
A Novel In Silico Benchmarked Pipeline Capable of Complete Protein Analysis: A Possible Tool for Potential Drug Discovery



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
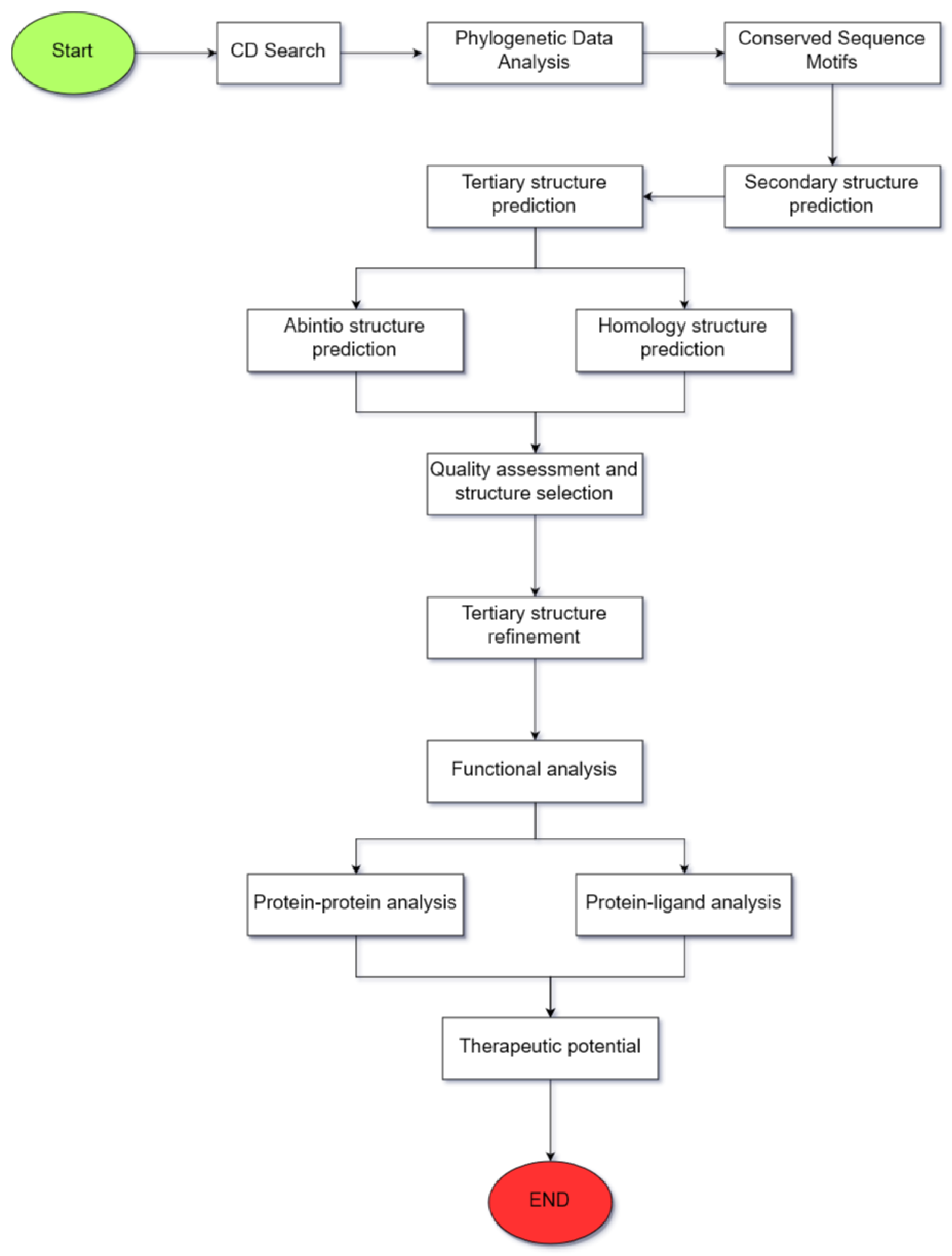
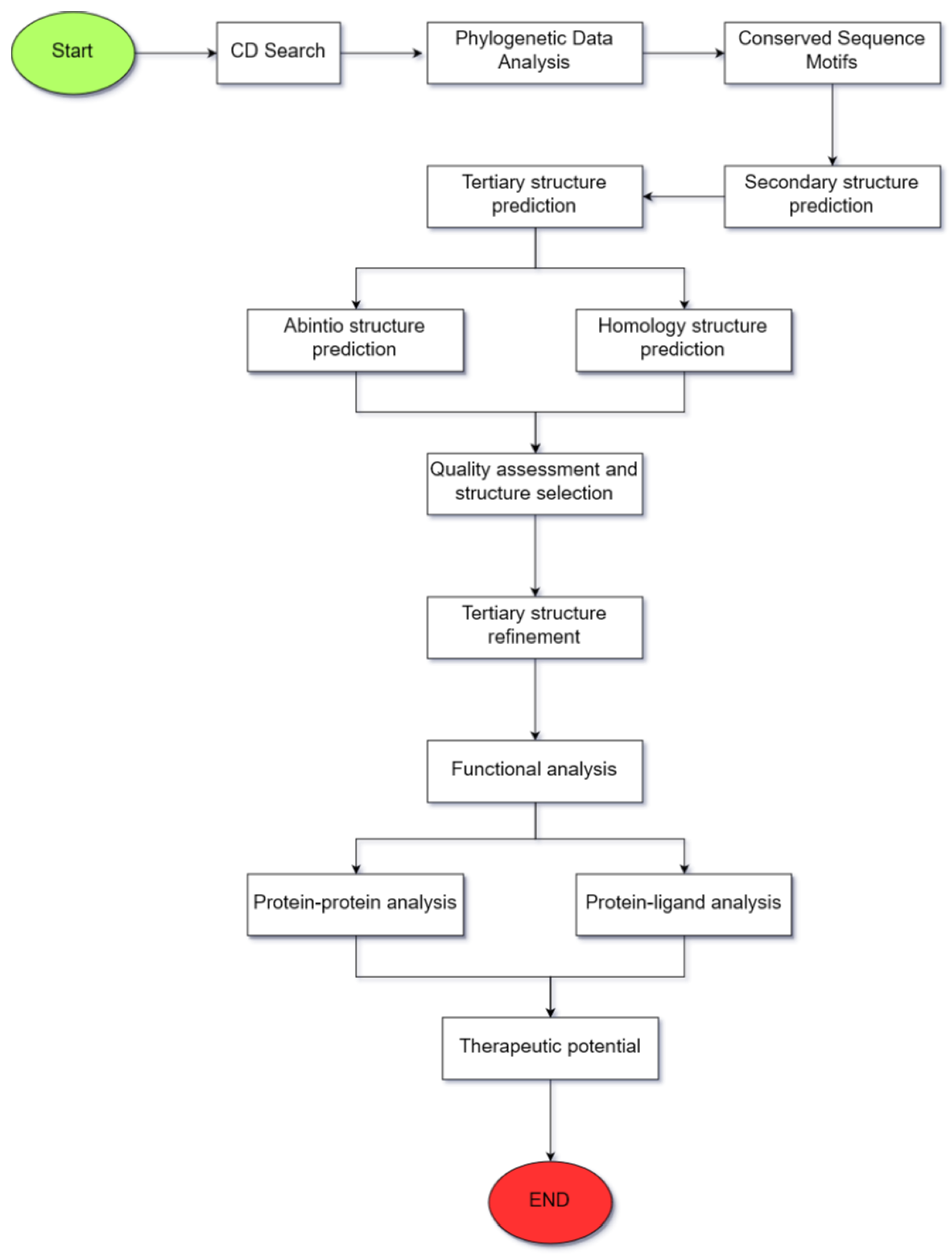
2. Materials and Methods
2.1. Validation of the Selection of the Proposed Computational Tools
2.2. Identification of Conserved Domains
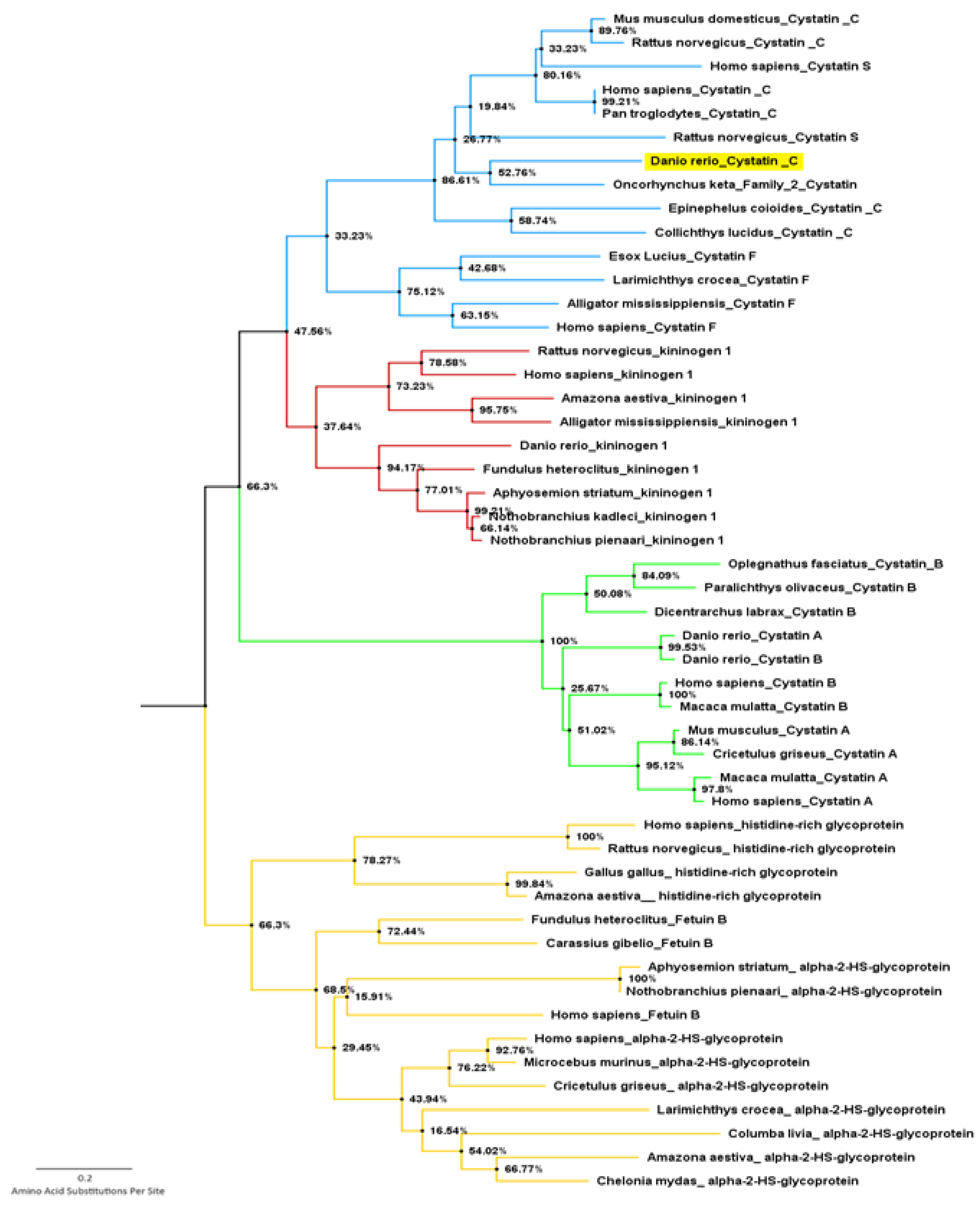
2.3. Phylogenetic Data Analysis
2.4. Identification of Conserved Sequence Motifs
2.5. Amino Acid Sequence and Secondary Structure Characterization
2.6. Tertiary Structure Prediction
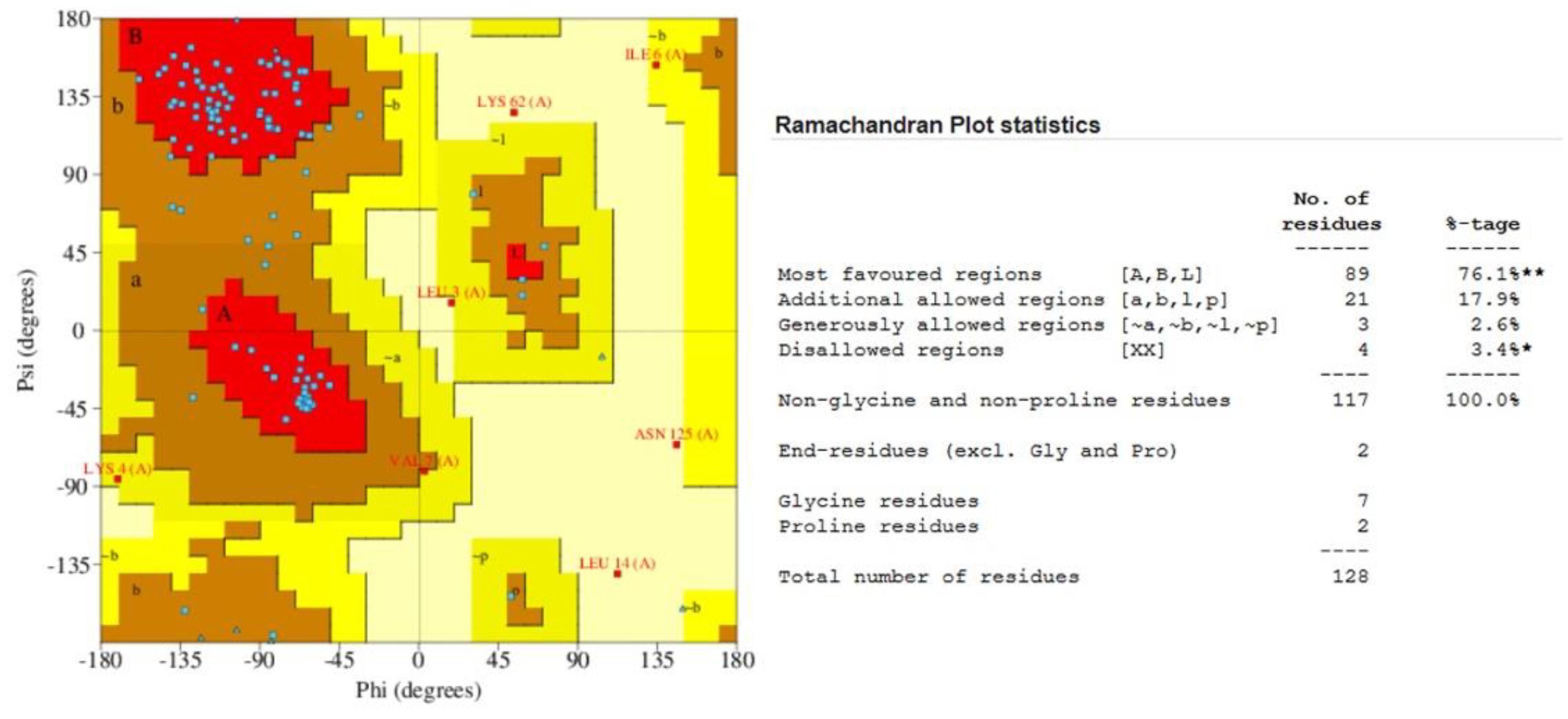
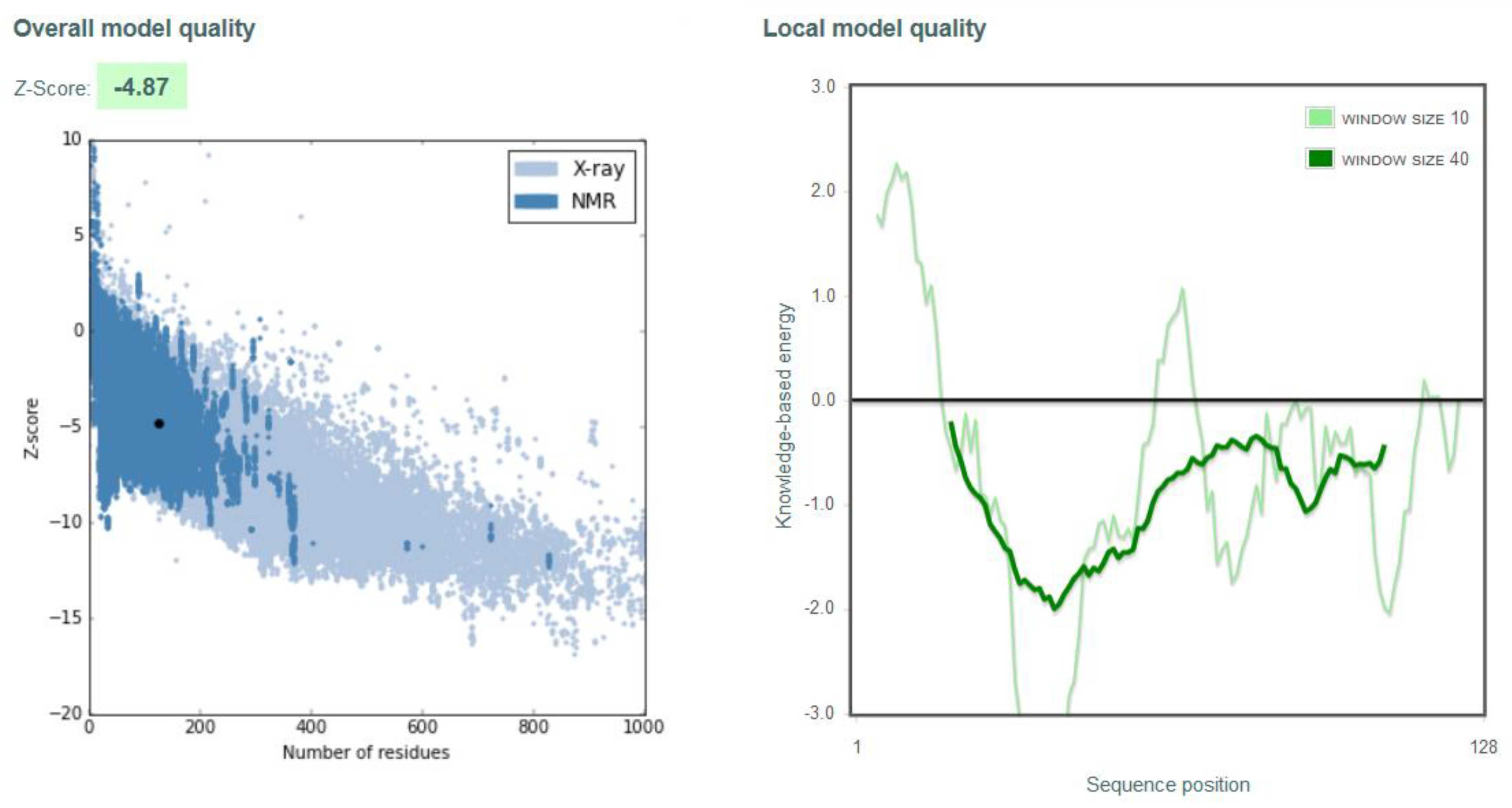
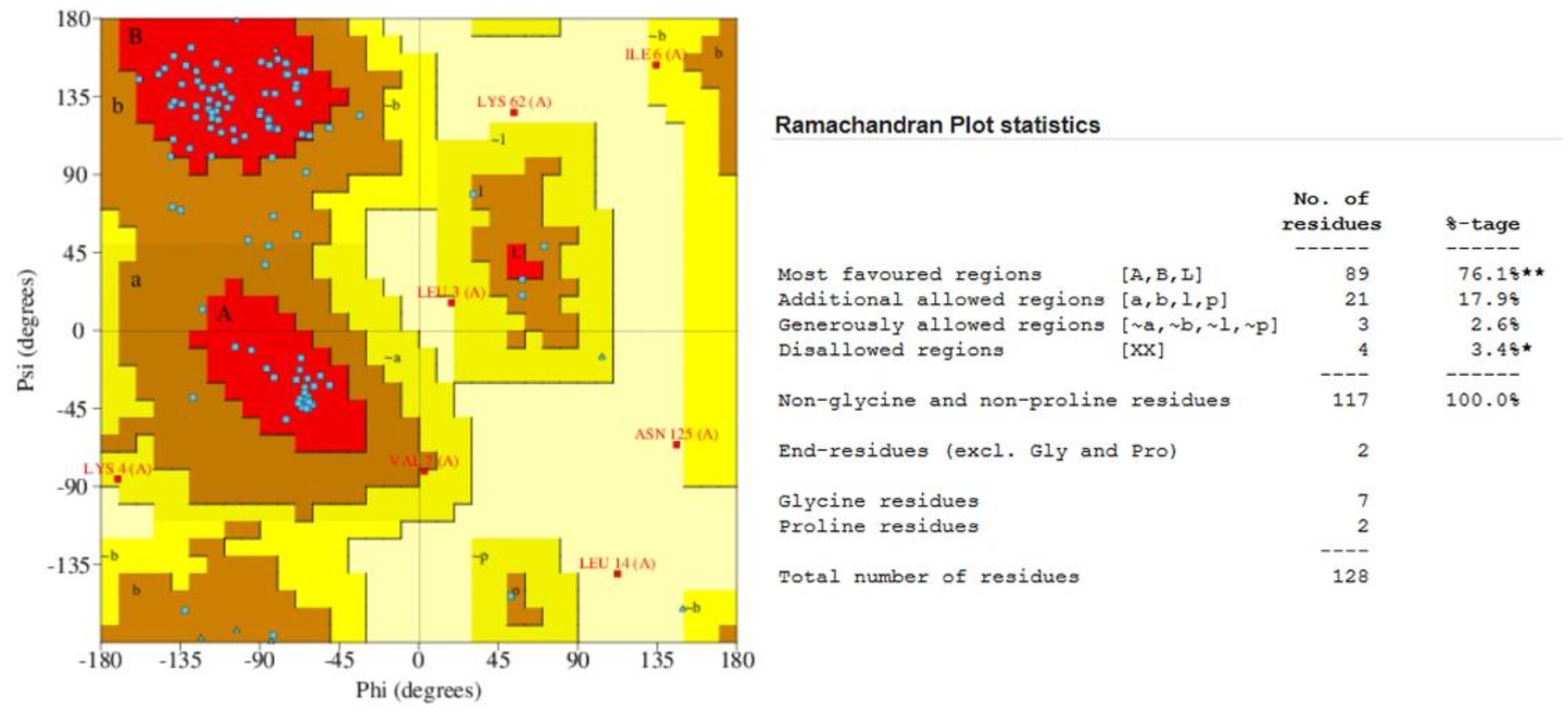
2.7. Quality Assessment of the Predicted Structure
2.8. Tertiary Structure Refinement
2.9. Surface Analysis of the Tertiary Structure
2.10. Functional Analysis through Molecular Docking
2.11. Analysis of the Therapeutic Potential
3. Results
3.1. Identification of Conserved Domains
3.2. Phylogenetic Data Analysis
3.3. Identification of Conserved Sequence Motifs
3.4. Secondary Structure Prediction
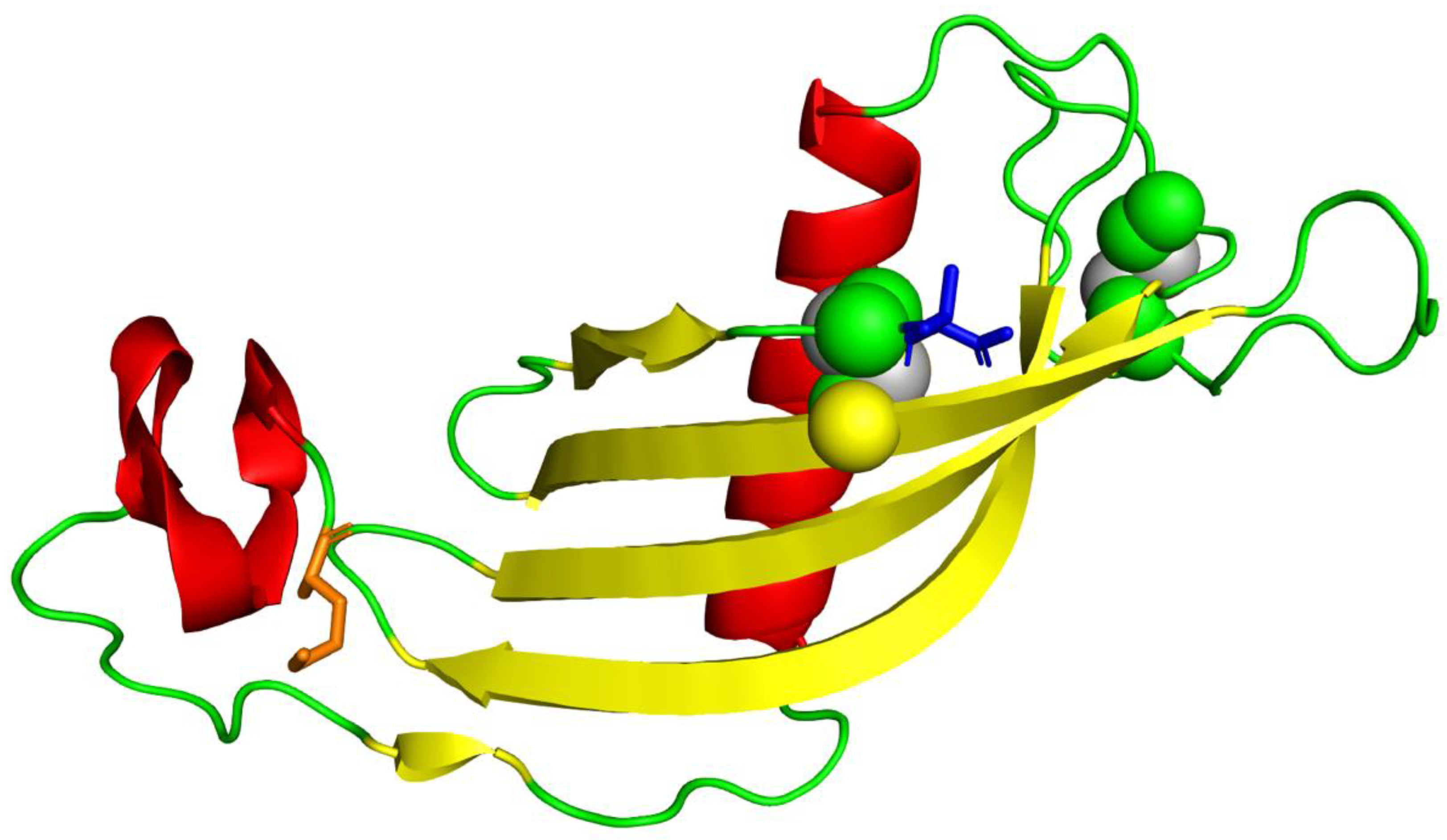
3.5. Protein Structure Prediction and Refinement
3.6. Comparative Analysis of Predicted Structure with Existing Structures
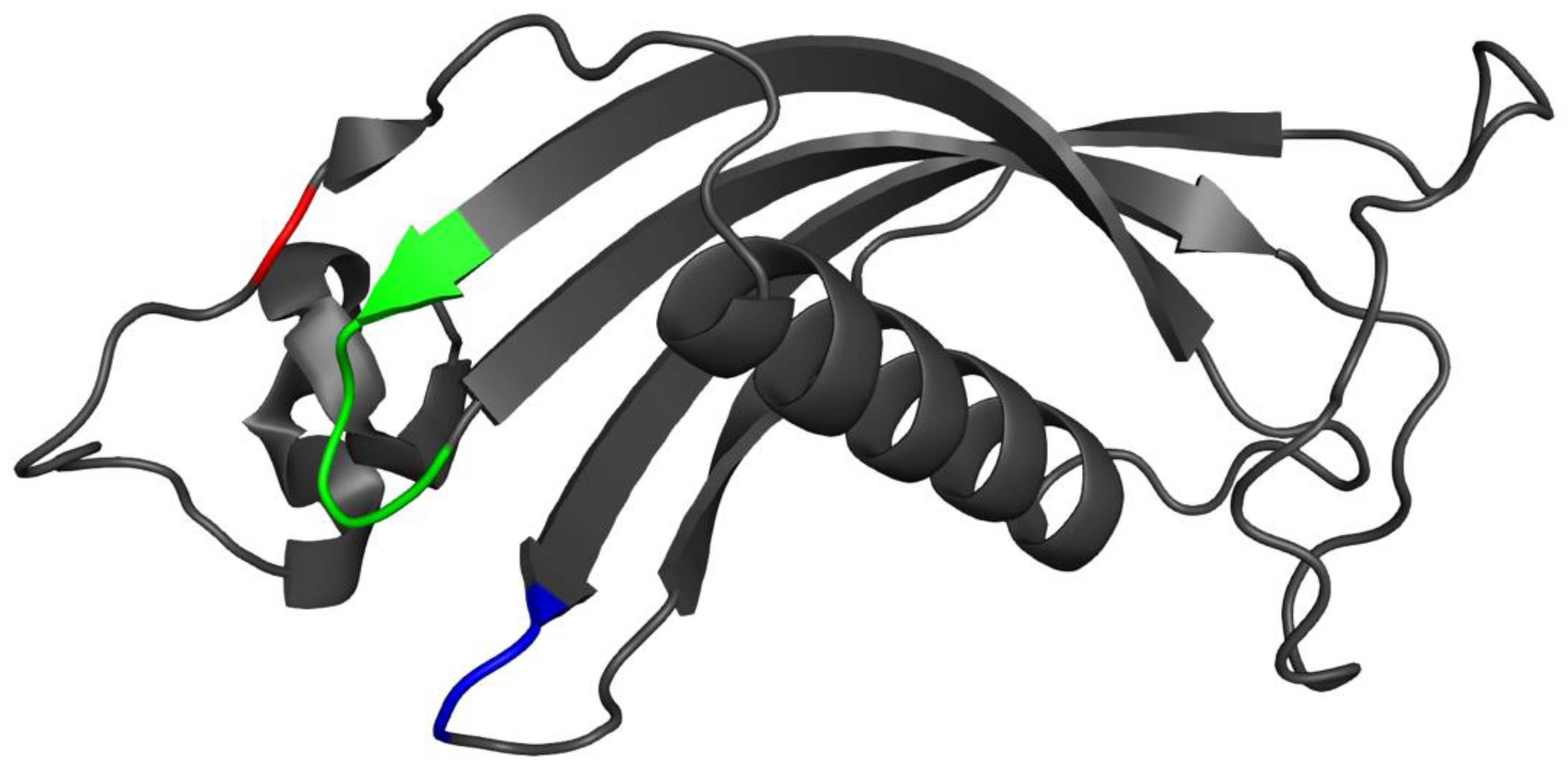
3.7. Placement of Evolutionarily Conserved Sequence Motifs
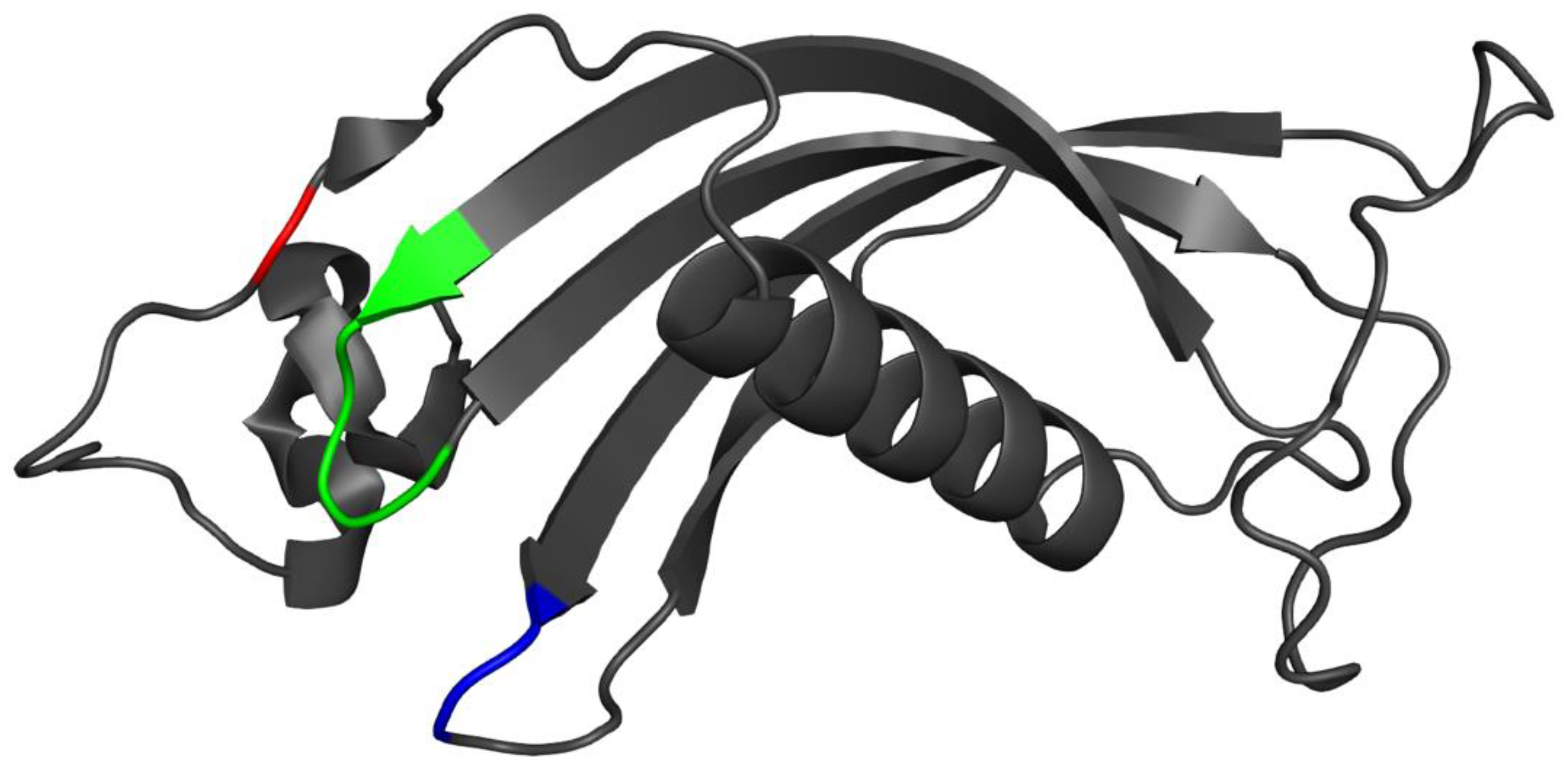
3.8. Surface Analysis of the Tertiary Structure
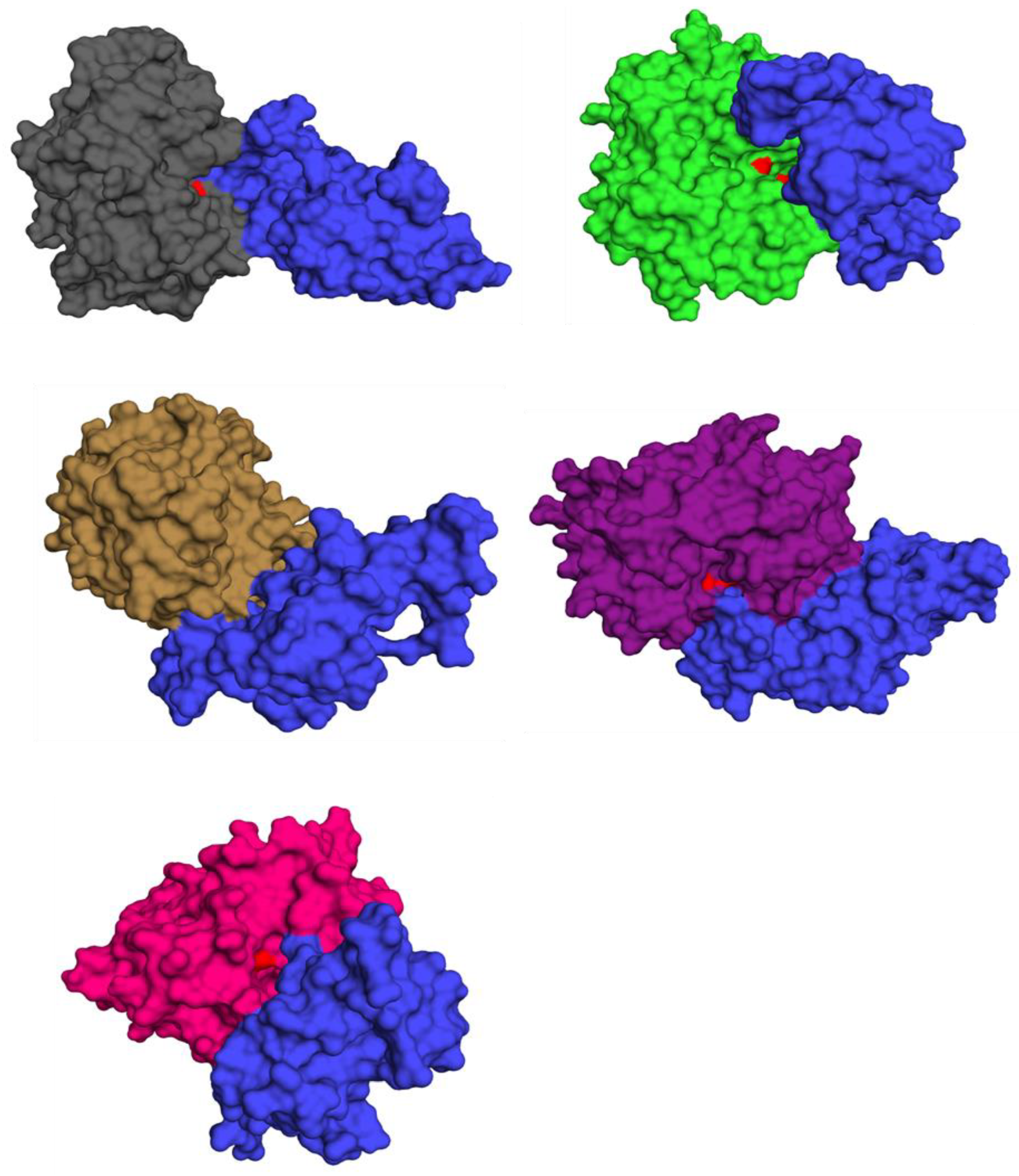
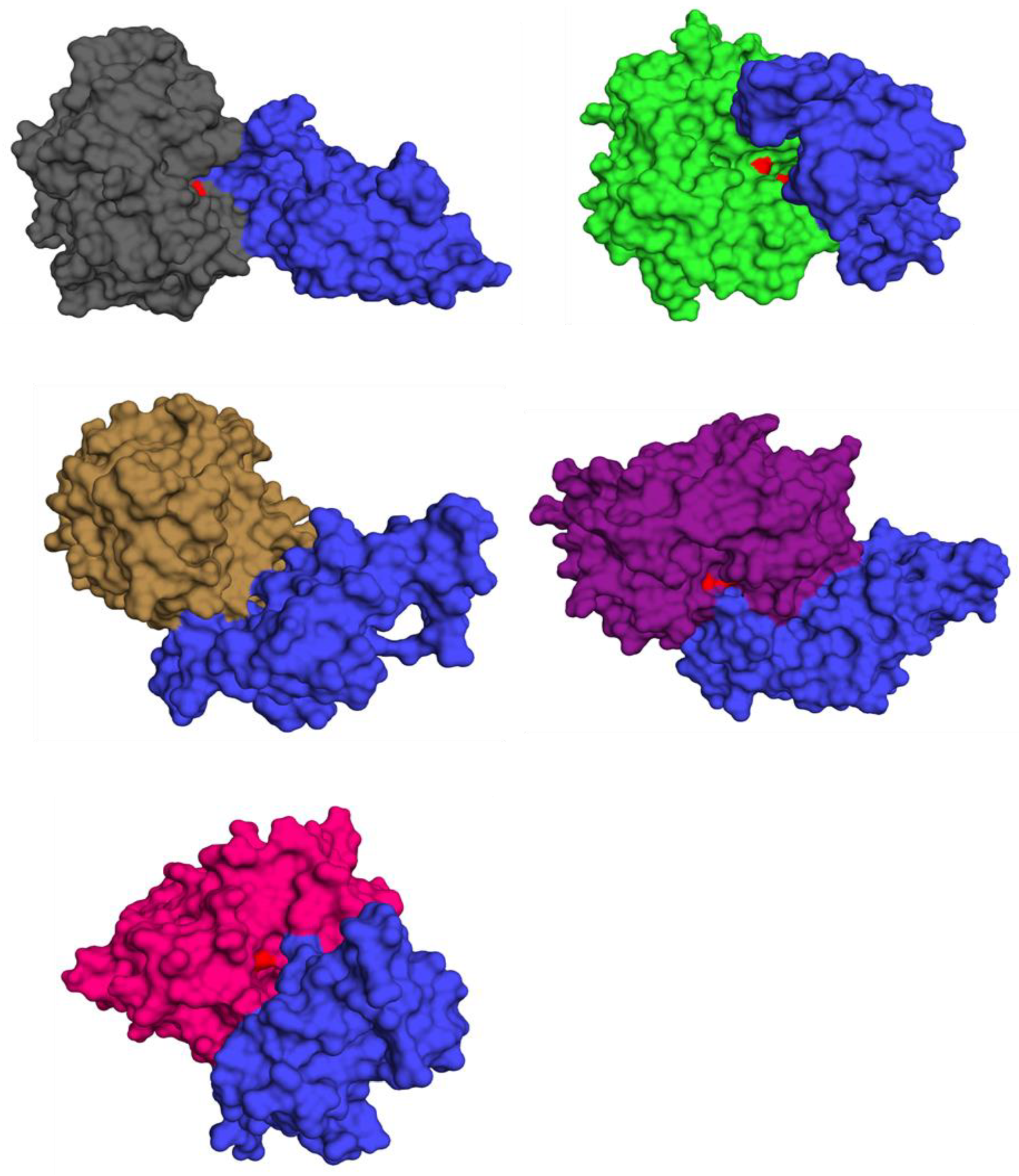
3.9. Functional Analysis via Virtual Screening
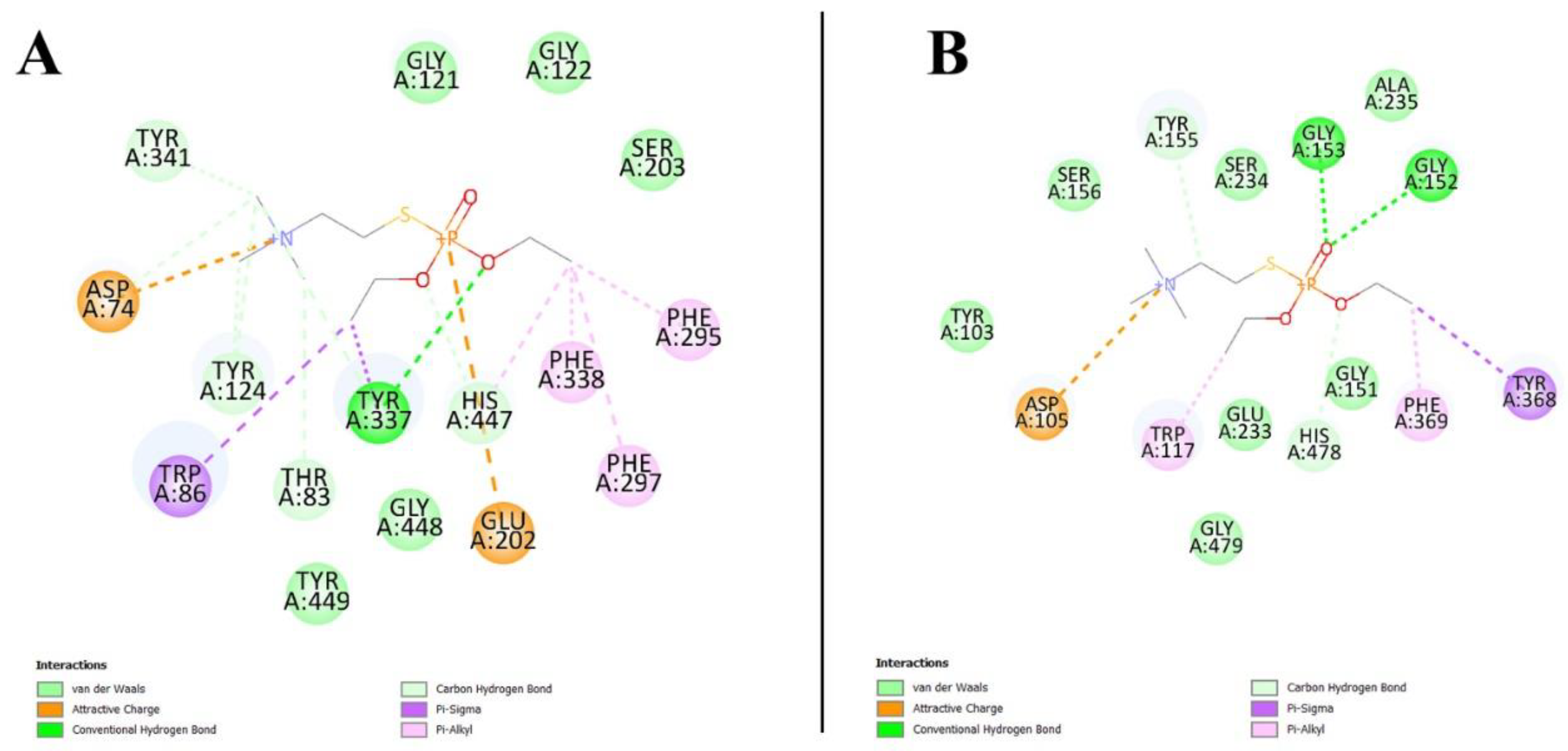
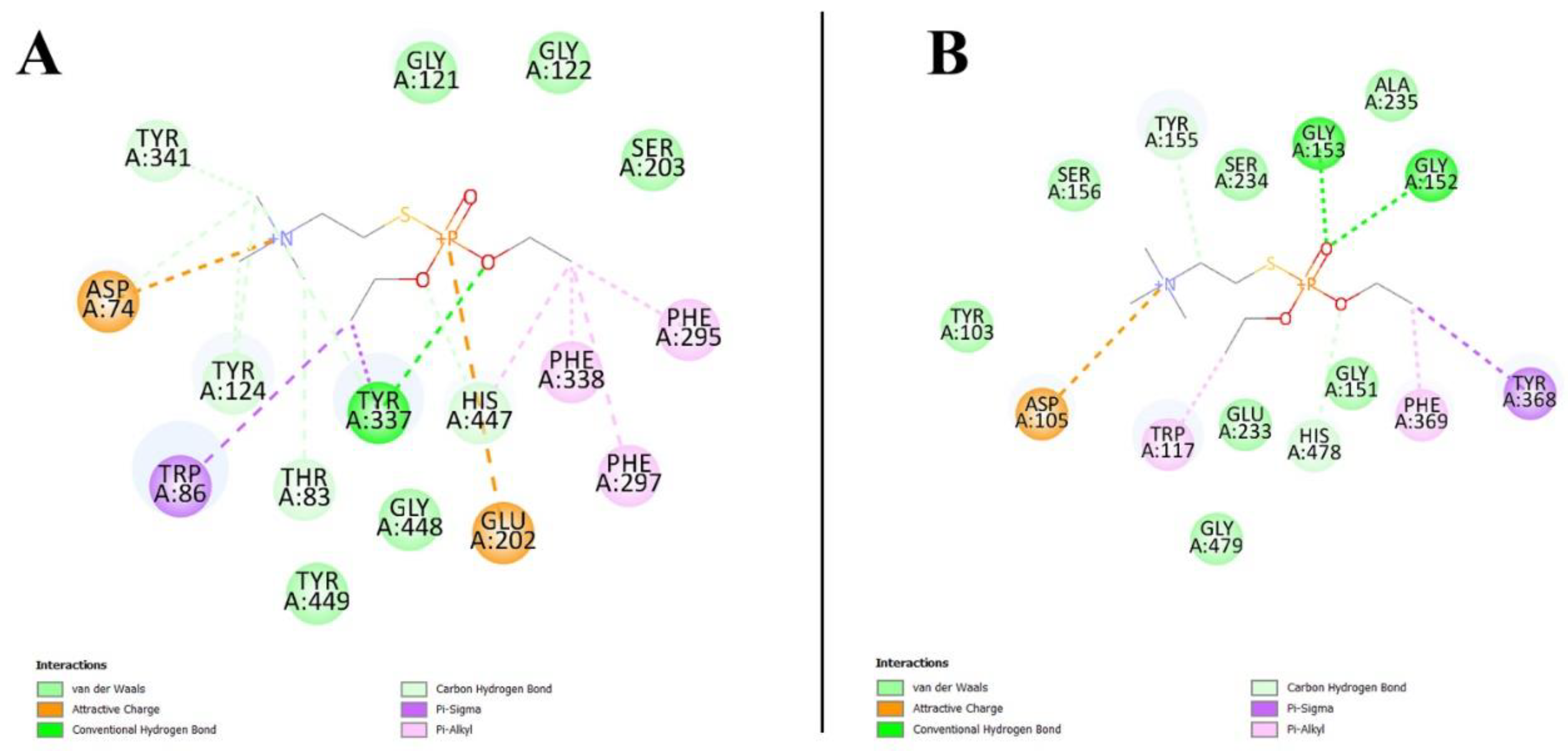
3.10. Virtual Screening Analysis for Protein–Ligand Interactions
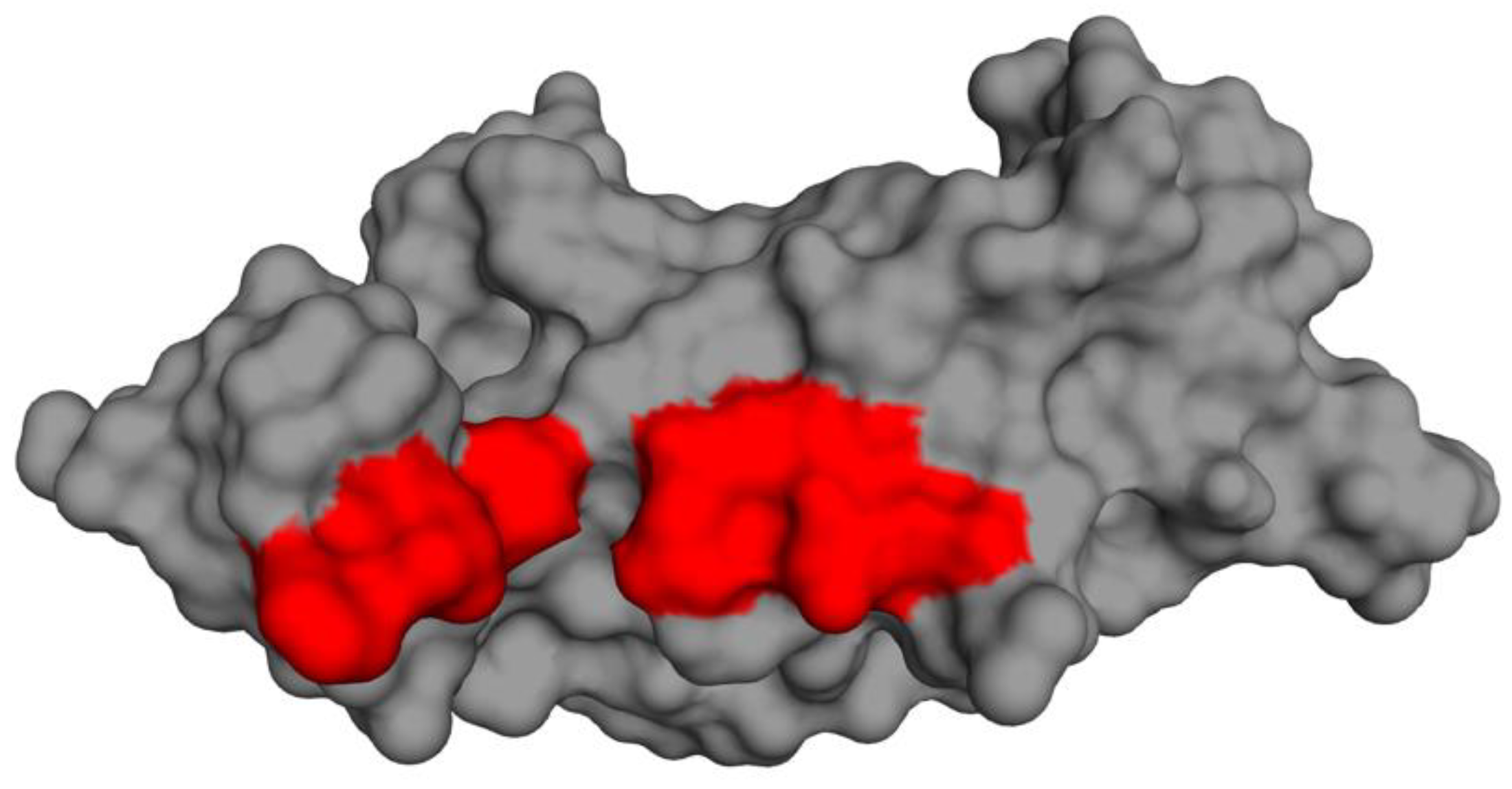
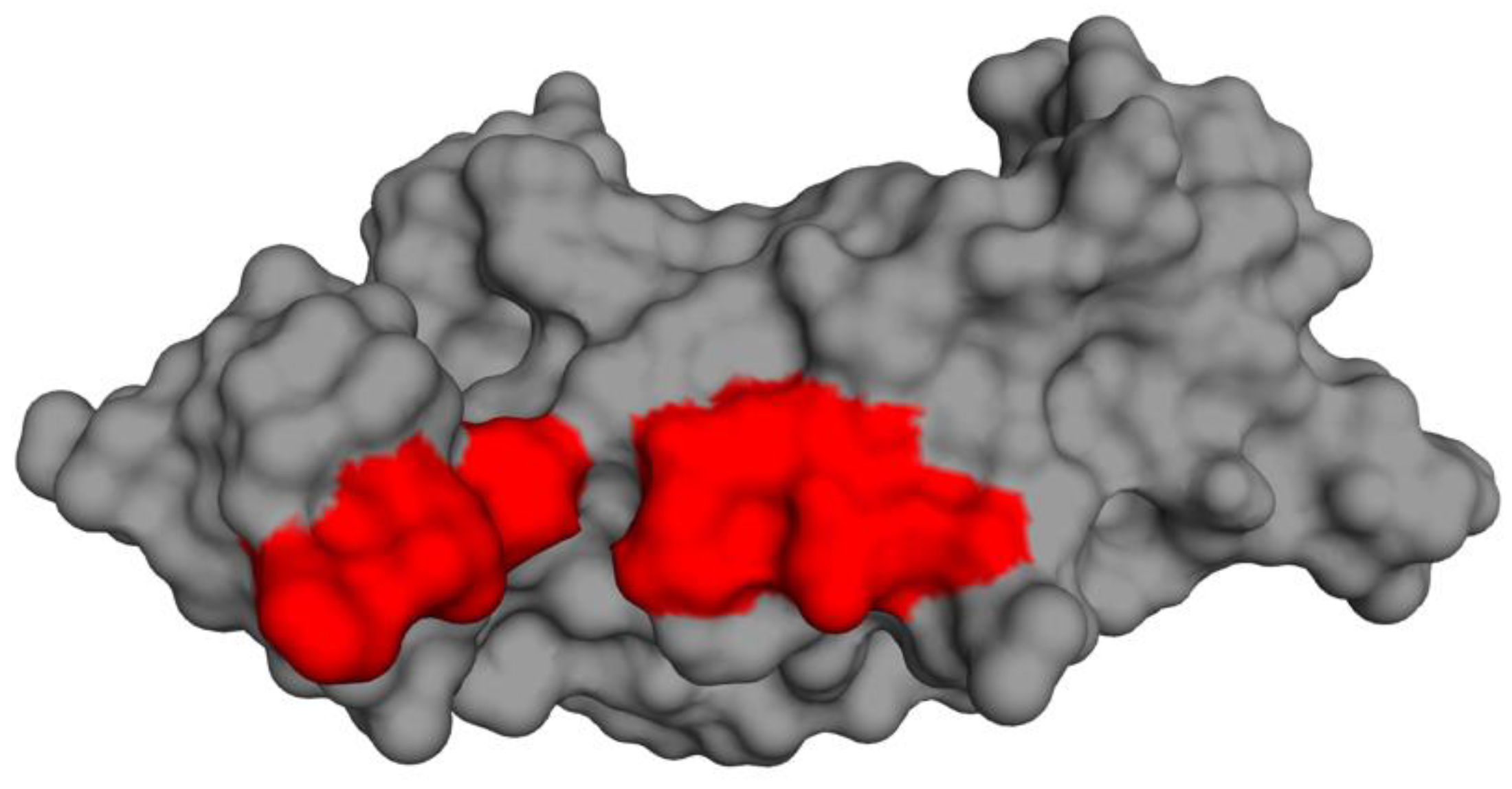
3.11. Prediction of Cystatin C Active Binding Site
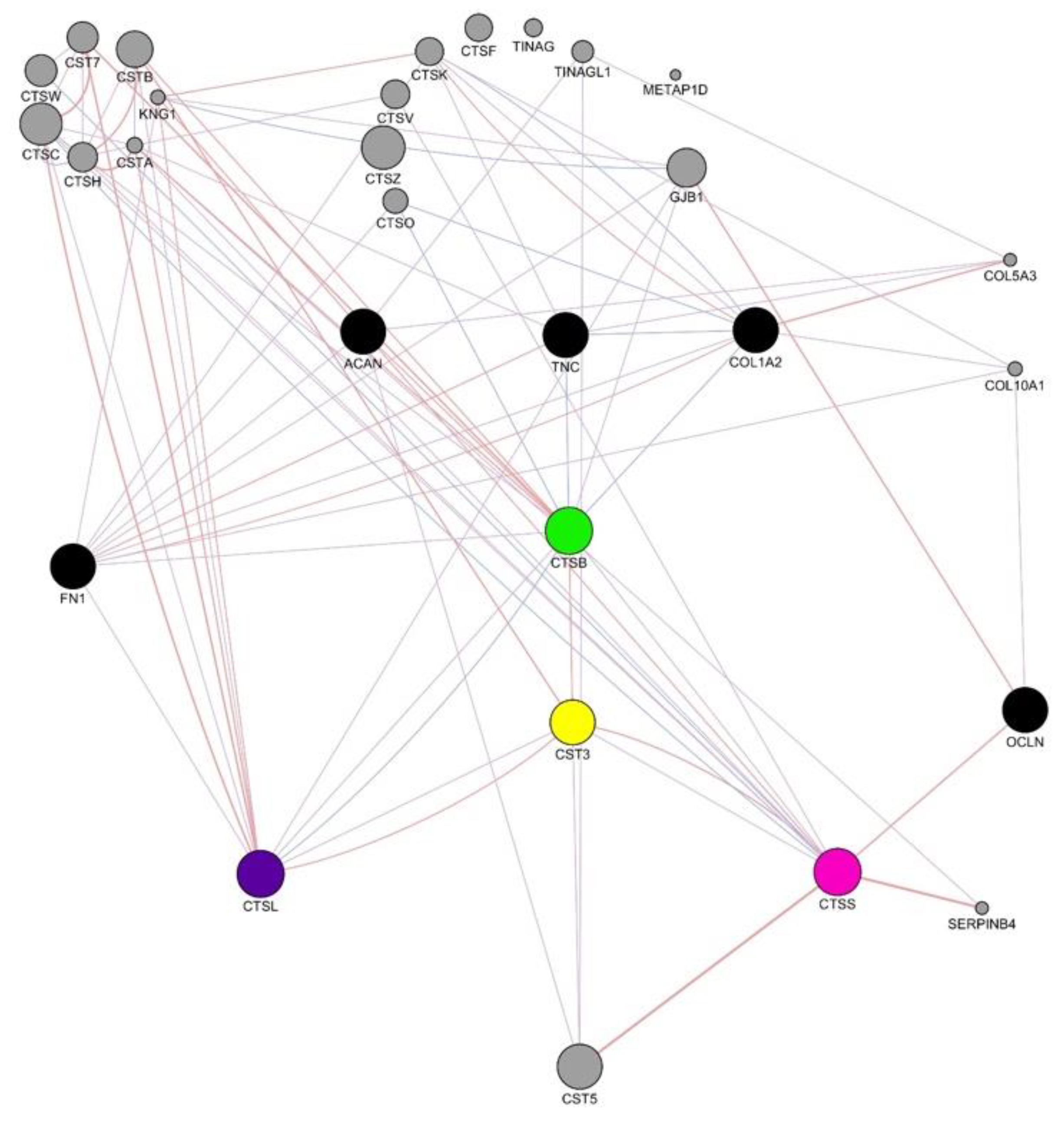
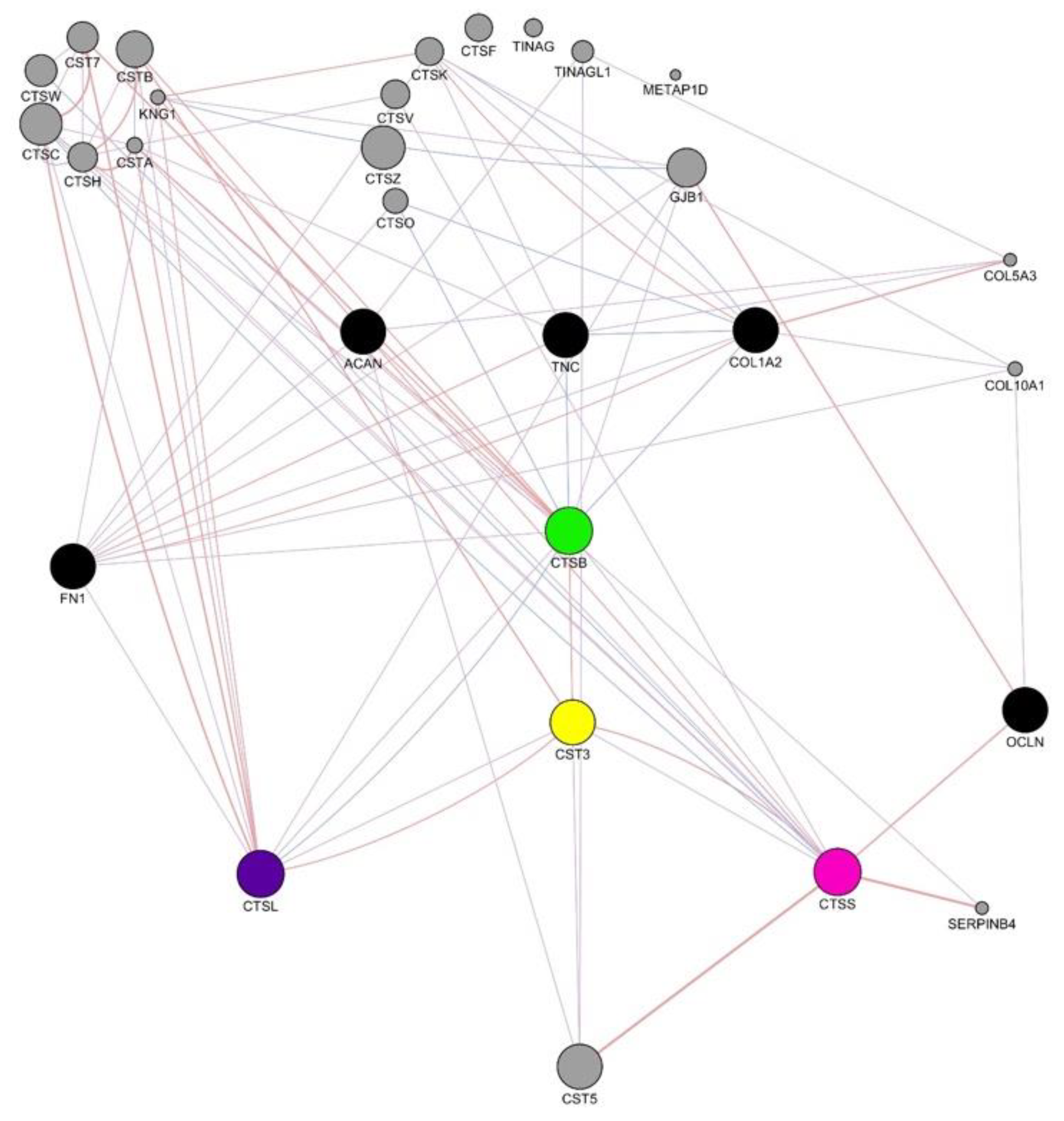
3.12. Human Gene Interaction Mapping of Cathepsin Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zarbafian, S.; Moghadasi, M.; Roshandelpoor, A.; Nan, F.; Li, K.; Vakli, P.; Vajda, S.; Kozakov, D.; Paschalidis, I.C. Protein docking refinement by convex underestimation in the low-dimensional subspace of encounter complexes. Sci. Rep. 2018, 8, 5896. [Google Scholar] [CrossRef] [Green Version]
- Godbey, W. Proteins. Introd. Biotechnol. 2014, 251, 9–33. [Google Scholar] [CrossRef]
- Skarzyńska, A.; Pawełkowicz, M.; Krzywkowski, T.; Świerkula, K.; Pląder, W.; Przybecki, Z. Bioinformatics pipeline for functional identification and characterization of proteins. Photonics Appl. Astron. Commun. Ind. High-Energy Phys. Exp. 2015, 9662, 96621M. [Google Scholar]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Margulies, E.H.; Blanchette, M.; Program, N.C.S.; Haussler, D.; Green, E.D. Identification and characterization of multi-species conserved sequences. Genome Res. 2003, 13, 2507–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.; Gehring, C.; Irving, H.R. Conserved functional motifs and homology modeling to predict hidden moonlighting functional sites. Front. Bioeng. Biotechnol. 2015, 3, 82. [Google Scholar] [CrossRef] [Green Version]
- Pruess, M.; Apweiler, R. Bioinformatics resources for in silico proteome analysis. J. Biomed. Biotechnol. 2003, 2003, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Balciunas, D.; Pogoda, H.-M.; Ding, Y.; Westcot, S.E.; Bedell, V.; Greenwood, T.M.; Urban, M.D.; Skuster, K.J.; Petzold, A.; et al. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat. Methods 2011, 8, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Ochieng, J.; Chaudhuri, G. Cystatin superfamily. J. Health Care Poor Underserved 2010, 21, 51–70. [Google Scholar] [CrossRef]
- Magister, Š.; Kos, J. Cystatins in immune system. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Z. TTD: Therapeutic target database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Dym, O.; Unger, T.; Toker, L.; Silman, I.; Sussman, J.; Center, I.S.P. Crystal structure of human acetylcholinesterase. Isr. Struct. Proteom. Cent. 2014. [Google Scholar] [CrossRef]
- Heendeniya, S.N.; Keerthirathna, L.; Manawadu, C.K.; Dissanayake, I.H.; Ali, R.; Mashhour, A.; Alzahrani, H.; Godakumbura, P.; Boudjelal, M.; Peiris, D.C. Therapeutic efficacy of Nyctanthes arbor-tristis flowers to inhibit proliferation of acute and chronic primary human leukemia cells, with adipocyte differentiation and in silico analysis of interactions between survivin protein and selected secondary metabolites. Biomolecules 2020, 10, 165. [Google Scholar] [CrossRef] [Green Version]
- Paoli, M.; Liddington, R.; Tame, J.; Wilkinson, A.; Dodson, G. Crystal structure of T state haemoglobin with oxygen bound at all four haems. J. Mol. Biol. 1996, 256, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruitt, K. NCBI reference sequence project: Update and current status. Nucleic Acids Res. 2003, 31, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; LeDoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2010, 39, D1005–D1010. [Google Scholar] [CrossRef] [Green Version]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2009, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.A.; Chilton, J.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ integrated into Galaxy. GigaScience 2015, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yang, J.; He, B.; Walker, S.E.; Zhang, H.; Govindarajoo, B.; Virtanen, J.; Xue, Z.; Shen, H.-B.; Zhang, Y. Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11. Proteins Struct. Funct. Bioinform. 2016, 84, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nat. Cell Biol. 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ritchie, D.W.; Venkatraman, V. Ultra-fast FFT protein docking on graphics processors. Bioinformatics 2010, 26, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Ghoorah, A.W.; Devignes, M.-D.; Smaïl-Tabbone, M.; Ritchie, D.W. Protein docking using case-based reasoning. Proteins Struct. Funct. Bioinform. 2013, 81, 2150–2158. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacIndoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.-D.; Ritchie, D.W. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, W445–W449. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Singh, H.; Srivastava, H.K.; Singh, S.; Kishore, G.; Raghava, G.P.S. Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinform. 2019, 19, 105–124. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Macarthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. Int. Tables Crystallogr. 2001, 21, 722–725. [Google Scholar]
- Shuid, A.N.; Kempster, R.; McGuffin, L.J. ReFOLD: A server for the refinement of 3D protein models guided by accurate quality estimates. Nucleic Acids Res. 2017, 45, W422–W428. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins Struct. Funct. Bioinform. 2014, 82, 1646–1655. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.; Vreven, T.; Janin, J.; Weng, Z. Protein-protein docking benchmark version 4.0. Proteins Struct. Funct. Bioinform. 2010, 78, 3111–3114. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, D.W. Evaluation of protein docking predictions usingHex 3.1 in CAPRI rounds 1 and 2. Proteins Struct. Funct. Bioinform. 2003, 52, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. ACS Pub. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Patel, M.; Patel, L.J. Design, synthesis, molecular docking, and antibacterial evaluation of some novel flouroquinolone derivatives as potent antibacterial agent. Sci. World J. 2014, 2014, 897187. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. The role of cystatins in cells of the immune system. FEBS Lett. 2006, 580, 6295–6301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Holmquist, M. Alpha beta-hydrolase fold enzymes structures, functions and mechanisms. Curr. Protein Pept. Sci. 2000, 1, 209–235. [Google Scholar] [CrossRef]
- Paraoan, L.; Hiscott, P.; Gosden, C.; Grierson, I. Cystatin C in macular and neuronal degenerations: Implications for mechanism(s) of age-related macular degeneration. Vis. Res. 2010, 50, 737–742. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, R.; Michalska, K.; Hernandez-Santoyo, A.; Wahlbom, M.; Grubb, A.; Jaskolski, M. Crystal structure of human cystatin C stabilized against amyloid formation. FEBS J. 2010, 277, 1726–1737. [Google Scholar] [CrossRef]
- Premachandra, H.; Wan, Q.; Elvitigala, D.A.S.; De Zoysa, M.; Choi, C.Y.; Whang, I.; Lee, J. Genomic characterization and expression profiles upon bacterial infection of a novel cystatin B homologue from disk abalone (Haliotis discus discus). Dev. Comp. Immunol. 2012, 38, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Björk, I.; Brieditis, I.; Raub-Segall, E.; Pol, E.; Håkansson, K.; Abrahamson, M. The importance of the second hairpin loop of cystatin C for proteinase binding. Characterization of the interaction of Trp-106 variants of the inhibitor with cysteine proteinases. Biochemistry 1996, 35, 10720–10726. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, A.; Ołdziej, S.; Liwo, A.; Scheraga, H.A. β-hairpin-forming peptides; models of early stages of protein folding. Biophys. Chem. 2010, 151, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fonović, M.; Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2560–2570. [Google Scholar] [CrossRef]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N. The refined 2.15 A X-ray crystal structure of human liver ca-thepsin B: The structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar] [CrossRef]
- Gunčar, G.; Podobnik, M.; Pungerčar, J.; BorutŠtrukelj, B.; Turk, V.; Turk, D. Crystal structure of porcine cathepsin H determined at 2.1 å resolution: Location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure 1998, 6, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Gunčar, G.; Pungercic, G.; Klemenčič, I.; Turk, V.; Turk, D. Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 1999, 18, 793–803. [Google Scholar] [CrossRef] [Green Version]
- McGrath, M.E.; Palmer, J.T.; Brömme, D.; Somoza, J.R. Crystal structure of human cathepsin S. Protein Sci. 1998, 7, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Axelsen, P.H.; Harel, M.; Silman, I.; Sussman, J.L. Structure and dynamics of the active site gorge of acetylcholinesterase: Synergistic use of molecular dynamics simulation and X-ray crystallography. Protein Sci. 1994, 3, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nye, D.B.; LeComte, J.T.J. Replacement of the distal histidine reveals a noncanonical heme binding site in a 2-on-2 hemoglobin. Biochemistry 2018, 57, 5785–5796. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Chakraborti, T.; Dhalla, N.S. Proteases in Human Diseases; Springer: Singapore, 2017. [Google Scholar] [CrossRef]
- Martin, T.A.; Jordan, N.; Davies, E.L.; Jiang, W. Metastasis to bone in human cancer is associated with loss of occludin expression. Anticancer. Res. 2016, 36, 1287–1293. [Google Scholar]
- Dutt, S.; Singh, V.; Marla, S.S.; Kumar, A. In silico analysis of sequential, structural and functional diversity of wheat cystatins and its implication in plant defense. Genom. Proteom. Bioinform. 2010, 8, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Stojanovic, N.; Florea, L.; Riemer, C.; Gumucio, D.; Slightom, J.; Goodman, M.; Miller, W.; Hardison, R. Comparison of five methods for finding conserved sequences in multiple alignments of gene regulatory regions. Nucleic Acids Res. 1999, 27, 3899–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordiš, D.; Turk, V. Phylogenomic analysis of the cystatin superfamily in eukaryotes and prokaryotes. BMC Evol. Biol. 2009, 9, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankun-Kelly, T.; Lindeman, A.D.; Bridges, S.M. Exploratory visual analysis of conserved domains on multiple sequence alignments. BMC Bioinform. 2009, 10, S7. [Google Scholar] [CrossRef] [Green Version]
- Abrahamson, M.; Alvarez-Fernandez, M.; Nathanson, C.-M. Cystatins. Biochem. Soc. Symp. 2003, 70, 179–199. [Google Scholar] [CrossRef]
- Zhang, Z. An Overview of Protein Structure Prediction: From Homology to Ab Initio. 2002. Available online: https://www.semanticscholar.org/paper/An-Overview-of-Protein-Structure-Prediction-%3A-From-Zhang/522af9cf5d1c3e4c1506d449286de6d3ebbd07ef (accessed on 26 October 2021).
- Zhang, Y.; Arakaki, A.K.; Skolnick, J. TASSER: An automated method for the prediction of protein tertiary structures in CASP6. Proteins Struct. Funct. Bioinform. 2005, 61, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Miklos, A.C.; Li, C.; Pielak, G.J. Using NMR-detected backbone amide 1H exchange to assess macromolecular crowding effects on globular-protein stability. Methods Enzymol. 2009, 466, 1–18. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein−protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Roy, U.K.; Halder, D. Protein Active Site Structure Prediction Strategy and Algorithm. Int. J. Curr. Eng. Technol. 2011, 2017, 1092–1096. [Google Scholar] [CrossRef] [Green Version]
- Jaimovich, A.; Rinott, R.; Schuldiner, M.; Margalit, H.; Friedman, N. Modularity and directionality in genetic interaction maps. Bioinformatics 2010, 26, i228–i236. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.; Saxena, V.S.N. Gene-gene interaction mapping of human cytomegalic virus through system biology approach. Biol. Syst. Open Access 2015, 4, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, D.D.B.D.; Perera, K.M.L.; Peiris, D.C. A Novel In Silico Benchmarked Pipeline Capable of Complete Protein Analysis: A Possible Tool for Potential Drug Discovery. Biology 2021, 10, 1113. https://doi.org/10.3390/biology10111113
Perera DDBD, Perera KML, Peiris DC. A Novel In Silico Benchmarked Pipeline Capable of Complete Protein Analysis: A Possible Tool for Potential Drug Discovery. Biology. 2021; 10(11):1113. https://doi.org/10.3390/biology10111113
Chicago/Turabian StylePerera, D. D. B. D., K. Minoli L. Perera, and Dinithi C. Peiris. 2021. "A Novel In Silico Benchmarked Pipeline Capable of Complete Protein Analysis: A Possible Tool for Potential Drug Discovery" Biology 10, no. 11: 1113. https://doi.org/10.3390/biology10111113
APA StylePerera, D. D. B. D., Perera, K. M. L., & Peiris, D. C. (2021). A Novel In Silico Benchmarked Pipeline Capable of Complete Protein Analysis: A Possible Tool for Potential Drug Discovery. Biology, 10(11), 1113. https://doi.org/10.3390/biology10111113