; Baranowski, M.; Zendzian-Piotrowska, M.; Brzezinska, Z.; Gorski, J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 2004, 53, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Ceramides: Nutrient Signals that Drive Hepatosteatosis. J. Lipid Atheroscler. 2020, 9, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Turpin-Nolan, S.M.; Hammerschmidt, P.; Chen, W.; Jais, A.; Timper, K.; Awazawa, M.; Brodesser, S.; Bruning, J.C. CerS1-Derived C18:0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin Resistance. Cell Rep. 2019, 26, 1–10.e17. [Google Scholar] [CrossRef] [PubMed]
- Tonks, K.T.; Coster, A.C.; Christopher, M.J.; Chaudhuri, R.; Xu, A.; Gagnon-Bartsch, J.; Chisholm, D.J.; James, D.E.; Meikle, P.J.; Greenfield, J.R.; et al. Skeletal muscle and plasma lipidomic signatures of insulin resistance and overweight/obesity in humans. Obesity 2016, 24, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Hallahan, N.L.; Brown, S.H.; Liu, M.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 2013, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Brown, S.H.; Lim, X.Y.; Fiveash, C.E.; Osborne, B.; Bentley, N.L.; Braude, J.P.; Mitchell, T.W.; Coster, A.C.; Don, A.S.; et al. Regulation of glucose homeostasis and insulin action by ceramide acyl-chain length: A beneficial role for very long-chain sphingolipid species. Biochim. Biophys. Acta 2016, 1861, 1828–1839. [Google Scholar] [CrossRef]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Boden, M.J.; Brandon, A.E.; Tid-Ang, J.D.; Preston, E.; Wilks, D.; Stuart, E.; Cleasby, M.E.; Turner, N.; Cooney, G.J.; Kraegen, E.W. Overexpression of manganese superoxide dismutase ameliorates high-fat diet-induced insulin resistance in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E798–E805. [Google Scholar] [CrossRef]
- Hoehn, K.L.; Salmon, A.B.; Hohnen-Behrens, C.; Turner, N.; Hoy, A.J.; Maghzal, G.J.; Stocker, R.; Van Remmen, H.; Kraegen, E.W.; Cooney, G.J.; et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 17787–17792. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Espinosa, A.; Gonzalez-Manan, D.; D’Espessailles, A.; Fernandez, V.; Videla, L.A.; Tapia, G. N-3 long-chain polyunsaturated fatty acid supplementation significantly reduces liver oxidative stress in high fat induced steatosis. PLoS ONE 2012, 7, e46400. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- Samocha-Bonet, D.; Chisholm, D.J.; Tonks, K.; Campbell, L.V.; Greenfield, J.R. Insulin-sensitive obesity in humans—A ‘favorable fat’ phenotype? Trends Endocrinol. Metab. 2012, 23, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Begaye, B.; Vinales, K.L.; Hollstein, T.; Ando, T.; Walter, M.; Bogardus, C.; Krakoff, J.; Piaggi, P. Impaired Metabolic Flexibility to High-Fat Overfeeding Predicts Future Weight Gain in Healthy Adults. Diabetes 2020, 69, 181–192. [Google Scholar] [CrossRef]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed]
- Rinnankoski-Tuikka, R.; Silvennoinen, M.; Torvinen, S.; Hulmi, J.J.; Lehti, M.; Kivela, R.; Reunanen, H.; Kainulainen, H. Effects of high-fat diet and physical activity on pyruvate dehydrogenase kinase-4 in mouse skeletal muscle. Nutr. Metab. 2012, 9, 53. [Google Scholar] [CrossRef]
- Reznick, J.; Preston, E.; Wilks, D.L.; Beale, S.M.; Turner, N.; Cooney, G.J. Altered feeding differentially regulates circadian rhythms and energy metabolism in liver and muscle of rats. Biochim. Biophys. Acta 2013, 1832, 228–238. [Google Scholar] [CrossRef]
- Kim, Y.I.; Lee, F.N.; Choi, W.S.; Lee, S.; Youn, J.H. Insulin regulation of skeletal muscle PDK4 mRNA expression is impaired in acute insulin-resistant states. Diabetes 2006, 55, 2311–2317. [Google Scholar] [CrossRef]
- Nellemann, B.; Vendelbo, M.H.; Nielsen, T.S.; Bak, A.M.; Hogild, M.; Pedersen, S.B.; Bienso, R.S.; Pilegaard, H.; Moller, N.; Jessen, N.; et al. Growth hormone-induced insulin resistance in human subjects involves reduced pyruvate dehydrogenase activity. Acta Physiol. 2014, 210, 392–402. [Google Scholar] [CrossRef]
- Lee, I.K. The role of pyruvate dehydrogenase kinase in diabetes and obesity. Diabetes Metab. J. 2014, 38, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Small, L.; Brandon, A.E.; Quek, L.E.; Krycer, J.R.; James, D.E.; Turner, N.; Cooney, G.J. Acute activation of pyruvate dehydrogenase increases glucose oxidation in muscle without changing glucose uptake. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E258–E266. [Google Scholar] [CrossRef]
- Wali, J.A.; Raubenheimer, D.; Senior, A.M.; Le Couteur, D.G.; Simpson, S.J. Cardio-Metabolic Consequences of Dietary Carbohydrates: Reconciling Contradictions Using Nutritional Geometry. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Batley, R.; Raubenheimer, D. Geometric analysis of macronutrient intake in humans: The power of protein? Appetite 2003, 41, 123–140. [Google Scholar] [CrossRef]
- Simpson, S.J.; Raubenheimer, D. Obesity: The protein leverage hypothesis. Obes. Rev. 2005, 6, 133–142. [Google Scholar] [CrossRef]
- Dussutour, A.; Simpson, S.J. Communal Nutrition in Ants. Curr. Biol. 2009, 19, 740–744. [Google Scholar] [CrossRef]
- Shariatmadari, F.; Forbes, J.M. Growth and food intake responses to diets of different protein contents and a choice between diets containing two concentrations of protein in broiler and layer strains of chicken. Br. Poult. Sci. 1993, 34, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.; Mayntz, D.; Raubenheimer, D.; Simpson, S.J. Protein-leverage in mice: The geometry of macronutrient balancing and consequences for fat deposition. Obesity 2008, 16, 566–571. [Google Scholar] [CrossRef]
- Gosby, A.K.; Conigrave, A.D.; Lau, N.S.; Iglesias, M.A.; Hall, R.M.; Jebb, S.A.; Brand-Miller, J.; Caterson, I.D.; Raubenheimer, D.; Simpson, S.J. Testing Protein Leverage in Lean Humans: A Randomised Controlled Experimental Study. PLoS ONE 2011, 6, e25929. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.P.; Klein, S. Pathogenesis and Pathophysiology of the Cardiometabolic Syndrome. J. Clin. Hypertens. 2009, 11, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Solon-Biet, S.M.; Mitchell, S.J.; Coogan, S.C.P.; Cogger, V.C.; Gokarn, R.; McMahon, A.C.; Raubenheimer, D.; de Cabo, R.; Simpson, S.J.; Le Couteur, D.G. Dietary Protein to Carbohydrate Ratio and Caloric Restriction: Comparing Metabolic Outcomes in Mice. Cell Rep. 2015, 11, 1529–1534. [Google Scholar] [CrossRef]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Harber, M.P.; Shrivastava, C.R.; Burant, C.F.; Horowitz, J.F. Improved insulin sensitivity after weight loss and exercise training is mediated by a reduction in plasma fatty acid mobilization, not enhanced oxidative capacity. J. Physiol. 2009, 587, 4949–4961. [Google Scholar] [CrossRef]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation 2014, 129, S102–S138. [Google Scholar] [CrossRef]
- Mann, J.I. Nutrition Recommendations for the Treatment and Prevention of Type 2 Diabetes and the Metabolic Syndrome: An Evidenced-Based Review. Nutr. Rev. 2006, 64, 422–427. [Google Scholar] [CrossRef]
- Goldstein, D.J. Beneficial health effects of modest weight loss. Int. J. Obes. Relat. Metab. Disord. 1992, 16, 397–415. [Google Scholar]
- Leidy, H.J.; Carnell, N.S.; Mattes, R.D.; Campbell, W.W. Higher Protein Intake Preserves Lean Mass and Satiety with Weight Loss in Pre-obese and Obese Women. Obesity 2007, 15, 421–429. [Google Scholar] [CrossRef] [PubMed]
- González-Salazar, L.E.; Pichardo-Ontiveros, E.; Palacios-González, B.; Vigil-Martínez, A.; Granados-Portillo, O.; Guizar-Heredia, R.; Flores-López, A.; Medina-Vera, I.; Heredia-G-Cantón, P.K.; Hernández-Gómez, K.G.; et al. Effect of the intake of dietary protein on insulin resistance in subjects with obesity: A randomized controlled clinical trial. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Abete, I.; Parra, D.; Martinez, J.A. Legume-, Fish-, or High-Protein-Based Hypocaloric Diets: Effects on Weight Loss and Mitochondrial Oxidation in Obese Men. J. Med. Food 2009, 12, 100–108. [Google Scholar] [CrossRef]
- Ratliff, J.; Mutungi, G.; Puglisi, M.J.; Volek, J.S.; Fernandez, M.L. Carbohydrate restriction (with or without additional dietary cholesterol provided by eggs) reduces insulin resistance and plasma leptin without modifying appetite hormones in adult men. Nutr. Res. 2009, 29, 262–268. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef]
- van Nielen, M.; Feskens, E.J.; Mensink, M.; Sluijs, I.; Molina, E.; Amiano, P.; Ardanaz, E.; Balkau, B.; Beulens, J.W.; Boeing, H.; et al. Dietary protein intake and incidence of type 2 diabetes in Europe: The EPIC-InterAct Case-Cohort Study. Diabetes Care 2014, 37, 1854–1862. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Suzuki, T.; Monno, I.; Kanasaki, K.; Watanabe, A.; Koya, D. A low-protein diet exerts a beneficial effect on diabetic status and prevents diabetic nephropathy in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Nutr. Metab. 2018, 15, 20. [Google Scholar] [CrossRef]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.-W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low Protein Intake Is Associated with a Major Reduction in IGF-1, Cancer, and Overall Mortality in the 65 and Younger but Not Older Population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Wilcox, W.R.; Cederbaum, S.D. Amino acid metabolism. In Emery and Rimoin’s Principles and Practice of Medical Genetics; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1–42. [Google Scholar]
- Wu, G.; Bazer, F.W.; Dai, Z.; Li, D.; Wang, J.; Wu, Z. Amino acid nutrition in animals: Protein synthesis and beyond. Annu. Rev. Anim. Biosci. 2014, 2, 387–417. [Google Scholar] [CrossRef]
- Richie, J., Jr.; Leutzinger, Y.; Parthasarathy, S.; Malloy, V.; Orentreich, N.; Zimmerman, J. Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J. 1994, 8, 1302–1307. [Google Scholar] [CrossRef]
- Sun, L.; Sadighi Akha, A.A.; Miller, R.A.; Harper, J.M. Life-Span Extension in Mice by Preweaning Food Restriction and by Methionine Restriction in Middle Age. J. Gerontology. Ser. Abiological Sci. Med. Sci. 2009, 64A, 711–722. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Reviews. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Ribeiro, R.; Senior, A.; Hsu, B.; Hirani, V.; Blyth, F.M.; Waite, L.M.; Simpson, S.J.; Naganathan, V.; Cumming, R.G.; et al. Branched chain amino acids, cardiometabolic risk factors and outcomes in older men: The Concord Health and Ageing in Men Project. J. Gerontol. Ser. Abiological Sci. Med. Sci. 2019. [Google Scholar] [CrossRef]
- Neinast, M.; Murashige, D.; Arany, Z. Branched Chain Amino Acids. Annu. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef]
- Bifari, F.; Nisoli, E. Branched-chain amino acids differently modulate catabolic and anabolic states in mammals: A pharmacological point of view. Br. J. Pharmacol. 2017, 174, 1366–1377. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef]
- Ribeiro, R.V.; Solon-Biet, S.M.; Pulpitel, T.; Senior, A.M.; Cogger, V.C.; Clark, X.; O’Sullivan, J.; Koay, Y.C.; Hirani, V.; Blyth, F.M.; et al. Of Older Mice and Men: Branched-Chain Amino Acids and Body Composition. Nutrients 2019, 11, 1882. [Google Scholar] [CrossRef]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef]
- Huffman, K.M.; Shah, S.H.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.; Slentz, C.A.; Tanner, C.J.; Kuchibhatla, M.; Houmard, J.A.; Newgard, C.B.; et al. Relationships Between Circulating Metabolic Intermediates and Insulin Action in Overweight to Obese, Inactive Men and Women. Diabetes Care 2009, 32, 1678–1683. [Google Scholar] [CrossRef]
- Connelly, M.A.; Wolak-Dinsmore, J.; Dullaart, R.P.F. Branched Chain Amino Acids Are Associated with Insulin Resistance Independent of Leptin and Adiponectin in Subjects with Varying Degrees of Glucose Tolerance. Metab. Syndr. Relat. Disord. 2017, 15, 183–186. [Google Scholar] [CrossRef]
- Elshorbagy, A.K.; Samocha-Bonet, D.; Jernerén, F.; Turner, C.; Refsum, H.; Heilbronn, L.K. Food Overconsumption in Healthy Adults Triggers Early and Sustained Increases in Serum Branched-Chain Amino Acids and Changes in Cysteine Linked to Fat Gain. J. Nutr. 2018, 148, 1073–1080. [Google Scholar] [CrossRef]
- Stöckli, J.; Fisher-Wellman, K.H.; Chaudhuri, R.; Zeng, X.-Y.; Fazakerley, D.J.; Meoli, C.C.; Thomas, K.C.; Hoffman, N.J.; Mangiafico, S.P.; Xirouchaki, C.E.; et al. Metabolomic analysis of insulin resistance across different mouse strains and diets. J. Biol. Chem. 2017, 292, 19135–19145. [Google Scholar] [CrossRef]
- Green, C.L.; Lamming, D.W. Regulation of metabolic health by essential dietary amino acids. Mech. Ageing Dev. 2019, 177, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Cummings, N.E.; Rastelli, A.L.; Gao, F.; Cava, E.; Bertozzi, B.; Spelta, F.; Pili, R.; Fontana, L. Restriction of dietary protein decreases mTORC1 in tumors and somatic tissues of a tumor-bearing mouse xenograft model. Oncotarget 2015, 6, 31233–31240. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Heblinski, M.; Wahl, D.; McMahon, A.C.; Warren, A.; Durrant-Whyte, J.; Walters, K.A.; Krycer, J.R.; et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab. 2016, 24, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Laeger, T.; Henagan, T.M.; Albarado, D.C.; Redman, L.M.; Bray, G.A.; Noland, R.C.; Münzberg, H.; Hutson, S.M.; Gettys, T.W.; Schwartz, M.W.; et al. FGF21 is an endocrine signal of protein restriction. J. Clin. Investig. 2014, 124, 3913–3922. [Google Scholar] [CrossRef]
- Maida, A.; Zota, A.; Sjoberg, K.A.; Schumacher, J.; Sijmonsma, T.P.; Pfenninger, A.; Christensen, M.M.; Gantert, T.; Fuhrmeister, J.; Rothermel, U.; et al. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J. Clin. Investig. 2016, 126, 3263. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Psaltopoulou, T.; Orfanos, P.; Hsieh, C.C.; Trichopoulos, D. Low-carbohydrate-high-protein diet and long-term survival in a general population cohort. Eur. J. Clin. Nutr. 2007, 61, 575–581. [Google Scholar] [CrossRef]
- Lagiou, P.; Sandin, S.; Weiderpass, E.; Lagiou, A.; Mucci, L.; Trichopoulos, D.; Adami, H.O. Low carbohydrate-high protein diet and mortality in a cohort of Swedish women. J. Intern. Med. 2007, 261, 366–374. [Google Scholar] [CrossRef]
- Fung, T.T.; van Dam, R.M.; Hankinson, S.E.; Stampfer, M.; Willett, W.C.; Hu, F.B. Low-carbohydrate diets and all-cause and cause-specific mortality: Two cohort studies. Ann. Intern. Med. 2010, 153, 289–298. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Winkvist, A.; Eliasson, M.; Jansson, J.H.; Hallmans, G.; Johansson, I.; Lindahl, B.; Lenner, P.; Van Guelpen, B. Low-carbohydrate, high-protein score and mortality in a northern Swedish population-based cohort. Eur. J. Clin. Nutr. 2012, 66, 694–700. [Google Scholar] [CrossRef]
- Willcox, B.J.; Willcox, D.C.; Todoriki, H.; Fujiyoshi, A.; Yano, K.; He, Q.; Curb, J.D.; Suzuki, M. Caloric restriction, the traditional Okinawan diet, and healthy aging. Ann. N. Y. Acad. Sci. 2007, 1114, 434–455. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, D.G.; Solon-Biet, S.; Wahl, D.; Cogger, V.C.; Willcox, B.J.; Willcox, D.C.; Raubenheimer, D.; Simpson, S.J. New Horizons: Dietary protein, ageing and the Okinawan ratio. Age Ageing 2016, 45, 443–447. [Google Scholar] [CrossRef]
- Astrup, A.; Grunwald, G.; Melanson, E.; Saris, W.; Hill, J. The role of low-fat diets in body weight control: A meta-analysis of ad libitum dietary intervention studies. Int. J. Obes. 2000, 24, 1545. [Google Scholar] [CrossRef]
- Simpson, S.J.; Le Couteur, D.G.; Raubenheimer, D. Putting the balance back in diet. Cell 2015, 161, 18–23. [Google Scholar] [CrossRef]
- Katz, D.L. Diets, diatribes, and a dearth of data. Circ. Cardiovasc. Qual. Outcomes 2014, 7, 809–811. [Google Scholar] [CrossRef]
- Mishra, B.N. Secret of eternal youth; Teaching from the centenarian hot spots (“blue zones”). Indian J. Community Med. Off. Publ. Indian Assoc. Prev. Soc. Med. 2009, 34, 273. [Google Scholar] [CrossRef]
- Willcox, D.C.; Willcox, B.J.; Hsueh, W.-C.; Suzuki, M. Genetic determinants of exceptional human longevity: Insights from the Okinawa Centenarian Study. Age 2006, 28, 313–332. [Google Scholar] [CrossRef]
- Seidelmann, S.B.; Claggett, B.; Cheng, S.; Henglin, M.; Shah, A.; Steffen, L.M.; Folsom, A.R.; Rimm, E.B.; Willett, W.C.; Solomon, S.D. Dietary carbohydrate intake and mortality: A prospective cohort study and meta-analysis. Lancet Public Health 2018, 3, e419–e428. [Google Scholar] [CrossRef]
- Kahleova, H.; Dort, S.; Holubkov, R.; Barnard, N. A Plant-Based High-Carbohydrate, Low-Fat Diet in Overweight Individuals in a 16-Week Randomized Clinical Trial: The Role of Carbohydrates. Nutrients 2018, 10, 1302. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Huang, T.; Zheng, J.; Mann, J.; Li, D. Effects of Macronutrient Distribution on Weight and Related Cardiometabolic Profile in Healthy Non-Obese Chinese: A 6-month, Randomized Controlled-Feeding Trial. EBioMedicine 2017, 22, 200–207. [Google Scholar] [CrossRef]
- Nordmann, A.J.; Nordmann, A.; Briel, M.; Keller, U.; Yancy, W.S.; Brehm, B.J.; Bucher, H.C. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2006, 166, 285–293. [Google Scholar] [CrossRef]
- Raubenheimer, D.; Simpson, S.J. Nutritional Ecology and Human Health. Annu. Rev. Nutr. 2016, 36, 603–626. [Google Scholar] [CrossRef] [PubMed]
- Raubenheimer, D.; Simpson, S.J. Protein Leverage: Theoretical Foundations and Ten Points of Clarification. Obesity 2019, 27, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Control, C.f.D. Prevention. Trends in intake of energy and macronutrients–United States, 1971–2000. Mmwr. Morb. Mortal. Wkly. Rep. 2004, 53, 80. [Google Scholar]
- Wright, J.D.; Wang, C.Y. Trends in intake of energy and macronutrients in adults from 1999–2000 through 2007–2008. NCHS Data Briefs 2010, 1–8. [Google Scholar]
- Cao, X.-P.; Tan, C.-C.; Yu, J.-T. Associations of fats and carbohydrates with cardiovascular disease and mortality—PURE and simple? Lancet 2018, 391, 1679–1680. [Google Scholar] [CrossRef]
- Zhai, F.; Wang, H.; Du, S.; He, Y.; Wang, Z.; Ge, K.; Popkin, B.M. Prospective study on nutrition transition in China. Nutr. Rev. 2009, 67, S56–S61. [Google Scholar] [CrossRef]
- Austin, G.L.; Ogden, L.G.; Hill, J.O. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971–2006. Am. J. Clin. Nutr. 2011, 93, 836–843. [Google Scholar] [CrossRef]
- Lieberman, H.R.; Fulgoni, V.L.; Agarwal, S.; Pasiakos, S.M.; Berryman, C.E. Protein intake is more stable than carbohydrate or fat intake across various US demographic groups and international populations. Am. J. Clin. Nutr. 2020. [Google Scholar] [CrossRef]
- Simpson, S.J.; Raubenheimer, D. The Nature of Nutrition. A Unifying Framework form Animal Adaption to Human Obesity; Princeton University Press: Princeton, NY, USA, 2012. [Google Scholar]
- Solon-Biet, S.M.; Walters, K.A.; Simanainen, U.K.; McMahon, A.C.; Ruohonen, K.; Ballard, J.W.; Raubenheimer, D.; Handelsman, D.J.; Le Couteur, D.G.; Simpson, S.J. Macronutrient balance, reproductive function, and lifespan in aging mice. Proc. Natl. Acad. Sci. USA 2015, 112, 3481–3486. [Google Scholar] [CrossRef]
- Lee, K.P.; Simpson, S.J.; Clissold, F.J.; Brooks, R.; Ballard, J.W.; Taylor, P.W.; Soran, N.; Raubenheimer, D. Lifespan and reproduction in Drosophila: New insights from nutritional geometry. Proc. Natl. Acad. Sci. USA 2008, 105, 2498–2503. [Google Scholar] [CrossRef]
- Piper, M.D.; Partridge, L.; Raubenheimer, D.; Simpson, S.J. Dietary restriction and aging: A unifying perspective. Cell Metab. 2011, 14, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Hoxha, S.; Carvalho, G.B.; Yamada, R.; Wang, H.-D.; Karayan, P.; He, S.; Brummel, T.; Kapahi, P.; William, W.J. High carbohydrate–low protein consumption maximizes Drosophila lifespan. Exp. Gerontol. 2013, 48, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Te Morenga, L.; Docherty, P.; Williams, S.; Mann, J. The Effect of a Diet Moderately High in Protein and Fiber on Insulin Sensitivity Measured Using the Dynamic Insulin Sensitivity and Secretion Test (DISST). Nutrients 2017, 9, 1291. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.M.; Nakagawa, S.; Raubenheimer, D.; Simpson, S.J. Global associations between macronutrient supply and age-specific mortality. Proc. Natl. Acad. Sci. USA 2020, 117, 30824–30835. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wali, J.A.; Solon-Biet, S.M.; Freire, T.; Brandon, A.E. Macronutrient Determinants of Obesity, Insulin Resistance and Metabolic Health. Biology 2021, 10, 336. https://doi.org/10.3390/biology10040336
Wali JA, Solon-Biet SM, Freire T, Brandon AE. Macronutrient Determinants of Obesity, Insulin Resistance and Metabolic Health. Biology. 2021; 10(4):336. https://doi.org/10.3390/biology10040336
Chicago/Turabian StyleWali, Jibran A., Samantha M. Solon-Biet, Therese Freire, and Amanda E. Brandon. 2021. "Macronutrient Determinants of Obesity, Insulin Resistance and Metabolic Health" Biology 10, no. 4: 336. https://doi.org/10.3390/biology10040336
APA StyleWali, J. A., Solon-Biet, S. M., Freire, T., & Brandon, A. E. (2021). Macronutrient Determinants of Obesity, Insulin Resistance and Metabolic Health. Biology, 10(4), 336. https://doi.org/10.3390/biology10040336