
Protection against Glucolipotoxicity by High Density Lipoprotein in Human PANC-1 Hybrid 1.1B4 Pancreatic Beta Cells: The Role of microRNA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Maintenance
2.2. Cellular Lipid Extraction and Analysis
2.3. Glucolipotoxicity Assays
2.4. Total RNA Isolation, Microchip Analysis and Q-PCR
2.5. Transfection with miRNA Mimics and Inhibitors
2.6. Bioinformatic Analysis
2.7. Statistical Analysis
3. Results
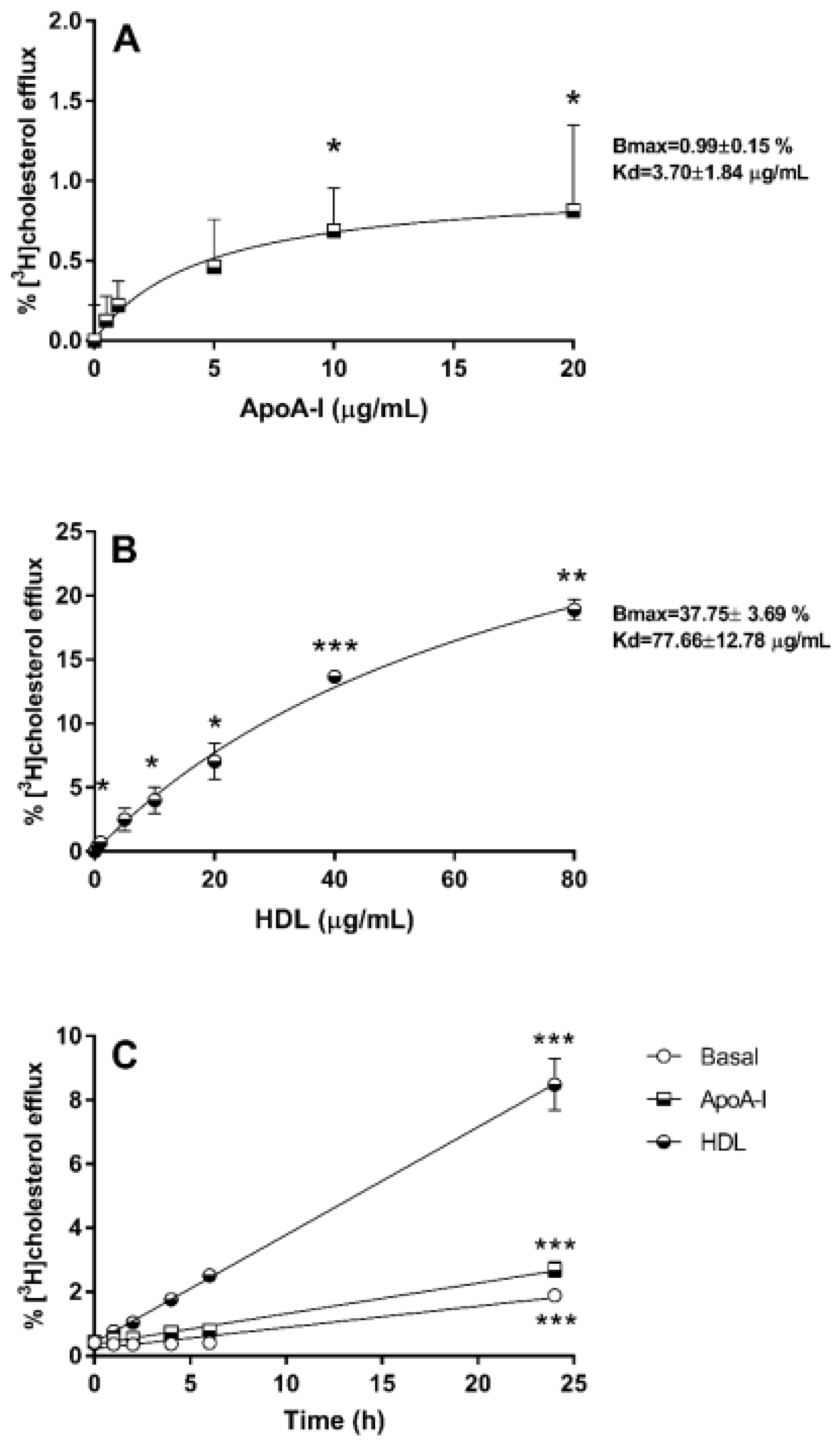
3.1. Cholesterol Efflux from 1.1B4 Cells
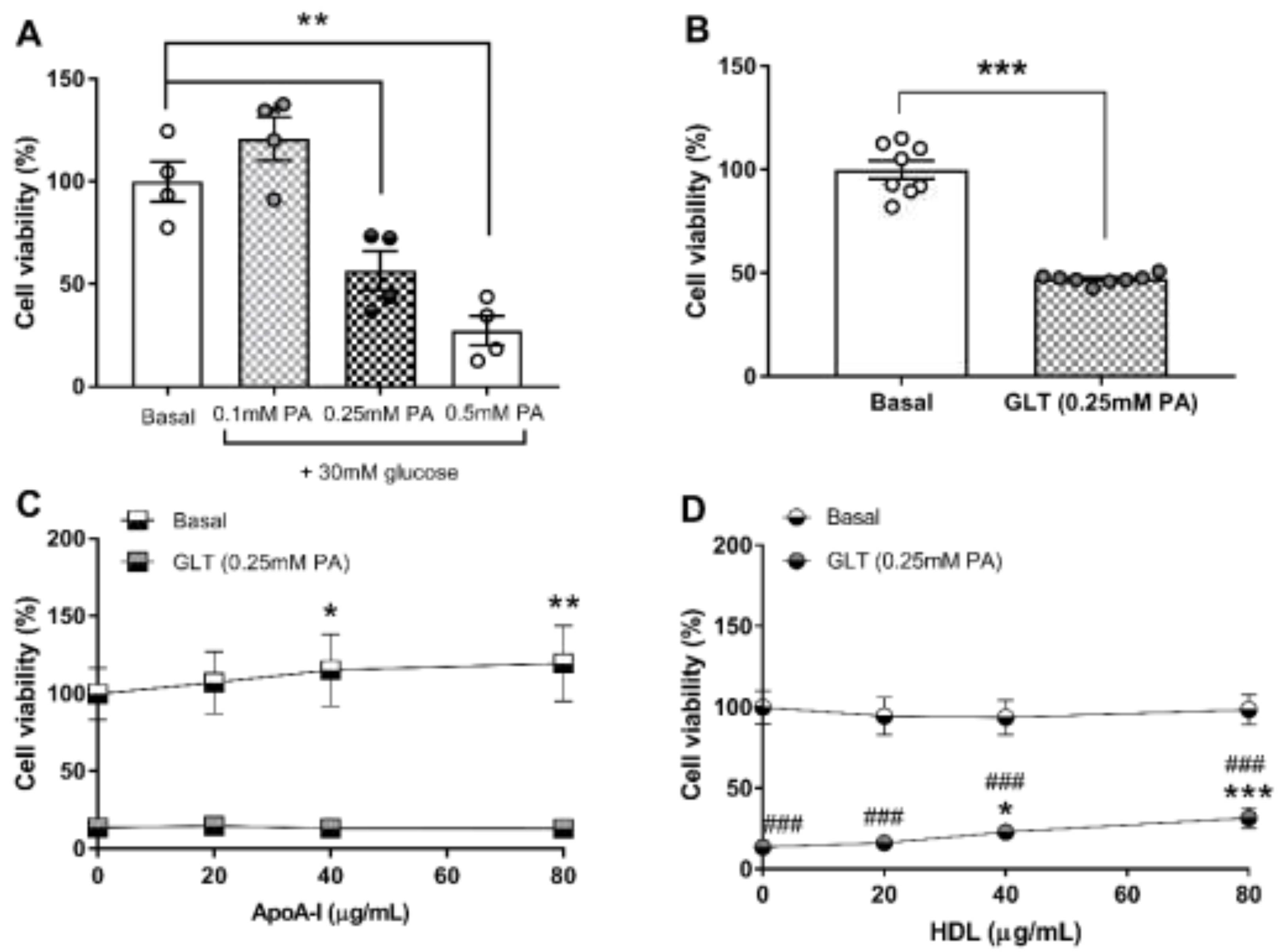
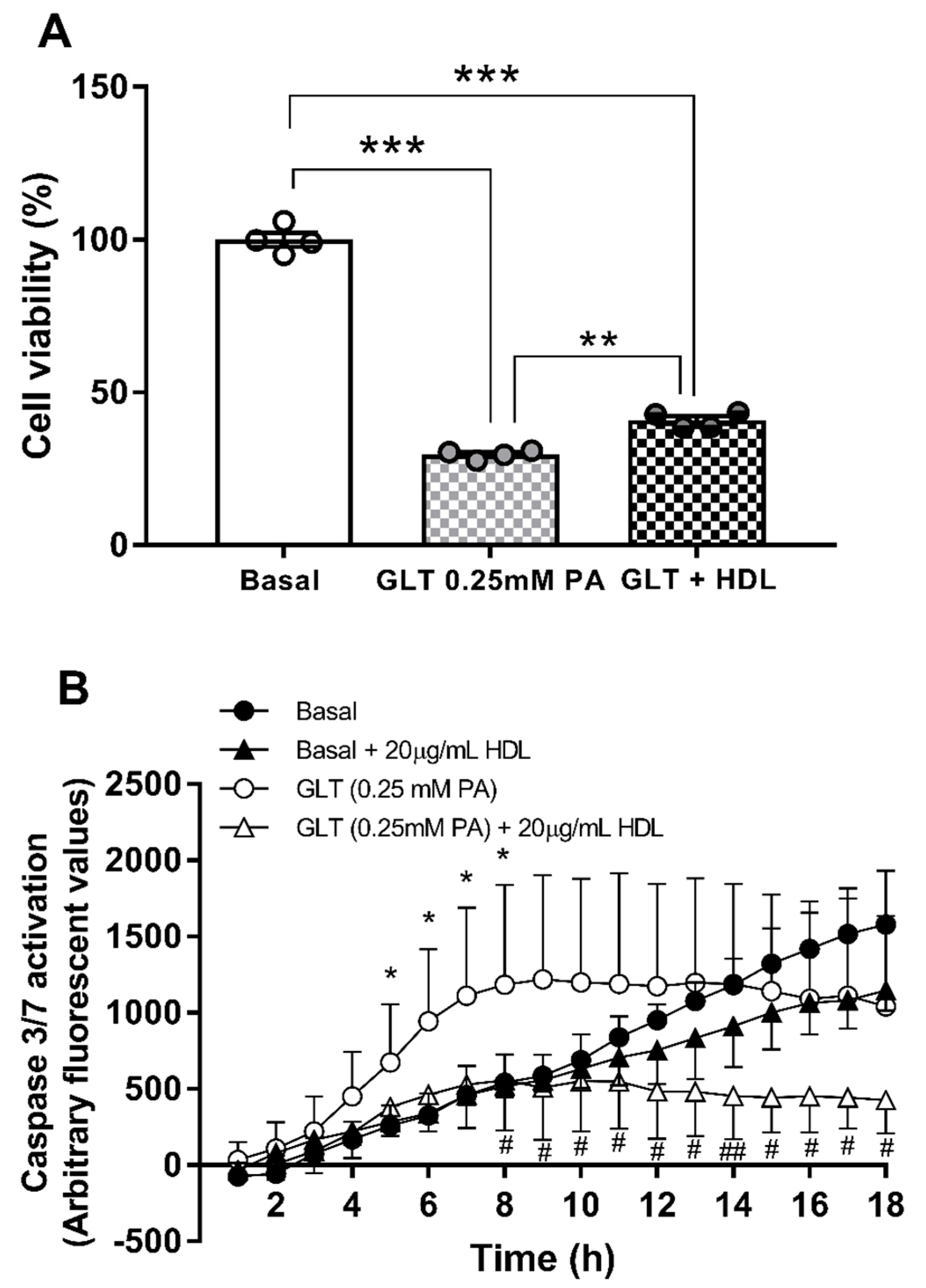
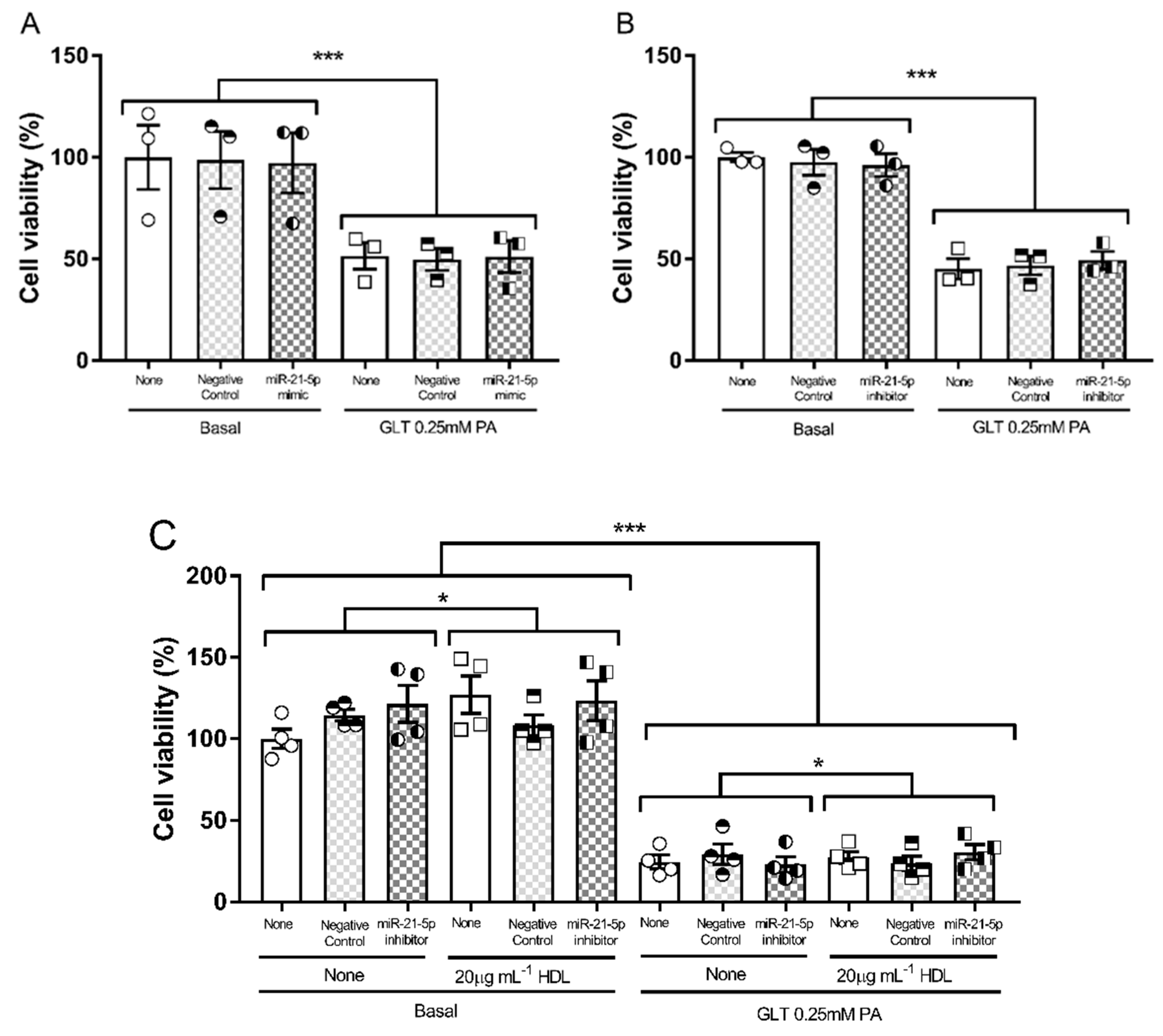
3.2. Glucolipotoxicity in 1.1B4 Cells
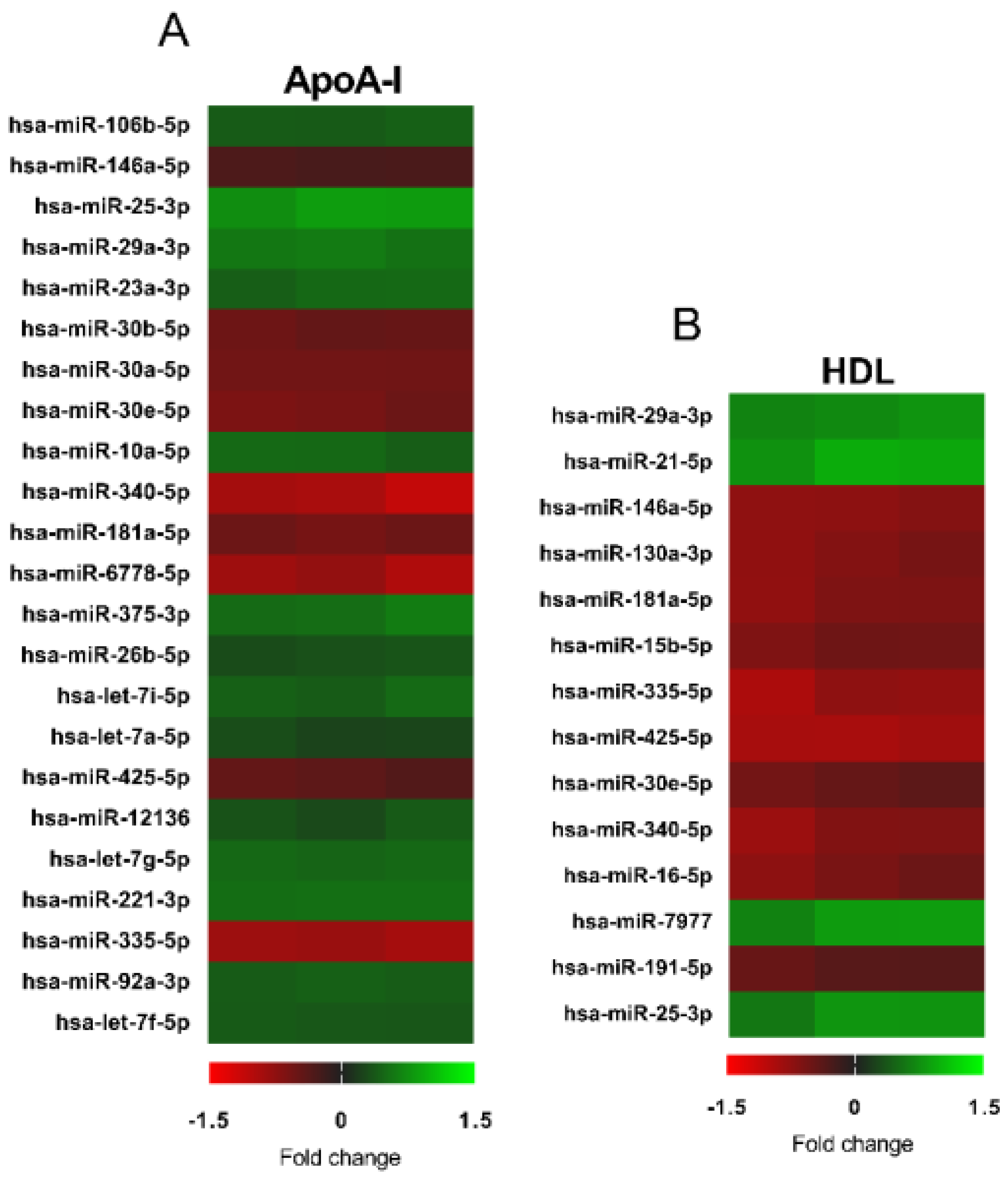
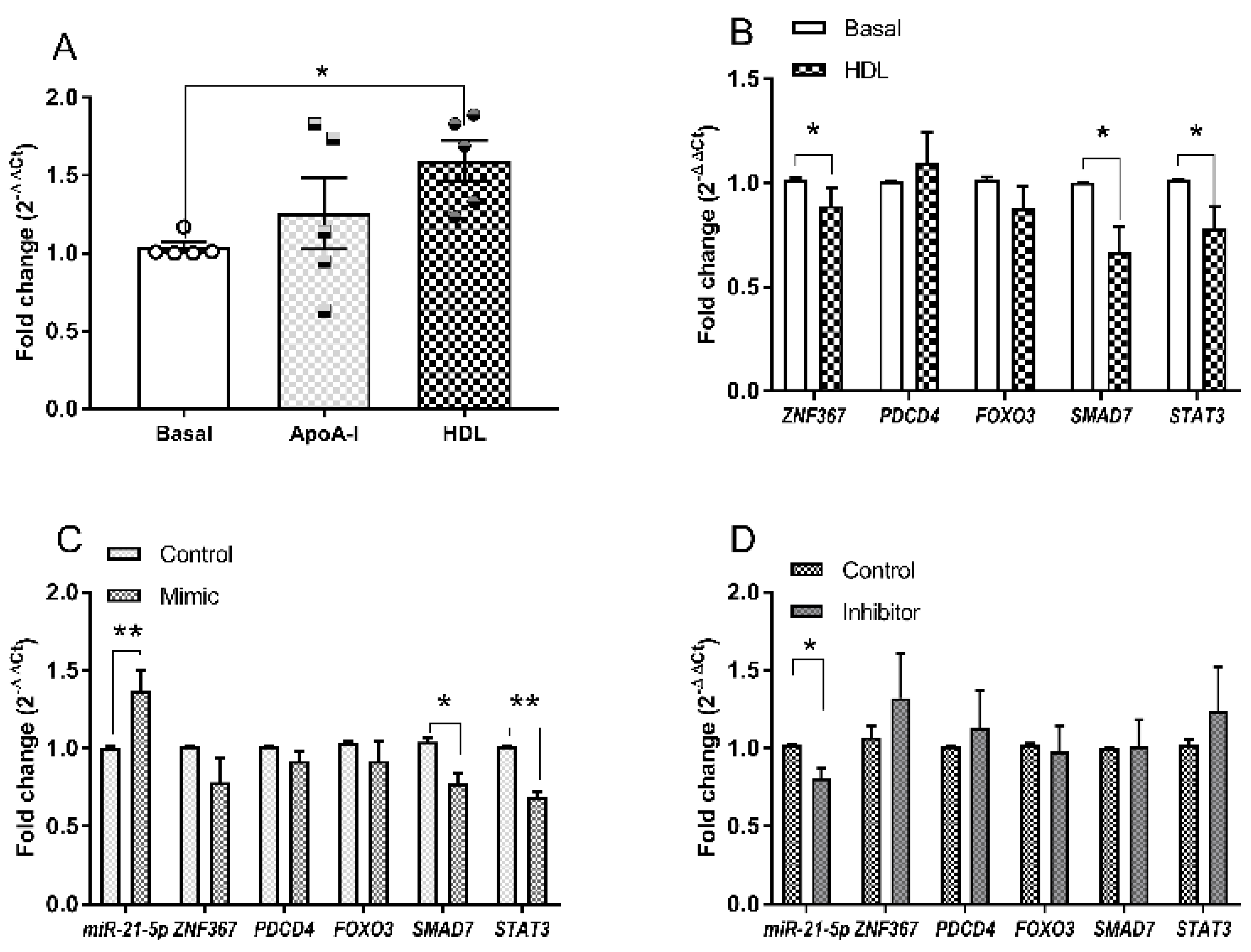
3.3. Changes in microRNA Expression Due to apoA-I and HDL in 1.1B4 Cells
3.4. Hsa-miR-21-5p and Glucolipotoxicity in 1.1B4 Cells
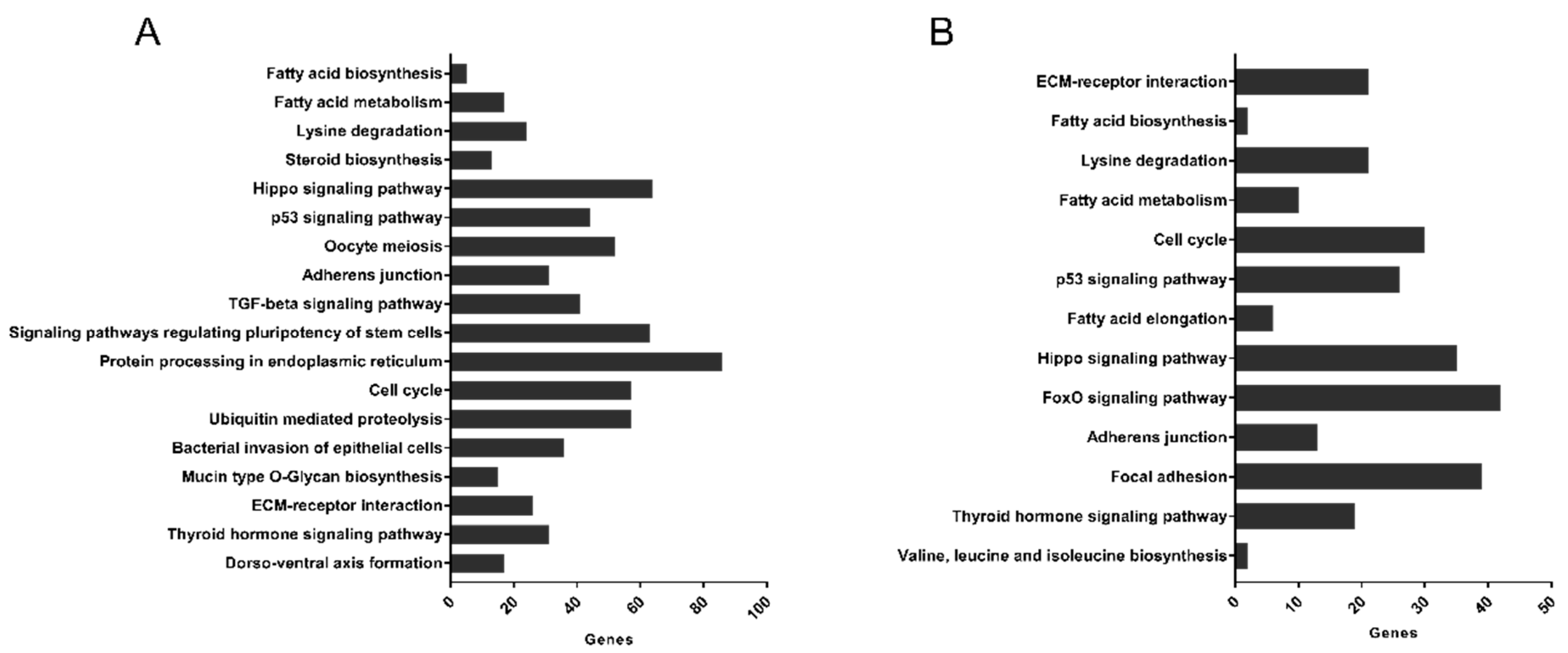
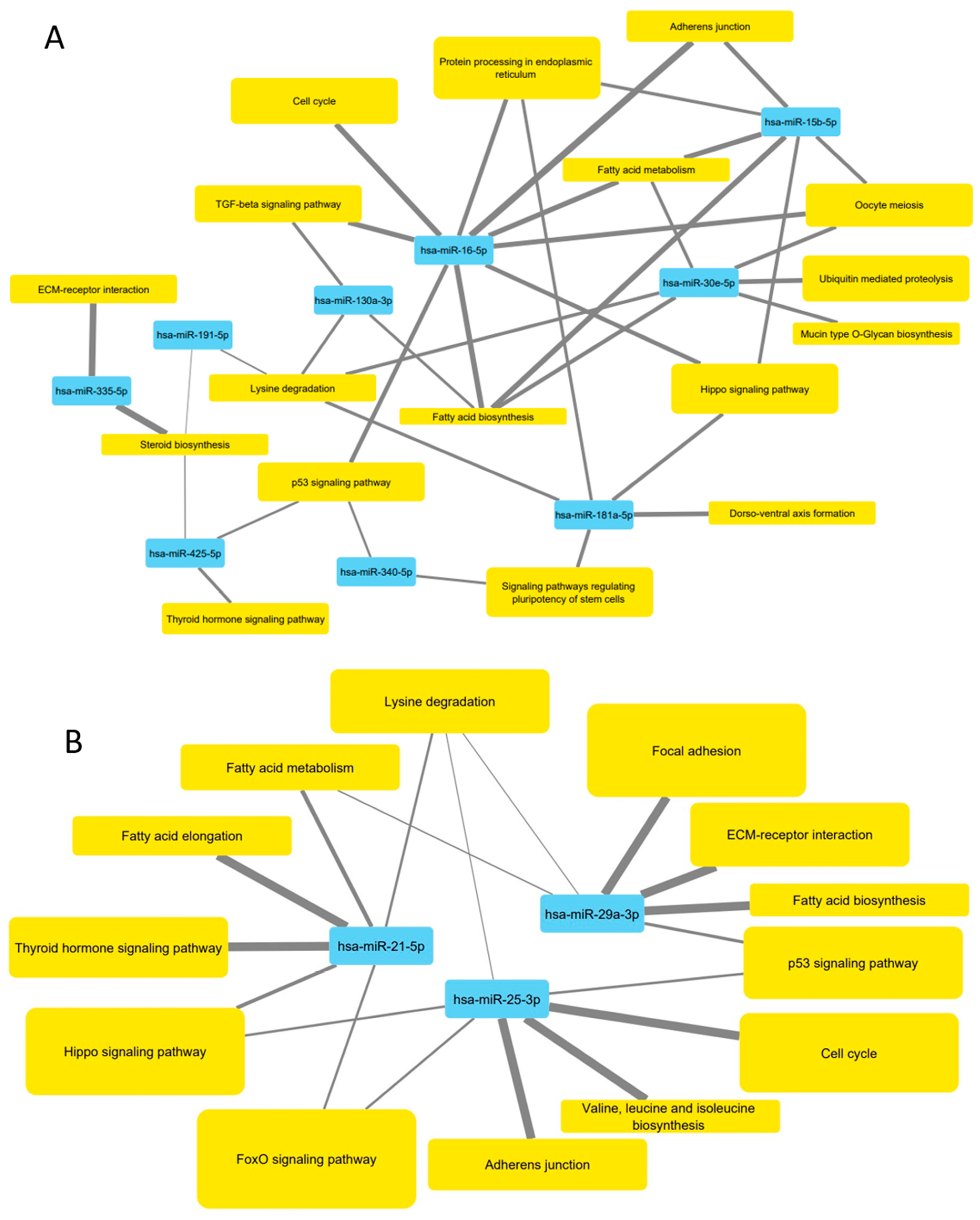
3.5. Pathways Targeted by microRNA Sequences Altered by HDL Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prentki, M.; Corkey, B.E. Are the β-Cell Signaling Molecules Malonyl-CoA and Cystolic Long-Chain Acyl-CoA Implicated in Multiple Tissue Defects of Obesity and NIDDM? Diabetes 1996, 45, 273–283. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Ghaddar, K.; Lopes, M.; Rosengre, V.; Piron, A.; Yi, X.; Johansson, H.; Lehto, J.; Iigoillo-Esteve, M.; Cunha, D.A.; et al. Combined transcriptomic and proteome profiling of the pancreatic β-cell re-sponse to palmitate unveils key pathways of β-cell lipotoxicity. BMC Genom. 2020, 21, 590. [Google Scholar] [CrossRef] [PubMed]
- Fryirs, M.; Barter, P.J.; Rye, K.-A. Cholesterol metabolism and pancreatic beta-cell function. Curr. Opin. Lipidol. 2009, 20, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Von Eckardstein, A.; Widmann, C. High-density lipoprotein, beta cells, and diabetes. Cardiovasc. Res. 2014, 103, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Rutti, S.; Ehses, J.A.; Sibler, R.A.; Prazak, R.; Rohrer, L.; Georgopoulos, S.; Meier, D.T.; Niclauss, N.; Berney, T.; Donath, M.Y.; et al. Low- and High-Density Lipoproteins Modulate Function, Apoptosis, and Proliferation of Primary Human and Murine Pancreatic β-Cells. J. Endocrinol. 2009, 150, 4521–4530. [Google Scholar] [CrossRef] [PubMed]
- Fryirs, M.A.; Barter, P.J.; Appavoo, M.; Tuch, B.E.; Tabet, F.; Heather, A.K.; Rye, K.-A. Effects of High-Density Lipoproteins on Pancreatic β-Cell Insulin Secretion. Arter. Thromb. Vasc. Biol. 2010, 30, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, M.; Kerksiek, A.; Gebert, K.; Annema, W.; Sibler, R.; Radosavljevic, S.; Luetjohann, D.; Rohrer, L.; von Eckardstein, A. HDL inhibits endoplasmic reticulum stress-induced apoptosis of pancreatic β-cells in vitro by activation of Smoothened. J. Lipid Res. 2020, 61, 492–504. [Google Scholar] [CrossRef]
- Cochran, B.J.; Bisoendial, R.J.; Hou, L.; Glaros, E.N.; Rossy, J.; Thomas, S.R.; Barter, P.J.; Rye, K.-A. Apolipoprotein A-I Increases Insulin Secretion and Production from Pancreatic β-Cells via a G-Protein-cAMP-PKA-FoxO1–Dependent Mechanism. Arter. Thromb. Vasc. Biol. 2014, 34, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Tang, S.; Wu, B.J.; Ong, K.-L.; Westerterp, M.; Barter, P.J.; Cochran, B.J.; Tabet, F.; Rye, K.-A. Apolipoprotein A-l improves pancreatic β-cell function independent of the ATP-binding cassette transporters ABCA1 and ABCG1. FASEB J. 2019, 33, 8479–8489. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.; Del Giudice, R.; Nagao, M.; Grönberg, C.; Eliasson, L.; Lagerstedt, J.O. Apolipoprotein A-I primes beta cells to increase glucose stimulated insulin secretion. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165613. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Pape, T.D.; Timmins, J.M.; Reuwer, A.Q.; Vasanji, Z.; Marsh, B.J.; Rodrigues, B.; Johnson, J.D.; Parks, J.S.; et al. β-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat. Med. 2007, 13, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Kruit, J.K.; Kremer, P.H.C.; Dai, L.; Tang, R.; Ruddle, P.; De Haan, W.; Brunham, L.R.; Verchere, C.B.; Hayden, M.R. Cholesterol efflux via ATP-binding cassette transporter A1 (ABCA1) and cholesterol uptake via the LDL receptor influences cholesterol-induced impairment of beta cell function in mice. Diabetology 2010, 53, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Kruit, J.K.; Wijesekara, N.; Fox, J.E.M.; Dai, X.-Q.; Brunham, L.R.; Searle, G.J.; Morgan, G.P.; Costin, A.J.; Tang, R.; Bhattacharjee, A.; et al. Islet Cholesterol Accumulation Due to Loss of ABCA1 Leads to Impaired Exocytosis of Insulin Granules. Diabetes 2011, 60, 3186–3196. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Zhang, L.-H.; Kang, M.H.; Abraham, T.; Bhattacharjee, A.; Warnock, G.L.; Verchere, C.B.; Hayden, M.R. miR-33a Modulates ABCA1 Expression, Cholesterol Accumulation, and Insulin Secretion in Pancreatic Islets. Diabetes 2012, 61, 653–658. [Google Scholar] [CrossRef]
- Kruit, J.K.; Wijesekara, N.; Westwell-Roper, C.; Vanmierlo, T.; De Haan, W.; Bhattacharjee, A.; Tang, R.; Wellington, C.L.; Lütjohann, D.; Johnson, J.D.; et al. Loss of Both ABCA1 and ABCG1 Results in Increased Disturbances in Islet Sterol Homeostasis, Inflammation, and Impaired -Cell Function. Diabetes 2012, 61, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Dullaart, R.P.; Annema, W.; de Boer, J.F.; Tietge, U.J. Pancreatic β-cell function relates positively to HDL functionality in well-controlled Type 2 diabetes mellitus. Atherosclerosis 2012, 222, 567–573. [Google Scholar] [CrossRef]
- Bardini, G.; Dicembrini, I.; Rotella, C.M.; Giannini, S. Correlation between HDL cholesterol levels and beta-cell function in subjects with various degree of glucose tolerance. Acta Diabetol. 2013, 50, 277–281. [Google Scholar] [CrossRef]
- Zheng, S.; Xu, H.; Zhou, H.; Ren, X.; Han, T.; Chen, Y.; Qiu, H.; Wu, P.; Zheng, J.; Wang, L.; et al. Associations of lipid profiles with insulin resistance and β cell function in adults with normal glucose tolerance and different categories of impaired glucose regulation. PLoS ONE 2017, 12, e0172221. [Google Scholar] [CrossRef]
- Kumar, H.; Mishra, M.; Bajpai, S.; Pokhria, D.; Arya, A.K.; Singh, R.K.; Tripathi, K. Correlation of insulin resistance, beta cell function and insulin sensitivity with serum sFas and sFasL in newly diagnosed type 2 diabetes. Acta Diabetol. 2011, 50, 511–518. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Succurro, E.; Marini, M.A.; Pedace, E.; Andreozzi, F.; Perticone, M.; Sciacqua, A.; Perticone, F.; Sesti, G. HDL cholesterol is an independent predictor of β-cell function decline and incident type 2 diabetes: A longitudinal study. Diabetes Metab. Res. Rev. 2020, 36, e3289. [Google Scholar] [CrossRef] [PubMed]
- Vergeer, M.; Brunham, L.R.; Koetsveld, J.; Kruit, J.K.; Verchere, C.B.; Kastelein, J.J.; Hayden, M.R.; Stroes, E.S. Carriers of Loss-of-Function Mutations in ABCA1 Display Pancreatic -Cell Dysfunction. Diabetes Care 2010, 33, 869–874. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rickels, M.R.; Goeser, E.S.; Fuller, C.; Lord, C.; Bowler, A.M.; Doliba, N.M.; Hegele, R.A.; Cuchel, M. Loss-of-Function Mutations inABCA1and Enhanced β-Cell Secretory Capacity in Young Adults. Diabetes 2014, 64, 193–199. [Google Scholar] [CrossRef]
- Ertek, S. High-density Lipoprotein (HDL) Dysfunction and the Future of HDL. Curr. Vasc. Pharmacol. 2018, 16, 490–498. [Google Scholar] [CrossRef]
- Plochberger, B.; Axmann, M.; Röhrl, C.; Weghuber, J.; Brameshuber, M.; Rossboth, B.K.; Mayr, S.; Ros, R.; Bittman, R.; Stangl, H.; et al. Direct observation of cargo transfer from HDL particles to the plasma membrane. Atheroscler. 2018, 277, 53–59. [Google Scholar] [CrossRef]
- Roehrich, M.-E.; Mooser, V.; Lenain, V.; Herz, J.; Nimpf, J.; Azhar, S.; Bideau, M.; Capponi, A.; Nicod, P.; Haefliger, J.-A.; et al. Insulin-secreting β-Cell Dysfunction Induced by Human Lipoproteins. J. Biol. Chem. 2003, 278, 18368–18375. [Google Scholar] [CrossRef]
- Kurano, M.; Hara, M.; Tsuneyama, K.; Sakoda, H.; Shimizu, T.; Tsukamoto, K.; Ikeda, H.; Yatomi, Y. Induction of insulin secretion by apolipoprotein M, a carrier for sphingosine 1-phosphate. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Osmai, M.; Osmai, Y.; Bangberthelsen, C.H.; Pallesen, E.M.H.; Vestergaard, A.L.; Novotny, G.W.; Pociot, F.; Mandrup-Poulsen, T. MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes/Metabolism Res. Rev. 2016, 32, 334–349. [Google Scholar] [CrossRef]
- McCluskey, J.T.; Hamid, M.; Guo-Parke, H.; McClenaghan, N.H.; Gomis, R.; Flatt, P.R. Development and functional characterization of insulin-releasing human pancreatic beta cell lines produced by electrofusion. J. Biol. Chem. 2011, 286, 21982–21992. [Google Scholar] [CrossRef] [PubMed]
- Nemecz, M.; Constantin, A.; Dumitrescu, M.; Alexandru, N.; Filippi, A.; Tanko, G.; Georgescu, A. The Distinct Effects of Palmitic and Oleic Acid on Pancreatic Beta Cell Function: The Elucidation of Associated Mechanisms and Effector Molecules. Front. Pharmacol. 2019, 9, 1554. [Google Scholar] [CrossRef]
- Honkimaa, A.; Kimura, B.; Sioofy-Khojine, A.-B.; Lin, J.; Laiho, J.; Oikarinen, S.; Hyöty, H. Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells. Microorganisms 2020, 8, 1790. [Google Scholar] [CrossRef]
- De la Llera Moya, M.; Atger, V.; Paul, J.L.; Fournier, N.; Moatti, N.; Giral, P.; Friday, K.E.; Rothblat, G. A cell culture system for screening human serum for ability to promote cellular cholesterol efflux. Relations between serum components and efflux, esterification, and transfer. Arter. Thromb 1994, 14, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef]
- Xu, M.; McCanna, D.J.; Sivak, J.G. Use of the viability reagent PrestoBlue in comparison with alamarBlue and MTT to assess the viability of human corneal epithelial cells. J. Pharmacol. Toxicol. Methods 2015, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jeelani, R.; Chatzicharalampous, C.; Kohan-Ghadr, H.-R.; Bai, D.; Morris, R.T.; Sliskovic, I.; Awonuga, A.; Abu-Soud, H.M. Hypochlorous acid reversibly inhibits caspase-3: A potential regulator of apoptosis. Free. Radic. Res. 2020, 54, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, L.; Cheng, L. Aberrant ERG expression associates with downregulation of miR-4638-5p and selected genomic alterations in a subset of diffuse large B-cell lymphoma. Mol. Carcinog. 2019, 58, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2014, 43, D153–D159. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2013, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Zhang, Y.-Y.; Zhu, B.-L.; Feng, F.-Z.; Yan, H.; Zhang, H.-Y.; Zhou, B. miR-21 regulates the proliferation and apoptosis of ovarian cancer cells through PTEN/PI3K/AKT. Eur. Rev. Med Pharmacol. Sci. 2019, 23, 4149–4155. [Google Scholar] [PubMed]
- Canfrán-Duque, A.; Rotllan, N.; Zhang, X.; Fernández-Fuertes, M.; Ramírez-Hidalgo, C.; Araldi, E.; Daimiel, L.; Busto, R.; Fernández-Hernando, C.; Suárez, Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017, 9, 1244–1262. [Google Scholar] [CrossRef]
- Hao, X.-J.; Xu, C.-Z.; Wang, J.-T.; Li, X.-J.; Wang, M.-M.; Gu, Y.-H.; Liang, Z.-G. miR-21 promotes proliferation and inhibits apoptosis of hepatic stellate cells through targeting PTEN/PI3K/AKT pathway. J. Recept. Signal Transduct. 2018, 38, 455–461. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, L.; Liu, W.; Xiao, Z. MiR-21 regulates the apoptosis of keloid fibroblasts by caspase-8 and the mitochondria-mediated apoptotic signaling pathway via targeting FasL. Biochem. Cell Biol. 2018, 96, 548–555. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.; Sun, G.; Mei, F.; Cui, Y.; Xu, H. Effect of miR-21 on Apoptosis in Lung Cancer Cell Through Inhibiting the PI3K/ Akt/NF-κB Signaling Pathway in Vitro and in Vivo. Cell. Physiol. Biochem. 2018, 46, 999–1008. [Google Scholar] [CrossRef]
- Ruan, Q.; Wang, T.; Kameswaran, V.; Wei, Q.; Johnson, D.S.; Matschinsky, F.; Shi, W.; Chen, Y.H. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 12030–12035. [Google Scholar] [CrossRef] [PubMed]
- Roggli, E.; Britan, A.; Gattesco, S.; Lin-Marq, N.; Abderrahmani, A.; Meda, P.; Regazzi, R. Involvement of MicroRNAs in the Cytotoxic Effects Exerted by Proinflammatory Cytokines on Pancreatic -Cells. Diabetes 2010, 59, 978–986. [Google Scholar] [CrossRef]
- Backe, M.B.; Novotny, G.W.; Christensen, D.P.; Grunnet, L.G.; Mandrup-Poulsen, T. Altering β-cell number through stable alteration of miR-21 and miR-34a expression. Islets 2014, 6, e27754. [Google Scholar] [CrossRef] [PubMed]
- Doghish, A.S.; Elsisi, A.M.; Amin, A.I.; Abulsoud, A.I. Circulating miR-148a-5p and miR-21-5p as Novel Diagnostic Biomarkers in Adult Egyptian Male Patients with Metabolic Syndrome. Can. J. Diabetes 2021, 5, 1499. [Google Scholar] [CrossRef]
- Guan, C.-Y.; Tian, S.; Cao, J.-L.; Wang, X.-Q.; Ma, X.; Xia, H.-F. Down-Regulated miR-21 in Gestational Diabetes Mellitus Placenta Induces PPAR-α to Inhibit Cell Proliferation and Infiltration. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3009–3034. [Google Scholar] [CrossRef]
- Hromadnikova, I.; Kotlabova, K.; Dvorakova, L.; Krofta, L. Diabetes Mellitus and Cardiovascular Risk Assessment in Mothers with a History of Gestational Diabetes Mellitus Based on Postpartal Expression Profile of MicroRNAs Associated with Diabetes Mellitus and Cardiovascular and Cerebrovascular Diseases. Int. J. Mol. Sci. 2020, 21, 2437. [Google Scholar] [CrossRef]
- Lv, Q.; Deng, J.; Chen, Y.; Wang, Y.; Liu, B.; Liu, J. Engineered Human Adipose Stem-Cell-Derived Exosomes Loaded with miR-21-5p to Promote Diabetic Cutaneous Wound Healing. Mol. Pharm. 2020, 17, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wan, G.; Peng, G.; Yan, P.; Qian, C.; Li, F. Long non-coding RNA XIST regulates hyperglycemia-associated apoptosis and migration in human retinal pigment epithelial cells. Biomed. Pharmacother. 2020, 125, 109959. [Google Scholar] [CrossRef]
- Re, A.; Caselle, M.; Bussolino, F. MicroRNA-mediated regulatory circuits—outlook and perspectives. Phys. Biol. 2017, 14, 045001. [Google Scholar] [CrossRef] [PubMed]
- Michell, D.L.; Vickers, K.C. HDL and microRNA therapeutics in cardiovascular disease. Pharmacol. Ther. 2016, 168, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Langi, G.; Szczerbinski, L.; Kretowski, A. Meta-Analysis of Differential miRNA Expression after Bariatric Surgery. J. Clin. Med. 2019, 8, 1220. [Google Scholar] [CrossRef]
- Mendes-Silva, A.P.; Pereira, K.S.; Tolentino-Araujo, G.T.; Nicolau, E.D.S.; Silva-Ferreira, C.M.; Teixeira, A.L.; Diniz, B.S. Shared Biologic Pathways Between Alzheimer Disease and Major Depression: A Systematic Review of MicroRNA Expression Studies. Am. J. Geriatr. Psychiatry 2016, 24, 903–912. [Google Scholar] [CrossRef]
- Pedretti, S.; Brulhart-Meynet, M.-C.; Montecucco, F.; Lecour, S.; James, R.W.; Frias, M.A. HDL protects against myocardial ischemia reperfusion injury via miR-34b and miR-337 expression which requires STAT3. PLoS ONE 2019, 14, e0218432. [Google Scholar] [CrossRef]
- Feuerborn, R.; Becker, S.; Poti, F.; Nagel, P.; Brodde, M.; Schmidt, H.; Christoffersen, C.; Ceglarek, U.; Burkhardt, R.; Nofer, J.-R. High density lipoprotein (HDL)-associated sphinosine-1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 ac-tivity and survivin expression. Atherosclerosis 2017, 257, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; James, R.W.; Gerber-Wicht, C.; Lang, U. Native and reconstituted HDL activate Stat3 in ventricular cardiomyocytes via ERK1/2: Role of sphingosine-1-phosphate. Cardiovasc. Res. 2009, 82, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Suzuki, K.; Remaley, A.T. HDL and sphingosine-1-phosphate activate stat3 in prostate cancer DU145 cells via ERK1/2 and S1P receptors, and promote cell migration and invasion. Prostate 2010, 71, 690–699. [Google Scholar] [CrossRef]
- Frias, M.A.; Lang, U.; Gerber-Wicht, C.; James, R.W. Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc. Res. 2009, 85, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; He, W.; Jia, Z.; Hao, S. Lycopene Improves Insulin Sensitivity through Inhibition of STAT3/Srebp-1c-Mediated Lipid Accumulation and Inflammation in Mice fed a High-Fat Diet. Exp. Clin. Endocrinol. Diabetes 2017, 125, 610–617. [Google Scholar] [CrossRef]
- Yang, X.; Jia, J.; Yu, Z.; Duanmu, Z.; He, H.; Chen, S.; Qu, C. Inhibition of JAK2/STAT3/SOCS3 signaling attenuates atherosclerosis in rabbit. BMC Cardiovasc. Disord. 2020, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2016, 9, a022095. [Google Scholar] [CrossRef]
- Wang, N.; Tall, A.R. Regulation and Mechanisms of ATP-Binding Cassette Transporter A1-Mediated Cellular Cholesterol Efflux. Arter. Thromb. Vasc. Biol. 2003, 23, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, M.V.; Joglekar, V.M.; Hardikar, A.A. Expression of islet-specific microRNAs during human pancreatic development. Gene Expr. Patterns 2009, 9, 109–113. [Google Scholar] [CrossRef]
- Jacovetti, C.; Rodriguez-Trejo, A.; Guay, C.; Sobel, J.; Gattesco, S.; Petrenko, V.; Saini, C.; Dibner, C.; Regazzi, R. MicroRNAs modulate core-clock gene expression in pancreatic islets during early postnatal life in rats. Diabetology 2017, 60, 2011–2020. [Google Scholar] [CrossRef]
- Lee, S.; Dong, H.H. FoxO integration of insulin signaling with glucose and lipid metabolism. J. Endocrinol. 2017, 233, R67–R79. [Google Scholar] [CrossRef]
- Oh, H.-M.; Yu, C.-R.; Dambuza, I.; Marrero, B.; Egwuagu, C.E. STAT3 Protein Interacts with Class O Forkhead Transcription Factors in the Cytoplasm and Regulates Nuclear/Cytoplasmic Localization of FoxO1 and FoxO3a Proteins in CD4+ T Cells. J. Biol. Chem. 2012, 287, 30436–30443. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarlton, J.M.R.; Lightbody, R.J.; Patterson, S.; Graham, A. Protection against Glucolipotoxicity by High Density Lipoprotein in Human PANC-1 Hybrid 1.1B4 Pancreatic Beta Cells: The Role of microRNA. Biology 2021, 10, 218. https://doi.org/10.3390/biology10030218
Tarlton JMR, Lightbody RJ, Patterson S, Graham A. Protection against Glucolipotoxicity by High Density Lipoprotein in Human PANC-1 Hybrid 1.1B4 Pancreatic Beta Cells: The Role of microRNA. Biology. 2021; 10(3):218. https://doi.org/10.3390/biology10030218
Chicago/Turabian StyleTarlton, Jamie M.R., Richard J. Lightbody, Steven Patterson, and Annette Graham. 2021. "Protection against Glucolipotoxicity by High Density Lipoprotein in Human PANC-1 Hybrid 1.1B4 Pancreatic Beta Cells: The Role of microRNA" Biology 10, no. 3: 218. https://doi.org/10.3390/biology10030218
APA StyleTarlton, J. M. R., Lightbody, R. J., Patterson, S., & Graham, A. (2021). Protection against Glucolipotoxicity by High Density Lipoprotein in Human PANC-1 Hybrid 1.1B4 Pancreatic Beta Cells: The Role of microRNA. Biology, 10(3), 218. https://doi.org/10.3390/biology10030218