
In Silico Multi-Target Approach Revealed Potential Lead Compounds as Scaffold for the Synthesis of Chemical Analogues Targeting SARS-CoV-2



Abstract
:Simple Summary
Abstract
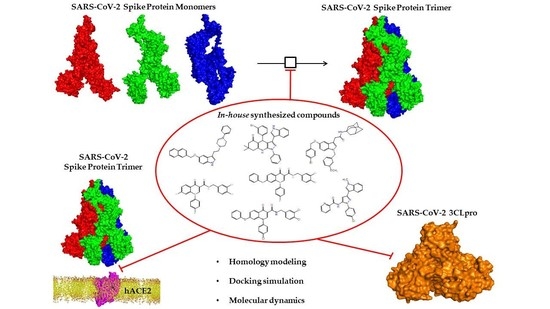
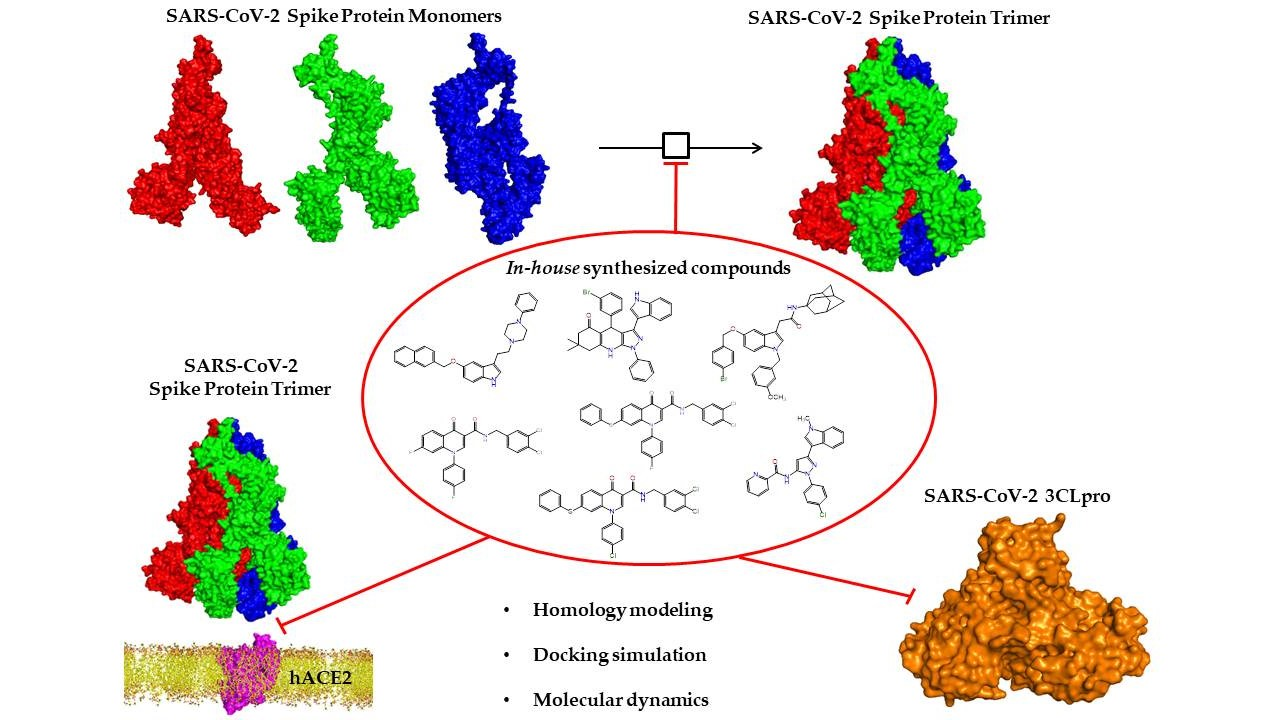{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. In Silico Methods
2.2.1. Optimization of Model 3D Structures
2.2.2. Energy Minimization and the Relaxation of Model 3D Structures
2.2.3. Docking Simulation
2.2.4. Classical Molecular Dynamics (cMD) Simulation
3. Results
3.1. SARS-CoV-2 S-Glycoprotein Trimerization Region: Virtual Screening and cMD
3.2. SARS-CoV-2 RBD/hACE2 Interaction Region: Virtual Screening and Cmd
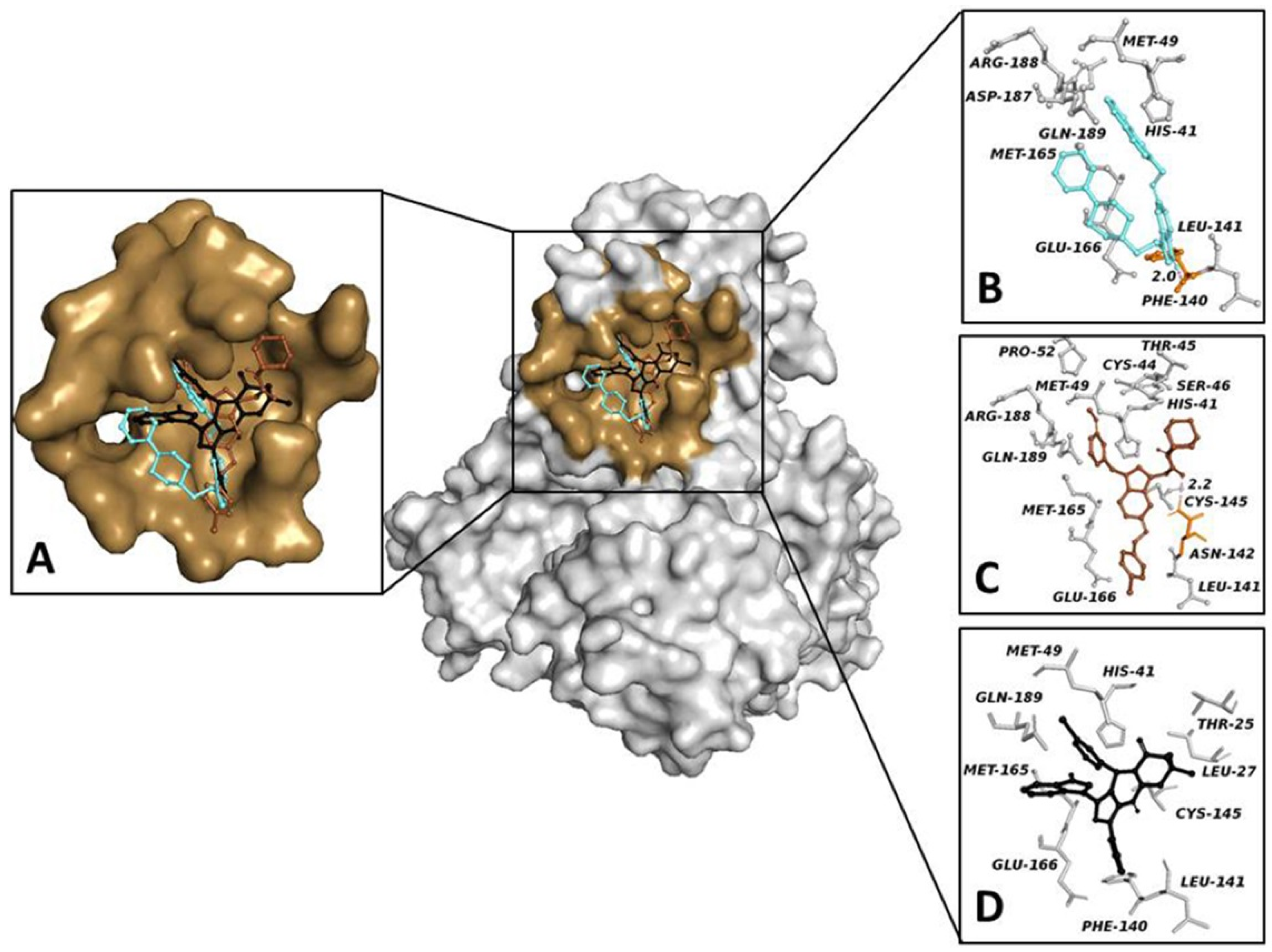
3.3. CLpro: Virtual Screening and cMD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delmas, B.; Laude, H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990, 64, 5367–5375. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2022, 181, 281–292. [Google Scholar] [CrossRef]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; ISBN 9781493924387. [Google Scholar]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Jayaweera, M.; Perera, H.; Gunawardana, B.; Manatunge, J. Transmission of COVID-19 virus by droplets and aerosols: A critical review on the unresolved dichotomy. Environ. Res. 2020, 188, 109819. [Google Scholar] [CrossRef]
- Huang, R.H.; Liu, D.J.; Tlili, A.; Yang, J.F.; Wang, H.H. Handbook on Facilitating Flexible Learning during Educational Disruption: The Chinese Experience in Maintaining Undisrupted Learning in COVID-19 Outbreak; Smart Learning Institute of Beijing Normal University, UNESCO: Beijing, China, 2020. [Google Scholar]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis. Eur. Respir. J. 2020, 55, 547. [Google Scholar] [CrossRef] [Green Version]
- Elshaboury, R.H.; Monk, M.M.; Bebell, L.M.; Bidell, M.R.; Adamsick, M.L.; Gandhi, R.G.; Paras, M.L.; Hohmann, E.L.; Letourneau, A.R. Remdesivir use and outcomes during the FDA COVID-19 emergency use authorization period. Ther. Adv. Infect. Dis. 2021, 8, 20499361211046669. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of lopinavir-ritonavir in adults hospitalized with severe COVID-19. N. Engl. J. Med. 2020, 328, 1787–1799. [Google Scholar] [CrossRef]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020, 146, 137–146. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Preliminary Report. N. Engl. J. Med. 2020, 383, 1813–1836. [Google Scholar] [CrossRef]
- Sun, J.K.; Chen, Y.T.; Fan, X.D.; Wang, X.Y.; Han, Q.Y.; Liu, Z.W. Advances in the use of chloroquine and hydroxychloroquine for the treatment of COVID-19. Postgrad. Med. 2020, 132, 604–613. [Google Scholar] [CrossRef]
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef] [PubMed]
- Alexpandi, R.; De Mesquita, J.F.; Pandian, S.K.; Ravi, A.V. Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in silico Analysis. Front. Microbiol. 2020, 11, 1796. [Google Scholar] [CrossRef]
- Rossi, F.; Tortora, C.; Argenziano, M.; Di Paola, A.; Punzo, F. Cannabinoid receptor type 2: A possible target in SARS-CoV-2 (CoV-19) infection? Int. J. Mol. Sci. 2020, 21, 3809. [Google Scholar] [CrossRef]
- Pasquini, S.; De Rosa, M.; Ligresti, A.; Mugnaini, C.; Brizzi, A.; Caradonna, N.P.; Cascio, M.G.; Bolognini, D.; Pertwee, R.G.; Di Marzo, V.; et al. Investigations on the 4-quinolone-3-carboxylic acid motif. 6. Synthesis and pharmacological evaluation of 7-substituted quinolone-3-carboxamide derivatives as high affinity ligands for cannabinoid receptors. Eur. J. Med. Chem. 2012, 58, 30–43. [Google Scholar] [CrossRef]
- Pasquini, S.; Mugnaini, C.; Brizzi, A.; Ligresti, A.; Di Marzo, V.; Ghiron, C.; Corelli, F. Rapid combinatorial access to a library of 1,5-disubstituted-3-indole-N- alkylacetamides as CB2 receptor ligands. J. Comb. Chem. 2009, 11, 795–798. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020, 525, 135–140. [Google Scholar] [CrossRef]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Zhang, L.K.; Ye, Y.; Jiang, H.L.; Xu, Y.C. Identification of a Novel Inhibitor of SARS-CoV-2 3CLpro; Acta Pharmacologica Sinica: London, UK, 2020. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, 204–212. [Google Scholar] [CrossRef]
- Janson, G.; Zhang, C.; Prado, M.G.; Paiardini, A. PyMod 2.0: Improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 2017, 33, 444–446. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Bongini, P.; Trezza, A.; Bianchini, M.; Spiga, O.; Niccolai, N. A possible strategy to fight COVID-19: Interfering with spike glycoprotein trimerization. Biochem. Biophys. Res. Commun. 2020, 528, 35–38. [Google Scholar] [CrossRef]
- Trezza, A.; Iovinelli, D.; Santucci, A.; Prischi, F.; Spiga, O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 13866. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.Y. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.M.; Cheng, V.C.C.; Hung, I.F.N.; Wong, M.M.L.; Chan, K.H.; Chan, K.S.; Kao, R.Y.T.; Poon, L.L.M.; Wong, C.L.P.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabi, Y.M.; Alothman, A.; Balkhy, H.H.; Al-Dawood, A.; AlJohani, S.; Al Harbi, S.; Kojan, S.; Al Jeraisy, M.; Deeb, A.M.; Assiri, A.M.; et al. Treatment of Middle East Respiratory Syndrome with a combination of lopinavir-ritonavir and interferon-β1b (MIRACLE trial): Study protocol for a randomized controlled trial. Trials 2018, 19, 81. [Google Scholar] [CrossRef]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) Structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, G.; Bedini, A.; Bartolucci, S.; Pala, D.; Mor, M.; Riccioni, T.; Borsini, F.; Cabri, W.; Celona, D.; Marzi, M.; et al. Towards the development of 5-HT7 ligands combining serotonin-like and arylpiperazine moieties. Eur. J. Med. Chem. 2014, 80, 8–35. [Google Scholar] [CrossRef]
- Singh, S.; Chauhan, P.; Ravi, M.; Yadav, P.P. Eosin Y-Yb(OTf)3 catalyzed visible light mediated electrocyclization/indole ring opening towards the synthesis of heterobiaryl-pyrazolo[3,4-b]pyridines. New J. Chem. 2018, 42, 6617–6620. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trezza, A.; Mugnaini, C.; Corelli, F.; Santucci, A.; Spiga, O. In Silico Multi-Target Approach Revealed Potential Lead Compounds as Scaffold for the Synthesis of Chemical Analogues Targeting SARS-CoV-2. Biology 2022, 11, 465. https://doi.org/10.3390/biology11030465
Trezza A, Mugnaini C, Corelli F, Santucci A, Spiga O. In Silico Multi-Target Approach Revealed Potential Lead Compounds as Scaffold for the Synthesis of Chemical Analogues Targeting SARS-CoV-2. Biology. 2022; 11(3):465. https://doi.org/10.3390/biology11030465
Chicago/Turabian StyleTrezza, Alfonso, Claudia Mugnaini, Federico Corelli, Annalisa Santucci, and Ottavia Spiga. 2022. "In Silico Multi-Target Approach Revealed Potential Lead Compounds as Scaffold for the Synthesis of Chemical Analogues Targeting SARS-CoV-2" Biology 11, no. 3: 465. https://doi.org/10.3390/biology11030465
APA StyleTrezza, A., Mugnaini, C., Corelli, F., Santucci, A., & Spiga, O. (2022). In Silico Multi-Target Approach Revealed Potential Lead Compounds as Scaffold for the Synthesis of Chemical Analogues Targeting SARS-CoV-2. Biology, 11(3), 465. https://doi.org/10.3390/biology11030465