
Emerging Roles of Metallothioneins in Beta Cell Pathophysiology: Beyond and above Metal Homeostasis and Antioxidant Response


Abstract
Simple Summary
Abstract
1. Introduction
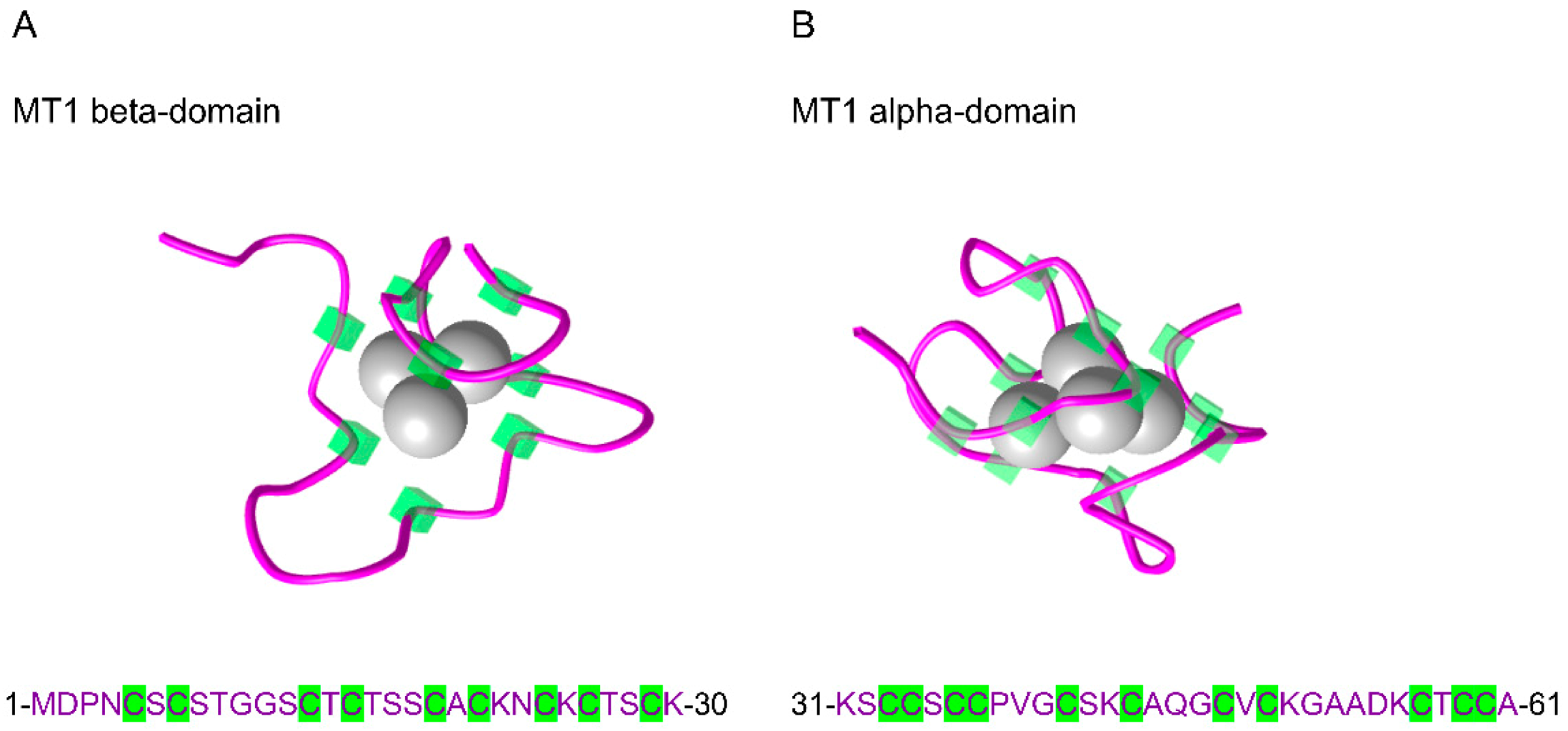
2. Metallothioneins: The Guardians of Metal and Redox Homeostasis
3. Metallothioneins in Pancreatic Beta Cells
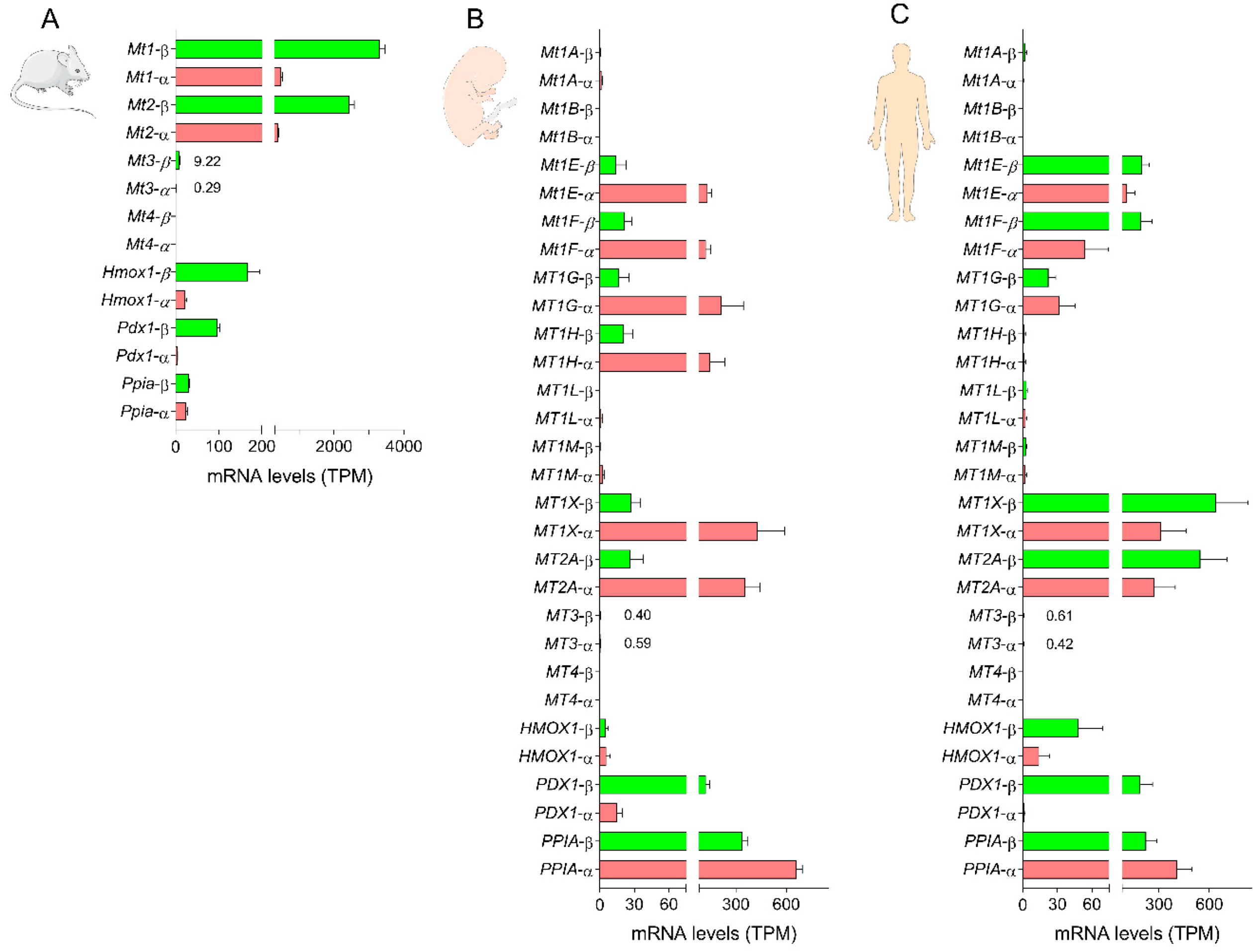
3.1. Expression of Metallothionein Genes in Mouse and Human Beta Cells
3.2. Physiological and Pathological Regulation of MT Gene Expression in Beta Cells
3.2.1. Regulation by Metals
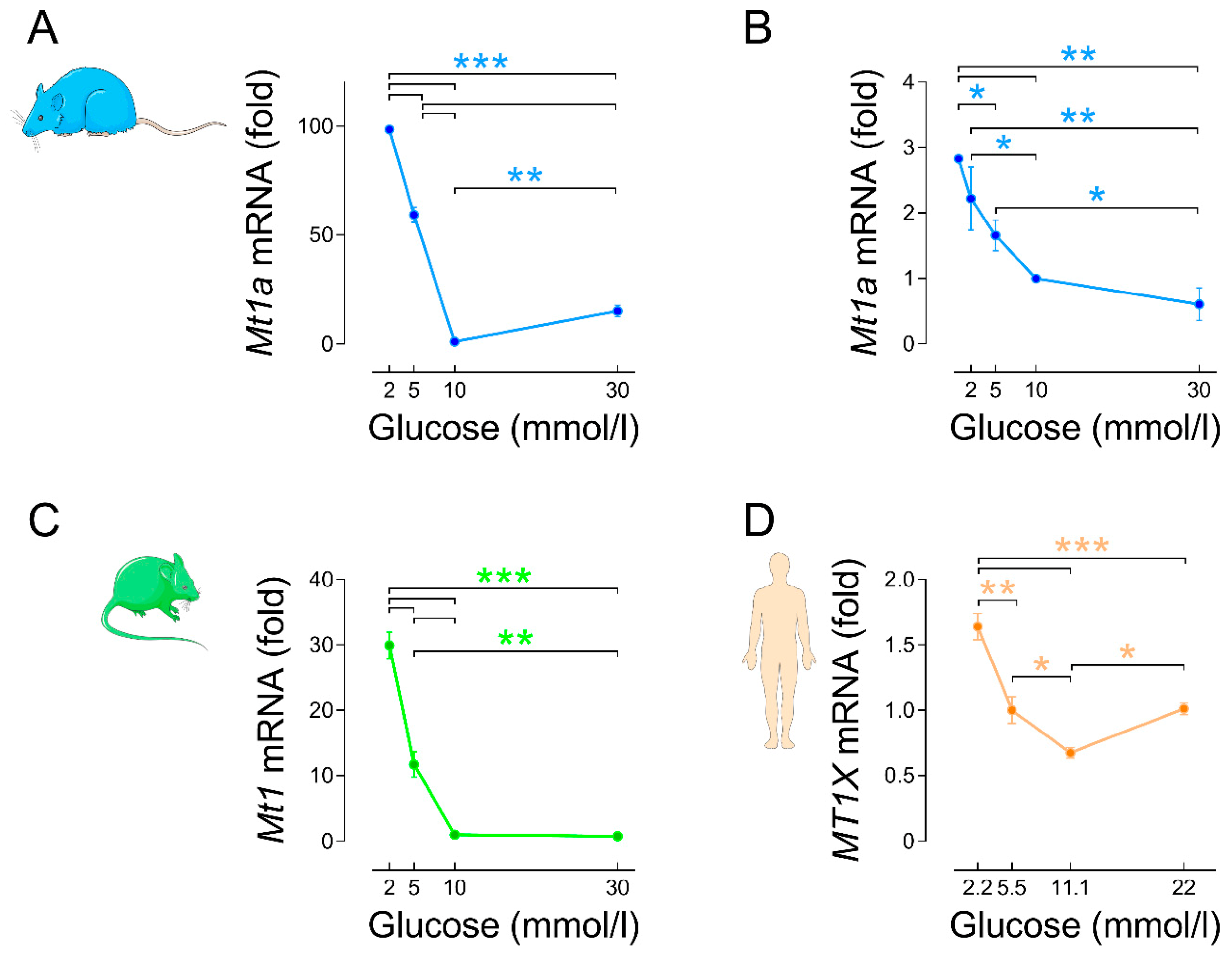
3.2.2. Regulation by Glucose
3.2.3. Regulation by Stress Stimuli
3.2.4. Regulation during Beta Cell Compensation and Failure
4. Adaptive Roles of MTs in Beta Cells
5. Negative Roles of MTs in Beta Cells
6. Unsettled Questions and Future Research Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53 (Suppl. 3), S16–S21. [Google Scholar] [CrossRef]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.-C. The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Utzschneider, K.M.; Kahn, S.E. Early beta cell dysfunction vs. insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 2020, 63, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Peyot, M.-L.; Masiello, P.; Madiraju, S.M. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Shi, Y.-C.; Chan, J.Y.; Laybutt, D.R.; Chae, H.; Abou-Samra, M.; Pappas, E.G.; Thomas, H.E.; Gilon, P.; Jonas, J.-C. Metallothionein 1 negatively regulates glucose-stimulated insulin secretion and is differentially expressed in conditions of beta cell compensation and failure in mice and humans. Diabetologia 2019, 62, 2273–2286. [Google Scholar] [CrossRef] [PubMed]
- Margoshes, M.; Vallee, B.L. A Cadmium Protein from Equine Kidney Cortex. J. Am. Chem. Soc. 1957, 79, 4813–4814. [Google Scholar] [CrossRef]
- Kagi, J.H.; Valee, B.L. Metallothionein: A cadmium- and zinc-containing protein from equine renal cortex. J. Biol. Chem. 1960, 235, 3460–3465. [Google Scholar] [CrossRef]
- Michalska, A.E.; Choo, K.H. Targeting and germ-line transmission of a null mutation at the metallothionein I and II loci in mouse. Proc. Natl. Acad. Sci. USA 1993, 90, 8088–8092. [Google Scholar] [CrossRef]
- Masters, B.A.; Kelly, E.J.; Quaife, C.J.; Brinster, R.L.; Palmiter, R.D. Targeted disruption of metallothionein I and II genes increases sensitivity to cadmium. Proc. Natl. Acad. Sci. USA 1994, 91, 584–588. [Google Scholar] [CrossRef]
- Kelly, E.J.; Palmiter, R.D. A murine model of Menkes disease reveals a physiological function of metallothionein. Nat. Genet. 1996, 13, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Habeebu, S.S.; Klaassen, C.D. Metallothionein-Null Mice Are Highly Susceptible to the Hematotoxic and Immunotoxic Effects of Chronic CdCl2 Exposure. Toxicol. Appl. Pharmacol. 1999, 159, 98–108. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Goyer, R.A.; Achanzar, W.; Waalkes, M.P. Metallothionein-I/II Null Mice Are More Sensitive than Wild-Type Mice to the Hepatotoxic and Nephrotoxic Effects of Chronic Oral or Injected Inorganic Arsenicals. Toxicol. Sci. 2000, 55, 460–467. [Google Scholar] [CrossRef]
- Waalkes, M.P.; Liu, J.; Goyer, R.A.; Diwan, B.A. Metallothionein-I/II Double Knockout Mice Are Hypersensitive to Lead-Induced Kidney Carcinogenesis: Role of inclusion body formation. Cancer Res. 2004, 64, 7766–7772. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Iszard, M.; Andrews, G.; Palmiter, R.; Klaassen, C. Transgenic Mice That Overexpress Metallothionein-I Are Protected from Cadmium Lethality and Hepatotoxicity. Toxicol. Appl. Pharmacol. 1995, 135, 222–228. [Google Scholar] [CrossRef]
- Richards, R.I.; Heguy, A.; Karin, M. Structural and functional analysis of the human metallothionein-IA gene: Differential induction by metal ions and glucocorticoids. Cell 1984, 37, 263–272. [Google Scholar] [CrossRef]
- Laukens, D.; Waeytens, A.; De Bleser, P.; Cuvelier, C.; De Vos, M. Human metallothionein expression under normal and pathological conditions: Mechanisms of gene regulation based on in silico promoter analysis. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 301–317. [Google Scholar] [CrossRef]
- Tohyama, C.; Shaikh, Z.A.; Nogawa, K.; Kobayashi, E.; Honda, R. Elevated urinary excretion of metallothionein due to environmental cadmiun exposure. Toxicology 1981, 20, 289–297. [Google Scholar] [CrossRef]
- Amiard, J.-C.; Amiard-Triquet, C.; Barka, S.; Pellerin, J.; Rainbow, P.S. Metallothioneins in aquatic invertebrates: Their role in metal detoxification and their use as biomarkers. Aquat. Toxicol. 2006, 76, 160–202. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Edwards, J.R. Early biomarkers of cadmium exposure and nephrotoxicity. BioMetals 2010, 23, 793–809. [Google Scholar] [CrossRef]
- Shariati, F.; Sari, A.E.; Mashinchian, A.; Pourkazemi, M. Metallothionein as Potential Biomarker of Cadmium Exposure in Persian Sturgeon (Acipenser persicus). Biol. Trace Element Res. 2011, 143, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Bitto, A.; Interdonato, M.; Galfo, F.; Irrera, N.; Mecchio, A.; Pallio, G.; Ramistella, V.; De Luca, F.; Minutoli, L.; et al. Oxidative stress and DNA repair and detoxification gene expression in adolescents exposed to heavy metals living in the Milazzo-Valle del Mela area (Sicily, Italy). Redox Biol. 2014, 2, 686–693. [Google Scholar] [CrossRef]
- Emdin, S.O.; Dodson, G.G.; Cutfield, J.M.; Cutfield, S.M. Role of zinc in insulin biosynthesis. Some possible zinc-insulin interactions in the pancreatic B-cell. Diabetologia 1980, 19, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.P.; Dauter, Z.; Dodson, E.J.; Dodson, G.G.; Dunn, M.F. X-ray structure of an unusual Ca2+ site and the roles of Zn2+ and Ca2+ in the assembly, stability, and storage of the insulin hexamer. Biochemistry 1991, 30, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.F. Zinc–Ligand Interactions Modulate Assembly and Stability of the Insulin Hexamer—A Review. BioMetals 2005, 18, 295–303. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the Zinc-Proteins Encoded in the Human Genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc Biochemistry: From a Single Zinc Enzyme to a Key Element of Life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Vašák, M. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1985, 827, 36–44. [Google Scholar] [CrossRef]
- Schwarz, M.A.; Lazo, J.S.; Yalowich, J.C.; Allen, W.P.; Whitmore, M.; Bergonia, H.A.; Tzeng, E.; Billiar, T.R.; Robbins, P.D.; Lancaster, J.R., Jr.; et al. Metallothionein protects against the cytotoxic and DNA-damaging effects of nitric oxide. Proc. Natl. Acad. Sci. USA 1995, 92, 4452–4456. [Google Scholar] [CrossRef]
- Kumari, M.R.; Hiramatsu, M.; Ebadi, M. Free radical scavenging actions of metallothionein isoforms I and II. Free. Radic. Res. 1998, 29, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Vallee, B.L. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc. Natl. Acad. Sci. USA 1998, 95, 3478–3482. [Google Scholar] [CrossRef]
- Cortese, M.M.; Suschek, C.V.; Wetzel, W.; Kröncke, K.-D.; Kolb-Bachofen, V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free. Radic. Biol. Med. 2008, 44, 2002–2012. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Kuroda, T. Transfer of copper and zinc from ionic and metallothionein-bound forms to Cu, Zn--superoxide dismutase. Res. Commun. Mol. Pathol. Pharmacol. 1995, 87, 287–296. [Google Scholar] [PubMed]
- Dalton, T.; Palmiter, R.D.; Andrews, G.K. Transcriptional induction of the mouse metallothionein-I gene in hydrogen peroxide-treated Hepa cells involves a composite major late transcription factor/antioxidant response element and metal response promoter elements. Nucleic Acids Res. 1994, 22, 5016–5023. [Google Scholar] [CrossRef] [PubMed]
- Campagne, M.V.L.; Thibodeaux, H.; Van Bruggen, N.; Cairns, B.; Lowe, D.G. Increased Binding Activity at an Antioxidant-Responsive Element in the Metallothionein-1 Promoter and Rapid Induction of Metallothionein-1 and -2 in Response to Cerebral Ischemia and Reperfusion. J. Neurosci. 2000, 20, 5200–5207. [Google Scholar] [CrossRef]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 Play Distinct Roles in Activation of Antioxidant Response Element-dependent Genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Muraoka, S.; Ogiso, T. Antioxidant activity of metallothionein compared with reduced glutathione. Life Sci. 1997, 60, 301–309. [Google Scholar] [CrossRef]
- Chubatsu, L.S.; Meneghini, R. Metallothionein protects DNA from oxidative damage. Biochem. J. 1993, 291 Pt 1, 193–198. [Google Scholar] [CrossRef]
- Lazo, J.S.; Kondo, Y.; Dellapiazza, D.; Michalska, A.E.; Choo, K.H.A.; Pitt, B.R. Enhanced Sensitivity to Oxidative Stress in Cultured Embryonic Cells from Transgenic Mice Deficient in Metallothionein I and II Genes. J. Biol. Chem. 1995, 270, 5506–5510. [Google Scholar] [CrossRef]
- Pitt, B.R.; Schwarz, M.; Woo, E.S.; Yee, E.; Wasserloos, K.; Tran, S.; Weng, W.; Mannix, R.J.; Watkins, S.A.; Tyurina, Y.Y.; et al. Overexpression of metallothionein decreases sensitivity of pulmonary endothelial cells to oxidant injury. Am. J. Physiol. Content 1997, 273, L856–L865. [Google Scholar] [CrossRef] [PubMed]
- Viarengo, A.; Burlando, B.; Cavaletto, M.; Marchi, B.; Ponzano, E.; Blasco, J. Role of metallothionein against oxidative stress in the mussel Mytilus galloprovincialis. Am. J. Physiol. Content 1999, 277, R1612–R1619. [Google Scholar] [CrossRef]
- Suzuki, Y.; Apostolova, M.D.; Cherian, M.G. Astrocyte cultures from transgenic mice to study the role of metallothionein in cytotoxicity of tert-butyl hydroperoxide. Toxicology 2000, 145, 51–62. [Google Scholar] [CrossRef]
- Chimienti, F.; Jourdan, E.; Favier, A.; Sève, M. Zinc resistance impairs sensitivity to oxidative stress in hela cells: Protection through metallothioneins expression. Free. Radic. Biol. Med. 2001, 31, 1179–1190. [Google Scholar] [CrossRef]
- Wang, G.W.; Klein, J.B.; Kang, Y.J. Metallothionein inhibits doxorubicin-induced mitochondrial cytochrome c release and caspase-3 activation in cardiomyocytes. J. Pharmacol. Exp. Ther. 2001, 298, 461–468. [Google Scholar] [PubMed]
- Liang, Q.; Carlson, E.C.; Donthi, R.V.; Kralik, P.M.; Shen, X.; Epstein, P.N. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes 2002, 51, 174–181. [Google Scholar] [CrossRef]
- Molinero, A.; Penkowa, M.; Hernández, J.; Camats, J.; Giralt, M.; Lago, N.; Carrasco, J.; Campbell, I.L.; Hidalgo, J. Metallothionein-I overexpression decreases brain pathology in transgenic mice with astrocyte-targeted expression of interleukin-6. J. Neuropathol. Exp. Neurol. 2003, 62, 315–328. [Google Scholar] [CrossRef]
- Sharma, S.K.; Ebadi, M. Metallothionein Attenuates 3-Morpholinosydnonimine (SIN-1)-Induced Oxidative Stress in Dopaminergic Neurons. Antioxidants Redox Signal. 2003, 5, 251–264. [Google Scholar] [CrossRef]
- Reinecke, F.; Levanets, O.; Olivier, Y.; Louw, R.; Semete, B.; Grobler, A.; Hidalgo, J.; Smeitink, J.; Olckers, A.; Van Der Westhuizen, F.H. Metallothionein isoform 2A expression is inducible and protects against ROS-mediated cell death in rotenone-treated HeLa cells. Biochem. J. 2006, 395, 405–415. [Google Scholar] [CrossRef][Green Version]
- Cavalca, E.; Cesani, M.; Gifford, J.C.; Esteves, M.S.; Terreni, M.R.; Leoncini, G.; Peviani, M.; Biffi, A.; Gifford, J.C. Metallothioneins are neuroprotective agents in lysosomal storage disorders. Ann. Neurol. 2018, 83, 418–432. [Google Scholar] [CrossRef]
- Wang, K.; Dai, X.; He, J.; Yan, X.; Yang, C.; Fan, X.; Sun, S.; Chen, J.; Xu, J.; Deng, Z.; et al. Endothelial Overexpression of Metallothionein Prevents Diabetes-Induced Impairment in Ischemia Angiogenesis Through Preservation of HIF-1α/SDF-1/VEGF Signaling in Endothelial Progenitor Cells. Diabetes 2020, 69, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.A.; Hidalgo, J.; Manso, Y.; Devisscher, L.; Laukens, D.; Lawrence, D.A. Metallothionein and stress combine to affect multiple organ systems. Cell Stress Chaperon 2014, 19, 605–611. [Google Scholar] [CrossRef]
- Subramanian Vignesh, K.; Deepe, G.S., Jr. Metallothioneins: Emerging Modulators in Immunity and Infection. Int. J. Mol. Sci. 2017, 18, 2197. [Google Scholar] [CrossRef]
- Robertson, R.P. Chronic Oxidative Stress as a Central Mechanism for Glucose Toxicity in Pancreatic Islet Beta Cells in Diabetes. J. Biol. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef]
- Bensellam, M.; Jonas, J.-C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Jabůrek, M.; Plecitá-Hlavatá, L. Contribution of Oxidative Stress and Impaired Biogenesis of Pancreatic β-Cells to Type 2 Diabetes. Antioxidants Redox Signal. 2019, 31, 722–751. [Google Scholar] [CrossRef]
- Roma, L.P.; Jonas, J.-C. Nutrient Metabolism, Subcellular Redox State, and Oxidative Stress in Pancreatic Islets and β-Cells. J. Mol. Biol. 2020, 432, 1461–1493. [Google Scholar] [CrossRef]
- Hansen, J.B.J.; Tonnesen, M.F.M.; Madsen, A.N.A.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.L.; Heller, R.S.R.; Nielsen, A.O.A.; Størling, J.; Baeyens, L.; et al. Divalent Metal Transporter 1 Regulates Iron-Mediated ROS and Pancreatic β Cell Fate in Response to Cytokines. Cell Metab. 2012, 16, 449–461. [Google Scholar] [CrossRef]
- Chimienti, F. Zinc, pancreatic islet cell function and diabetes: New insights into an old story. Nutr. Res. Rev. 2013, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gilon, P.; Chae, H.-Y.; Rutter, G.A.; Ravier, M.A. Calcium signaling in pancreatic β-cells in health and in Type 2 diabetes. Cell Calcium 2014, 56, 340–361. [Google Scholar] [CrossRef]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular zinc in insulin secretion and action: A determinant of diabetes risk? Proc. Nutr. Soc. 2015, 75, 61–72. [Google Scholar] [CrossRef]
- Farooq, M. Zinc Deficiency is Associated with Poor Glycemic Control. J. Coll. Physicians Surg. Pak. 2019, 29, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, X.; Li, S.; Zhu, Y.; Chen, S.; Li, P.; Luo, C.; Huang, Y.; Li, X.; Hu, X.; et al. Interactions between plasma copper concentrations and SOD1 gene polymorphism for impaired glucose regulation and type 2 diabetes. Redox Biol. 2019, 24, 101172. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.M., Jr.; Uriu-Hare, J.Y.; Olin, K.L.; Oster, M.H.; Anawalt, B.D.; Critchfield, J.W.; Keen, C.L. Copper, Zinc, Manganese, and Magnesium Status and Complications of Diabetes Mellitus. Diabetes Care 1991, 14, 1050–1056. [Google Scholar] [CrossRef]
- Garg, V.K.; Gupta, R.; Goyal, R.K. Hypozincemia in diabetes mellitus. J. Assoc. Physicians India 1994, 42, 720–721. [Google Scholar]
- Taylor, C.G. Zinc, the Pancreas, and Diabetes: Insights from Rodent Studies and Future Directions. BioMetals 2005, 18, 305–312. [Google Scholar] [CrossRef]
- Viktorínová, A.; Tošerová, E.; Križko, M.; Ďuračková, Z. Altered metabolism of copper, zinc, and magnesium is associated with increased levels of glycated hemoglobin in patients with diabetes mellitus. Metabolism 2009, 58, 1477–1482. [Google Scholar] [CrossRef]
- Basaki, M.; Saeb, M.; Nazifi, S.; Shamsaei, H.A. Zinc, Copper, Iron, and Chromium Concentrations in Young Patients with Type 2 Diabetes Mellitus. Biol. Trace Element Res. 2012, 148, 161–164. [Google Scholar] [CrossRef]
- Jansen, J.; Rosenkranz, E.; Overbeck, S.; Warmuth, S.; Mocchegiani, E.; Giacconi, R.; Weiskirchen, R.; Karges, W.; Rink, L. Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J. Nutr. Biochem. 2012, 23, 1458–1466. [Google Scholar] [CrossRef]
- Giacconi, R.; Bonfigli, A.R.; Testa, R.; Sirolla, C.; Cipriano, C.; Marra, M.; Muti, E.; Malavolta, M.; Costarelli, L.; Piacenza, F.; et al. +647 A/C and +1245 MT1A polymorphisms in the susceptibility of diabetes mellitus and cardiovascular complications. Mol. Genet. Metab. 2008, 94, 98–104. [Google Scholar] [CrossRef]
- Yang, L.; Li, H.; Yu, T.; Zhao, H.; Cherian, M.G.; Cai, L.; Liu, Y. Polymorphisms in metallothionein-1 and -2 genes associated with the risk of type 2 diabetes mellitus and its complications. Am. J. Physiol. Metab. 2008, 294, E987–E992. [Google Scholar] [CrossRef] [PubMed]
- Raudenska, M.; Gumulec, J.; Podlaha, O.; Sztalmachova, M.; Babula, P.; Eckschlager, T.; Adam, V.; Kizek, R.; Masarik, M. Metallothionein polymorphisms in pathological processes. Metallomics 2014, 6, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Yau, E.T.; Mennear, J.H. Pancreatic metallothionein: Protection against cadmium-induced inhibition of insulin secretory activity. Toxicol. Appl. Pharmacol. 1977, 39, 515–520. [Google Scholar] [CrossRef]
- Zimny, S.; Gogolin, F.; Abel, J.; Gleichmann, H. Metallothionein in isolated pancreatic islets of mice: Induction by zinc and streptozotocin, a naturally occurring diabetogen. Arch. Toxicol. 1993, 67, 61–65. [Google Scholar] [CrossRef]
- Ohly, P.; Dohle, C.; Abel, J.; Seissler, J.; Gleichmann, H. Zinc sulphate induces metallothionein in pancreatic islets of mice and protects against diabetes induced by multiple low doses of streptozotocin. Diabetologia 2000, 43, 1020–1030. [Google Scholar] [CrossRef]
- Tomita, T.; Matsubara, O. Immunocytochemical Localization of Metallothionein in Human Pancreatic Islets. Pancreas 2000, 20, 21–24. [Google Scholar] [CrossRef]
- DiGruccio, M.R.; Mawla, A.M.; Donaldson, C.J.; Noguchi, G.M.; Vaughan, J.; Cowing-Zitron, C.; van der Meulen, T.; Huising, M.O.; van der Meulen, T. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 2016, 5, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, D.M.; Nowosielska, A.; Afik, S.; Pechhold, S.; Cura, A.J.; Kennedy, N.J.; Kim, S.; Kucukural, A.; Davis, R.J.; Kent, S.C.; et al. Novel Observations From Next-Generation RNA Sequencing of Highly Purified Human Adult and Fetal Islet Cell Subsets. Diabetes 2015, 64, 3172–3181. [Google Scholar] [CrossRef]
- El Muayed, M.; Raja, M.R.; Zhang, X.; MacRenaris, K.W.; Bhatt, S.; Chen, X.; Urbanek, M.; O’Halloran, T.V.; Lowe, J.W.L., Jr. Accumulation of cadmium in insulin-producing β cells. Islets 2012, 4, 405–416. [Google Scholar] [CrossRef]
- Andrews, G.K.; Kage, K.; Palmiter-Thomas, P.; Sarras, M.P. Metal Ions Induce Expression Pancreatic Exocrine and of Met allot hionein in Endocrine Cells. Pancreas 1990, 5, 548–554. [Google Scholar] [CrossRef]
- Ohly, P.; Gleichmann, H. Metallothionein: In Vitro Induction With Zinc and Streptozotocin in Pancreatic Islets of Mice. Exp. Clin. Endocrinol. Diabetes 1995, 103 (Suppl. 2), 79–82. [Google Scholar] [CrossRef]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose Regulates Free Cytosolic Zn2+ Concentration, Slc39 (ZiP), and Metallothionein Gene Expression in Primary Pancreatic Islet β-Cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef] [PubMed]
- Duprez, J.; Roma, L.P.; Close, A.-F.; Jonas, J.-C. Protective Antioxidant and Antiapoptotic Effects of ZnCl2 in Rat Pancreatic Islets Cultured in Low and High Glucose Concentrations. PLoS ONE 2012, 7, e46831. [Google Scholar] [CrossRef]
- Nygaard, S.B.; Larsen, A.; Knuhtsen, A.; Rungby, J.; Smidt, K. Effects of zinc supplementation and zinc chelation on in vitro β-cell function in INS-1E cells. BMC Res. Notes 2014, 7, 84. [Google Scholar] [CrossRef]
- Hinke, S.A.; Hellemans, K.; Schuit, F.C. Plasticity of the β cell insulin secretory competence: Preparing the pancreatic β cell for the next meal. J. Physiol. 2004, 558, 369–380. [Google Scholar] [CrossRef]
- Jonas, J.-C.; Bensellam, M.; Duprez, J.; Elouil, H.; Guiot, Y.; Pascal, S.M.A. Glucose regulation of islet stress responses and β-cell failure in type 2 diabetes. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 65–81. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Van Lommel, L.; Overbergh, L.; Schuit, F.C.; Jonas, J.-C. Cluster analysis of rat pancreatic islet gene mRNA levels after culture in low-, intermediate- and high-glucose concentrations. Diabetologia 2009, 52, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A. Isolated mouse pancreatic islets in culture: Effects of serum and different culture media on the insulin production of the islets. Diabetologia 1978, 14, 397–404. [Google Scholar] [CrossRef]
- Ling, Z.; Pipeleers, D.G. Preservation of glucose-responsive islet beta-cells during serum-free culture. Endocrinol. 1994, 134, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Efanova, I.B.; Zaitsev, S.V.; Zhivotovsky, B.; Köhler, M.; Efendić, S.; Orrenius, S.; Berggren, P.-O. Glucose and Tolbutamide Induce Apoptosis in Pancreatic β-Cells. A process dependent on intracellular Ca2+ concentration. J. Biol. Chem. 1998, 273, 33501–33507. [Google Scholar] [CrossRef]
- Bensellam, M.; Duvillié, B.; Rybachuk, G.; Laybutt, D.R.; Magnan, C.; Guiot, Y.; Pouysségur, J.; Jonas, J.-C. Glucose-Induced O2 Consumption Activates Hypoxia Inducible Factors 1 and 2 in Rat Insulin-Secreting Pancreatic Beta-Cells. PLoS ONE 2012, 7, e29807. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Korbutt, G.S.; Hellerström, C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the beta-cell function. J. Clin. Investig. 1992, 90, 1263–1268. [Google Scholar] [CrossRef]
- Ling, Z.; Pipeleers, D.G. Prolonged exposure of human beta cells to elevated glucose levels results in sustained cellular activation leading to a loss of glucose regulation. J. Clin. Investig. 1996, 98, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Schnedl, W.J.; Ferber, S.; Johnson, J.H.; Newgard, C.B. STZ Transport and Cytotoxicity: Specific Enhancement in GLUT2-Expressing Cells. Diabetes 1994, 43, 1326–1333. [Google Scholar] [CrossRef]
- Takasu, N.; Komiya, I.; Asawa, T.; Nagasawa, Y.; Yamada, T. Streptozocin- and Alloxan-Induced H2O2 Generation and DNA Fragmentation in Pancreatic Islets: H2O2 as Mediator for DNA Fragmentation. Diabetes 1991, 40, 1141–1145. [Google Scholar] [CrossRef]
- Gille, L.; Schott-Ohly, P.; Friesen, N.; Walde, S.S.I.; Udilova, N.; Nohl, H.; Gleichmann, H. Generation of Hydroxyl Radicals Mediated by Streptozotocin in Pancreatic Islets of Mice in vitro. Pharmacol. Toxicol. 2002, 90, 317–326. [Google Scholar] [CrossRef]
- Buchau, A.S.; Schott-Ohly, P.; Lgssiar, A.; Gleichmann, H.; Friesen, N.T.E. Generation of hydrogen peroxide and failure of antioxidative responses in pancreatic islets of male C57BL/6 mice are associated with diabetes induced by multiple low doses of streptozotocin. Diabetologia 2004, 47, 676–685. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2007, 51, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Laychock, S.G.; Duzen, J.; Simpkins, C.O. Metallothionein induction in islets of Langerhans and insulinoma cells. Mol. Cell. Endocrinol. 2000, 165, 179–187. [Google Scholar] [CrossRef]
- Cardozo, A.K.; Kruhøffer, M.; Leeman, R.; Ørntoft, T.; Eizirik, D.L. Identification of Novel Cytokine-Induced Genes in Pancreatic -Cells by High-Density Oligonucleotide Arrays. Diabetes 2001, 50, 909–920. [Google Scholar] [CrossRef]
- Ortis, F.; Naamane, N.; Flamez, D.; Ladrière, L.; Moore, F.; Cunha, D.A.; Colli, M.L.; Thykjaer, T.; Thorsen, K.; Ørntoft, T.F.; et al. Cytokines Interleukin-1 and Tumor Necrosis Factor- Regulate Different Transcriptional and Alternative Splicing Networks in Primary -Cells. Diabetes 2009, 59, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Mandrup-Poulsen, T.; Zumsteg, U.; Reimers, J.; Pociot, F.; Mørch, L.; Helqvist, S.; Dinarello, C.A.; Nerup, J. Involvement of interleukin 1 and interleukin 1 antagonist in pancreatic β-cell destruction in insulin-dependent diabetes mellitus. Cytokine 1993, 5, 185–191. [Google Scholar] [CrossRef]
- Tabatabaie, T.; Vasquez-Weldon, A.; Moore, D.R.; Kotake, Y. Free Radicals and the Pathogenesis of Type 1 Diabetes: -Cell Cytokine-Mediated Free Radical Generation Via Cyclooxygenase-2. Diabetes 2003, 52, 1994–1999. [Google Scholar] [CrossRef]
- Lee, D.K.; Carrasco, J.; Hidalgo, J.; Andrews, G.K. Identification of a signal transducer and activator of transcription (STAT) binding site in the mouse metallothionein-I promoter involved in interleukin-6-induced gene expression. Biochem. J. 1999, 337 Pt 1, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Haslinger, A.; Holtgreve, H.; Richards, R.I.; Krauter, P.; Westphal, H.M.; Beato, M. Characterization of DNA sequences through which cadmium and glucocorticoid hormones induce human metallothionein-IIA gene. Nat. Cell Biol. 1984, 308, 513–519. [Google Scholar] [CrossRef]
- Kelly, E.J.; Sandgren, E.P.; Brinster, R.L.; Palmiter, R.D. A pair of adjacent glucocorticoid response elements regulate expression of two mouse metallothionein genes. Proc. Natl. Acad. Sci. USA 1997, 94, 10045–10050. [Google Scholar] [CrossRef]
- Sargsyan, E.; Cen, J.; Roomp, K.; Schneider, R.; Bergsten, P. Identification of early biological changes in palmitate-treated isolated human islets. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Bikopoulos, G.; da Silva Pimenta, A.; Lee, S.C.; Lakey, J.R.; Der, S.D.; Chan, C.B.; Ceddia, R.B.; Wheeler, M.B.; Rozakis-Adcock, M. Ex vivo transcriptional profiling of human pancreatic islets following chronic exposure to monounsaturated fatty acids. J. Endocrinol. 2007, 196, 455–464. [Google Scholar] [CrossRef]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-Generated Hydrogen Peroxide as Important Mediator of Lipotoxicity in Insulin-Producing Cells. Diabetes 2010, 60, 200–208. [Google Scholar] [CrossRef]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.A.; Bellomo, E.A.; Hodson, D.J.; Meur, G.; Solomou, A.; Mitchell, R.K.; Hollinshead, M.; Chimienti, F.; Bosco, M.; Hughes, S.J.; et al. Hypoxia lowers SLC30A8/ZnT8 expression and free cytosolic Zn2+ in pancreatic beta cells. Diabetologia 2014, 57, 1635–1644. [Google Scholar] [CrossRef]
- Chan, J.Y.; Luzuriaga, J.; Bensellam, M.; Biden, T.J.; Laybutt, D.R. Failure of the Adaptive Unfolded Protein Response in Islets of Obese Mice Is Linked with Abnormalities in β-Cell Gene Expression and Progression to Diabetes. Diabetes 2013, 62, 1557–1568. [Google Scholar] [CrossRef]
- Shafrir, E.; Ziv, E.; Mosthaf, L. Nutritionally induced insulin resistance and receptor defect leading to beta-cell failure in animal models. Ann. New York Acad. Sci. 1999, 892, 223–246. [Google Scholar] [CrossRef]
- Kjørholt, C.; Åkerfeldt, M.C.; Biden, T.J.; Laybutt, D.R. Chronic Hyperglycemia, Independent of Plasma Lipid Levels, Is Sufficient for the Loss of -Cell Differentiation and Secretory Function in the db/db Mouse Model of Diabetes. Diabetes 2005, 54, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Marselli, L.; Thorne, J.; Dahiya, S.; Sgroi, D.C.; Sharma, A.; Bonner-Weir, S.; Marchetti, P.; Weir, G.C. Gene Expression Profiles of Beta-Cell Enriched Tissue Obtained by Laser Capture Microdissection from Subjects with Type 2 Diabetes. PLoS ONE 2010, 5, e11499. [Google Scholar] [CrossRef] [PubMed]
- Roat, R.; Rao, V.; Doliba, N.M.; Matschinsky, F.M.; Tobias, J.W.; Garcia, E.; Ahima, R.S.; Imai, Y. Alterations of Pancreatic Islet Structure, Metabolism and Gene Expression in Diet-Induced Obese C57BL/6J Mice. PLoS ONE 2014, 9, e86815. [Google Scholar] [CrossRef]
- Hatanaka, M.; Anderson-Baucum, E.; Lakhter, A.; Kono, T.; Maier, B.; Tersey, S.A.; Tanizawa, Y.; Evans-Molina, C.; Mirmira, R.G.; Sims, E.K. Chronic high fat feeding restricts islet mRNA translation initiation independently of ER stress via DNA damage and p53 activation. Sci. Rep. 2017, 7, 3758. [Google Scholar] [CrossRef]
- Maret, W. Zinc in Pancreatic Islet Biology, Insulin Sensitivity, and Diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef]
- Raz, I.; Karsai, D.; Katz, M. The influence of zinc supplementation on glucose homeostasis in NIDDM. Diabetes Res. (Edinburgh, Scotland) 1989, 11, 73–79. [Google Scholar]
- Minami, T.; Shimizu, M.; Tanaka, H.; Okazaki, Y.; Cherian, M. Metallothionein does not protect mouse endocrine cells from damage induced by alloxan injection. Toxicology 1999, 132, 33–41. [Google Scholar] [CrossRef]
- De Sena, K.C.M.; Arrais, R.F.; das Gracas Almeida, M.; De Araújo, A.D.M.; Dos Santos, M.M.; De Lima, V.T.; Pedrosa, L.D.F.C. Effects of Zinc Supplementation in Patients with Type 1 Diabetes. Biol. Trace Element Res. 2005, 105, 1–9. [Google Scholar] [CrossRef]
- El Dib, R.; Gameiro, O.L.F.; Ogata, M.S.P.; Módolo, N.S.P.; Braz, L.G.; Jorge, E.C.; Junior, P.D.N.; Beletate, V.; Nascimento, P.D. Zinc supplementation for the prevention of type 2 diabetes mellitus in adults with insulin resistance. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Chen, H.; Carlson, E.C.; Pellet, L.; Moritz, J.T.; Epstein, P.N. Overexpression of metallothionein in pancreatic beta-cells reduces streptozotocin-induced DNA damage and diabetes. Diabetes 2001, 50, 2040–2046. [Google Scholar] [CrossRef]
- Chen, S.; Han, J.; Liu, Y. Dual Opposing Roles of Metallothionein Overexpression in C57BL/6J Mouse Pancreatic β-Cells. PLoS ONE 2015, 10, e0137583. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, H.; Epstein, P.N. Metallothionein Protects Islets from Hypoxia and Extends Islet Graft Survival by Scavenging Most Kinds of Reactive Oxygen Species. J. Biol. Chem. 2004, 279, 765–771. [Google Scholar] [CrossRef]
- Lim, K.S.; Won, Y.-W.; Park, Y.S.; Kim, Y.-H. Preparation and functional analysis of recombinant protein transduction domain-metallothionein fusion proteins. Biochimie 2010, 92, 964–970. [Google Scholar] [CrossRef]
- Park, L.; Min, D.; Kim, H.; Chung, H.-Y.; Lee, C.-H.; Park, I.-S.; Kim, Y.; Park, Y. Tat-enhanced delivery of metallothionein can partially prevent the development of diabetes. Free. Radic. Biol. Med. 2011, 51, 1666–1674. [Google Scholar] [CrossRef]
- Park, L.; Min, D.; Kim, H.; Park, J.; Choi, S.; Park, Y. The combination of metallothionein and superoxide dismutase protects pancreatic β cells from oxidative damage. Diabetes/Metabolism Res. Rev. 2011, 27, 802–808. [Google Scholar] [CrossRef]
- Jung, H.S.; Lim, K.S.; Kim, M.J.; Hwang, Y.H.; Yoo, C.; Lee, Y.-K.; Kim, Y.-H.; Lee, D.Y. Hypoxic resistance of hypodermically transplanted pancreatic islets by using cell-absorbable antioxidant Tat-metallothionein. J. Control. Release 2013, 172, 1092–1101. [Google Scholar] [CrossRef]
- Kim, M.J.; Hwang, Y.H.; Kim, Y.H.; Lee, D.Y. Immunomodulation of cell-penetrating tat-metallothionein for successful outcome of xenotransplanted pancreatic islet. J. Drug Target. 2016, 25, 350–359. [Google Scholar] [CrossRef]
- Li, X.; Chen, H.; Epstein, P.N. Metallothionein and Catalase Sensitize to Diabetes in Nonobese Diabetic Mice: Reactive Oxygen Species May Have a Protective Role in Pancreatic -Cells. Diabetes 2006, 55, 1592–1604. [Google Scholar] [CrossRef]
- Huang, S.H.; Chu, C.H.; Yu, J.C.; Chuang, W.C.; Lin, G.J.; Chen, P.L.; Chou, F.-C.; Chau, L.Y.; Sytwu, H.K. Transgenic expression of haem oxygenase-1 in pancreatic beta cells protects non-obese mice used as a model of diabetes from autoimmune destruction and prolongs graft survival following islet transplantation. Diabetologia 2010, 53, 2389–2400. [Google Scholar] [CrossRef][Green Version]
- Chou, F.-C.; Sytwu, H.-K. Overexpression of thioredoxin in islets transduced by a lentiviral vector prolongs graft survival in autoimmune diabetic NOD mice. J. Biomed. Sci. 2009, 16, 71. [Google Scholar] [CrossRef]
- Bertera, S.; Crawford, M.L.; Alexander, A.M.; Papworth, G.D.; Watkins, S.C.; Robbins, P.D.; Trucco, M. Gene transfer of manganese superoxide dismutase extends islet graft function in a mouse model of autoimmune diabetes. Diabetes 2003, 52, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.-R.; Choi, J.A.; Koh, J.-Y. The role of metallothionein-3 in streptozotocin-induced beta-islet cell death and diabetes in mice. Metallomics 2014, 6, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Sandgren, E.P.; Koeller, D.M.; Brinster, R.L. Distal regulatory elements from the mouse metallothionein locus stimulate gene expression in transgenic mice. Mol. Cell. Biol. 1993, 13, 5266–5275. [Google Scholar] [CrossRef] [PubMed]
- Iszard, M.; Liu, J.; Liu, Y.; Dalton, T.; Andrews, G.; Palmiter, R.; Klaassen, C. Characterization of Metallothionein-I-Transgenic Mice. Toxicol. Appl. Pharmacol. 1995, 133, 305–312. [Google Scholar] [CrossRef]
- Artells, E.; Palacios, Ò.; Capdevila, M.; Atrian, S. Mammalian MT1 and MT2 metallothioneins differ in their metal binding abilities. Metallomics 2013, 5, 1397–1410. [Google Scholar] [CrossRef]
- Atrian, S.; Capdevila, M. Metallothionein-protein interactions. Biomol. Concepts 2013, 4, 143–160. [Google Scholar] [CrossRef]
- Knipp, M.; Meloni, G.; Roschitzki, A.B.; Vasák, M. Zn7Metallothionein-3 and the Synaptic Vesicle Cycle: Interaction of Metallothionein-3 with the Small GTPase Rab3A†. Biochemistry 2005, 44, 3159–3165. [Google Scholar] [CrossRef]
- Guo, W.; Grant, A.; Novick, P. Exo84p Is an Exocyst Protein Essential for Secretion. J. Biol. Chem. 1999, 274, 23558–23564. [Google Scholar] [CrossRef]
- El Ghazi, I.; Martin, B.L.; Armitage, I.M. New Proteins Found Interacting with Brain Metallothionein-3 Are Linked to Secretion. Int. J. Alzheimer Dis. 2011, 2011, 208634. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Qu, W.; Cooper, R.N.; Goyer, R.A.; Diwan, B.A.; Waalkes, M.P. Potential Role of α-Synuclein and Metallothionein in Lead-Induced Inclusion Body Formation. Toxicol. Sci. 2009, 111, 100–108. [Google Scholar] [CrossRef]
- Geng, X.; Lou, H.; Wang, J.; Li, L.; Swanson, A.L.; Sun, M.; Beers-Stolz, D.; Watkins, S.; Perez, R.G.; Drain, P. α-Synuclein binds the KATP channel at insulin-secretory granules and inhibits insulin secretion. Am. J. Physiol. Metab. 2011, 300, E276–E286. [Google Scholar] [CrossRef]
- Steneberg, P.; Bernardo, L.; Edfalk, S.; Lundberg, L.; Backlund, F.; Östenson, C.-G.; Edlund, H. The Type 2 Diabetes-Associated Gene Ide Is Required for Insulin Secretion and Suppression of -Synuclein Levels in -Cells. Diabetes 2013, 62, 2004–2014. [Google Scholar] [CrossRef]
- Rodriguez-Araujo, G.; Nakagami, H.; Takami, Y.; Katsuya, T.; Akasaka, H.; Saitoh, S.; Shimamoto, K.; Morishita, R.; Rakugi, H.; Kaneda, Y. Low alpha-synuclein levels in the blood are associated with insulin resistance. Sci. Rep. 2015, 5, 12081. [Google Scholar] [CrossRef]
- Butler, M.G.; Wang, K.; Naggert, J.K.; Rethmeyer, J.A.; Gunewardena, S.S.; Manzardo, A.M.; Marshall, J.D. Coding and noncoding expression patterns associated with rare obesity-related disorders: Prader–Willi and Alström syndromes. Adv. Genom. Genet. 2015, 5, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Juntti-Berggren, L.; Lindh, U.; Berggren, P.-O.; Frankel, B.J. Elemental Composition in the Pancreatic B Cell Is Normal in the Prediabetic Chinese Hamster. Pancreas 1992, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Juntti-Berggren, L.; Lindh, U.; Berggren, P.-O.; Berglund, O.; Frankel, B.J. Diabetes is not associated with a change in the elemental composition of the pancreatic B cell in diabetic C57BL KsJ-db/db mice. Biosci. Rep. 1990, 10, 217–223. [Google Scholar] [CrossRef] [PubMed]
- De Samber, B.; Bensellam, M.; Van Malderen, S.J.M.; Seiboth, F.; Brückner, D.; Garrevoet, J.; Falkenberg, G.; Jonas, J.-C.; Vincze, L. Proof-of-concept for 2D/CT element analysis of entire cryofrozen islets of Langerhans using a cryoloop synchrotron X-ray fluorescence setup. J. Anal. At. Spectrom. 2020, 35, 1368–1379. [Google Scholar] [CrossRef]
- Takahashi, S. Positive and negative regulators of the metallothionein gene (Review). Mol. Med. Rep. 2012, 12, 795–799. [Google Scholar] [CrossRef]
- Roma, L.P.; Duprez, J.; Jonas, J.-C. Glucokinase activation is beneficial or toxic to cultured rat pancreatic islets depending on the prevailing glucose concentration. Am. J. Physiol. Metab. 2015, 309, E632–E639. [Google Scholar] [CrossRef]
- Martens, G.A.; Cai, Y.; Hinke, S.A.; Stangé, G.; Van De Casteele, M.; Pipeleers, D. Glucose Suppresses Superoxide Generation in Metabolically Responsive Pancreatic β Cells. J. Biol. Chem. 2005, 280, 20389–20396. [Google Scholar] [CrossRef]
- Roma, L.P.; Pascal, S.M.; Duprez, J.; Jonas, J.-C. Mitochondrial oxidative stress contributes differently to rat pancreatic islet cell apoptosis and insulin secretory defects after prolonged culture in a low non-stimulating glucose concentration. Diabetologia 2012, 55, 2226–2237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deglasse, J.-P.; Roma, L.P.; Pastor-Flores, D.; Gilon, P.; Dick, T.P.; Jonas, J.-C. Glucose Acutely Reduces Cytosolic and Mitochondrial H2O2 in Rat Pancreatic Beta Cells. Antioxidants Redox Signal. 2019, 30, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Goginashvili, A.; Zhang, Z.; Erbs, E.; Spiegelhalter, C.; Kessler, P.; Mihlan, M.; Pasquier, A.; Krupina, K.; Schieber, N.; Cinque, L.; et al. Insulin secretory granules control autophagy in pancreatic cells. Science 2015, 347, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Car, N.; Car, A.; Granic, M.; Skrabalo, Z.; Momčilović, B. Zinc and copper in the serum of diabetic patients. Biol. Trace Element Res. 1992, 32, 325–329. [Google Scholar] [CrossRef]
- Squitti, R.; Negrouk, V.; Perera, M.; Llabre, M.M.; Ricordi, C.; Rongioletti, M.C.A.; Mendez, A.J. Serum copper profile in patients with type 1 diabetes in comparison to other metals. J. Trace Elements Med. Biol. 2019, 56, 156–161. [Google Scholar] [CrossRef]
- El Refaey, H.; Ebadi, M.; Kuszynski, C.A.; Sweeney, J.; Hamada, F.M.; Hamed, A. Identification of metallothionein receptors in human astrocytes. Neurosci. Lett. 1997, 231, 131–134. [Google Scholar] [CrossRef]
- Klassen, R.B.; Crenshaw, K.; Kozyraki, R.; Verroust, P.J.; Tío, L.; Atrian, S.; Allen, P.L.; Hammond, T.G. Megalin mediates renal uptake of heavy metal metallothionein complexes. Am. J. Physiol. Physiol. 2004, 287, F393–F403. [Google Scholar] [CrossRef]
- Yin, X.; Knecht, D.A.; Lynes, M.A. Metallothionein mediates leukocyte chemotaxis. BMC Immunol. 2005, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Abouhamed, M.; Verroust, P.J.; Thévenod, F. Megalin-Dependent Internalization of Cadmium-Metallothionein and Cytotoxicity in Cultured Renal Proximal Tubule Cells. J. Pharmacol. Exp. Ther. 2006, 318, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.S.; Penkowa, M.; Dittmann, J.; King, C.E.; Bartlett, C.; Asmussen, J.W.; Hidalgo, J.; Carrasco, J.; Leung, Y.K.J.; Walker, A.K.; et al. Redefining the Role of Metallothionein within the Injured Brain: Extracellular metallothioneins play an important role in the astrocyte-neuron response to injury. J. Biol. Chem. 2008, 283, 15349–15358. [Google Scholar] [CrossRef]
- Devisscher, L.; Hindryckx, P.; Lynes, M.A.; Waeytens, A.; Cuvelier, C.; De Vos, F.; Vanhove, C.; De Vos, M.; Laukens, D. Role of metallothioneins as danger signals in the pathogenesis of colitis. J. Pathol. 2014, 233, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Lee, K.; Maxwell, E.L.; Liang, C.; Laybutt, D.R. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia 2019, 62, 993–999. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | ID | AA | Protein Sequences |
|---|---|---|---|
| MT1A | P04731 | 61 | MDPNCSCAT-GGSCTCTGSCKCKECKCTSCKKSCCSCCPMSCAKCAQGCICKGAS------EKCSCCA |
| MT1B | P07438 | 61 | MDPNCSCTT-GGSCACAGSCKCKECKCTSCKKCCCSCCPVGCAKCAQGCVCKGSS------EKCRCCA |
| MT1E | P04732 | 61 | MDPNCSCA-TGGSCTCAGSCKCKECKCTSCKKSCCSCCPVGCAKCAQGCVCKGAS------EKCSCCA |
| MT1F | P04733 | 61 | MDPNCSCA-AGVSCTCAGSCKCKECKCTSCKKSCCSCCPVGCSKCAQGCVCKGAS------EKCSCCD |
| MT1G | P13640 | 62 | MDPNCSCAAAGVSCTCASSCKCKECKCTSCKKSCCSCCPVGCAKCAQGCICKGAS------EKCSCCA |
| MT1H | P80294 | 61 | MDPNCSCEA-GGSCACAGSCKCKKCKCTSCKKSCCSCCPLGCAKCAQGCICKGAS------EKCSCCA |
| MT1L | Q93083 | 61 | MDPNCSCAT-GGSCSCASSCKCKECKCTSCKKSCCSCCPMGCAKCAQGCVCKGAS------EKCSCCA |
| MT1M | Q8N339 | 61 | MDPNCSCTT-GVSCACTGSCTCKECKCTSCKKSCCSCCPVGCAKCAHGCVCKGTL------ENCSCCA |
| MT1X | P80297 | 61 | MDPNCSCSPV-GSCACAGSCKCKECKCTSCKKSCCSCCPVGCAKCAQGCICKGTS------DKCSCCA |
| MT2A | P02795 | 61 | MDPNCSCA-AGDSCTCAGSCKCKECKCTSCKKSCCSCCPVGCAKCAQGCICKGAS------DKCSCCA |
| MT3 | P25713 | 68 | MDPETCPCPSGGSCTCADSCKCEGCKCTSCKKSCCSCCPAECEKCAKDCVCKGGEAAEAEAEKCSCCQ |
| MT4 | P47944 | 62 | MDPRECVCMSGGICMCGDNCKCTTCNCKTYWKSCCPCCPPGCAKCARGCICKGGS------DKCSCCP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bensellam, M.; Laybutt, D.R.; Jonas, J.-C. Emerging Roles of Metallothioneins in Beta Cell Pathophysiology: Beyond and above Metal Homeostasis and Antioxidant Response. Biology 2021, 10, 176. https://doi.org/10.3390/biology10030176
Bensellam M, Laybutt DR, Jonas J-C. Emerging Roles of Metallothioneins in Beta Cell Pathophysiology: Beyond and above Metal Homeostasis and Antioxidant Response. Biology. 2021; 10(3):176. https://doi.org/10.3390/biology10030176
Chicago/Turabian StyleBensellam, Mohammed, D. Ross Laybutt, and Jean-Christophe Jonas. 2021. "Emerging Roles of Metallothioneins in Beta Cell Pathophysiology: Beyond and above Metal Homeostasis and Antioxidant Response" Biology 10, no. 3: 176. https://doi.org/10.3390/biology10030176
APA StyleBensellam, M., Laybutt, D. R., & Jonas, J.-C. (2021). Emerging Roles of Metallothioneins in Beta Cell Pathophysiology: Beyond and above Metal Homeostasis and Antioxidant Response. Biology, 10(3), 176. https://doi.org/10.3390/biology10030176