Preparation of Chitosan-Coated Poly(L-Lactic Acid) Fibers for Suture Threads

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
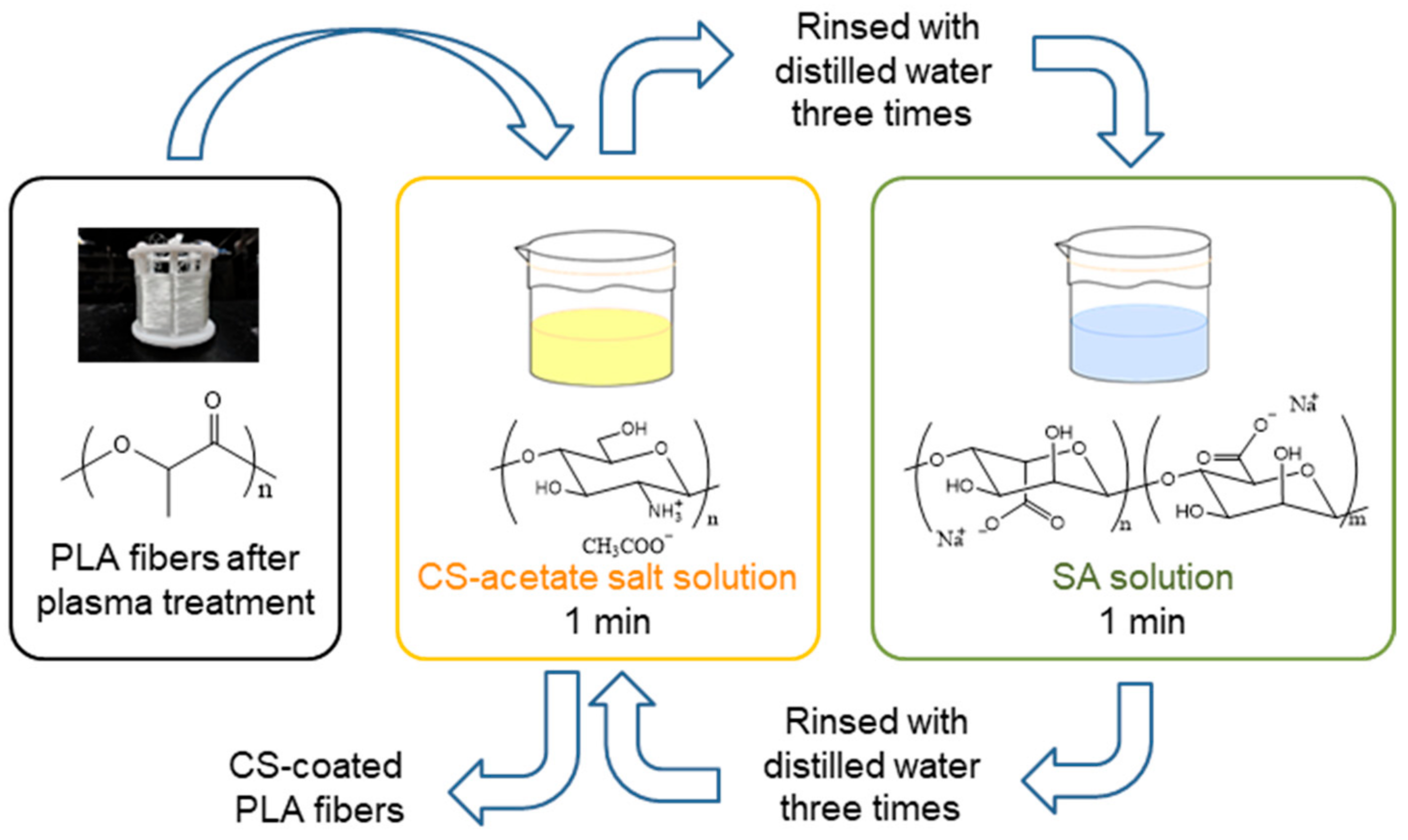
2.2. Methods
2.3. Tensile Testing
2.4. Surface Characterization
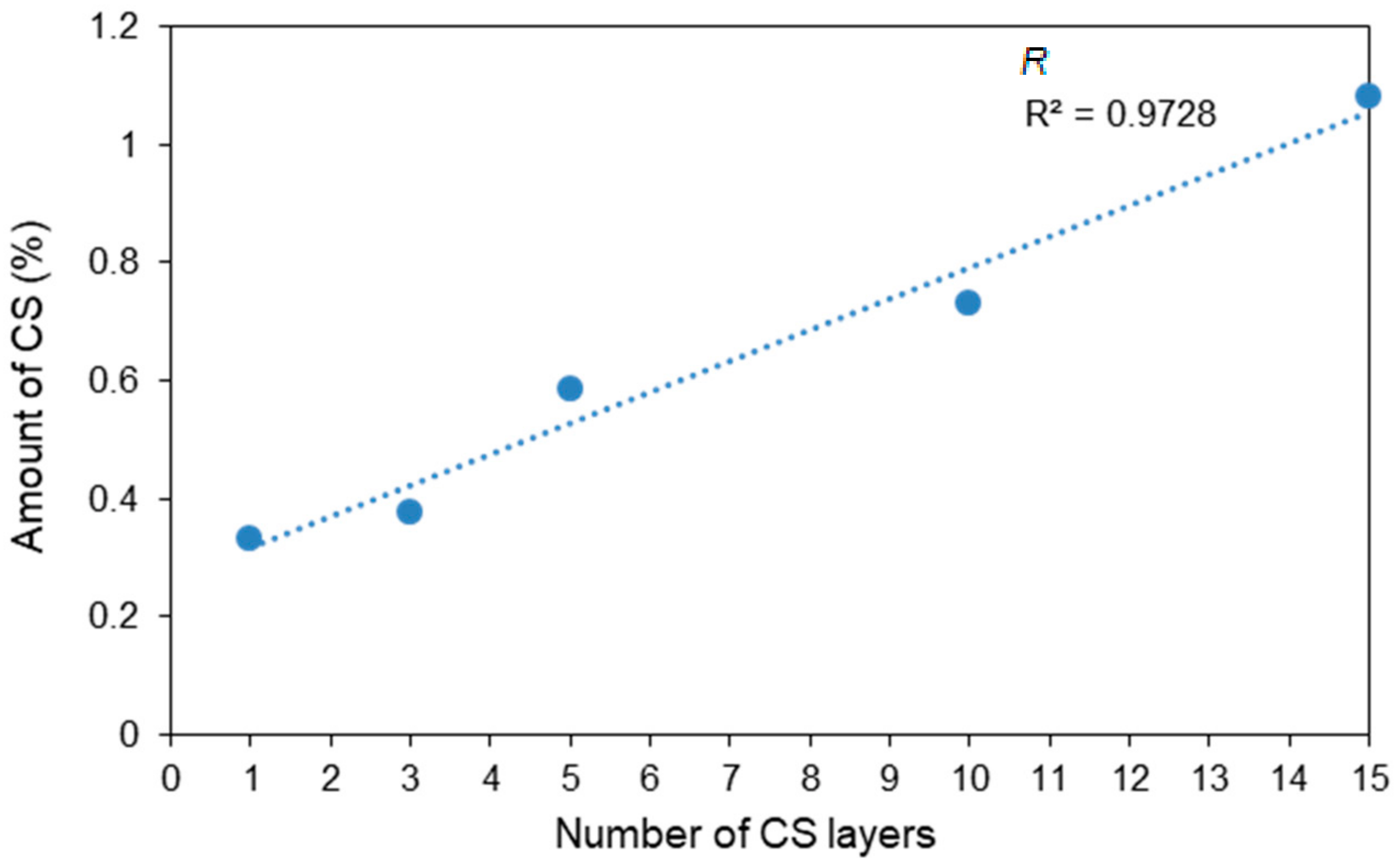
2.5. Determining the Amount of CS
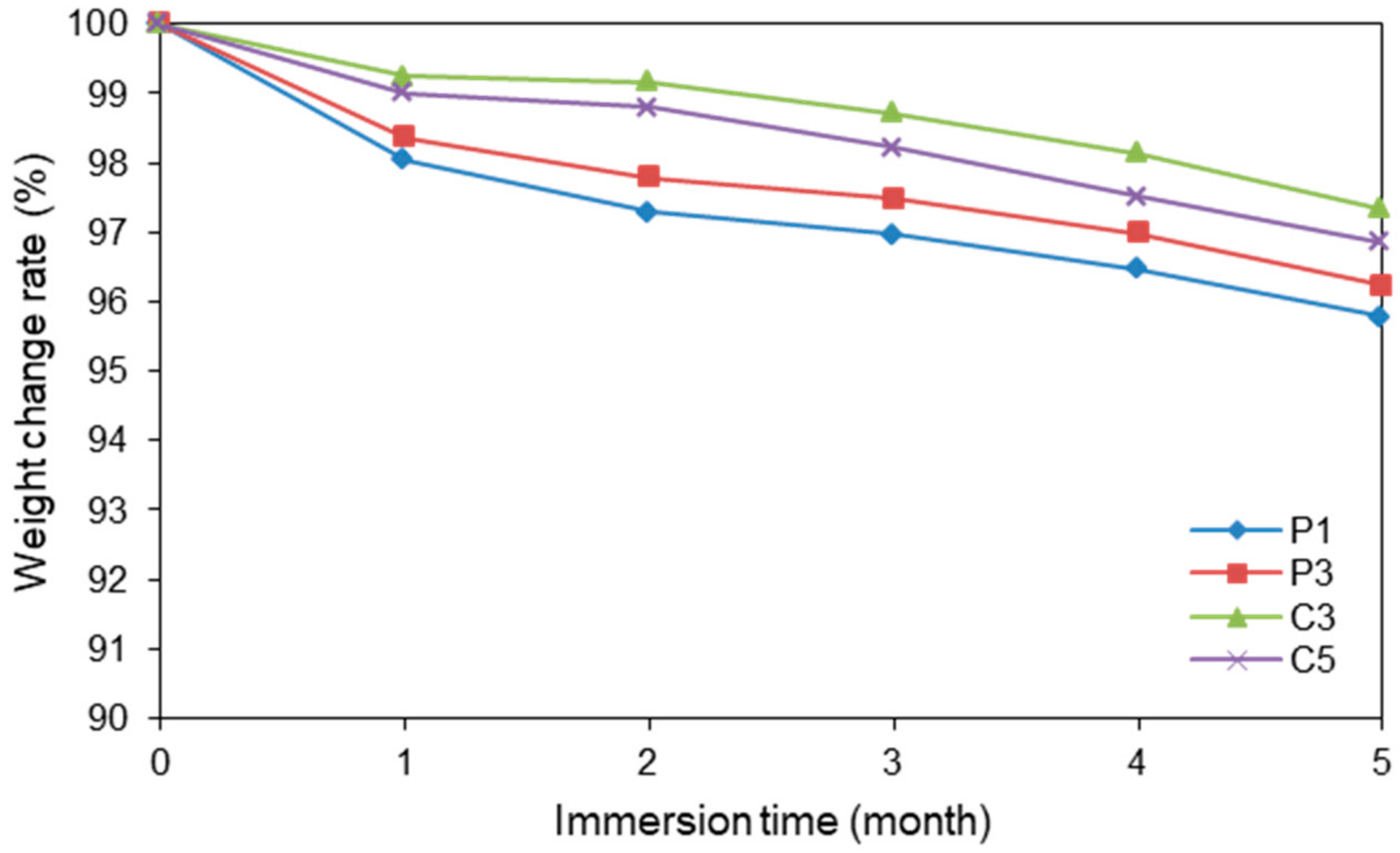
2.6. Degradation Testing in PBS Solution
3. Results and Discussion
3.1. Tensile Testing
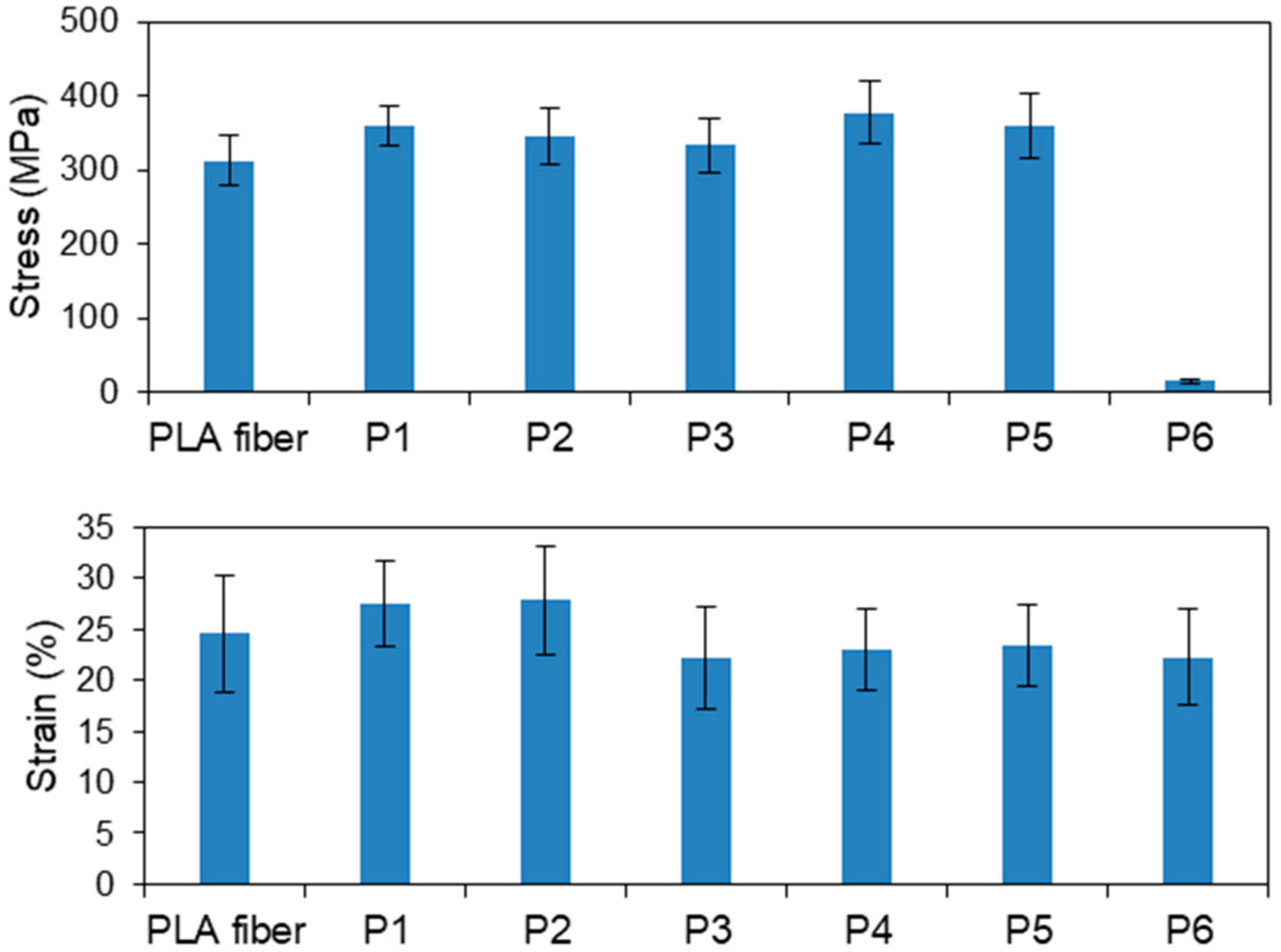
3.1.1. Plasma-Treated PLA Fibers
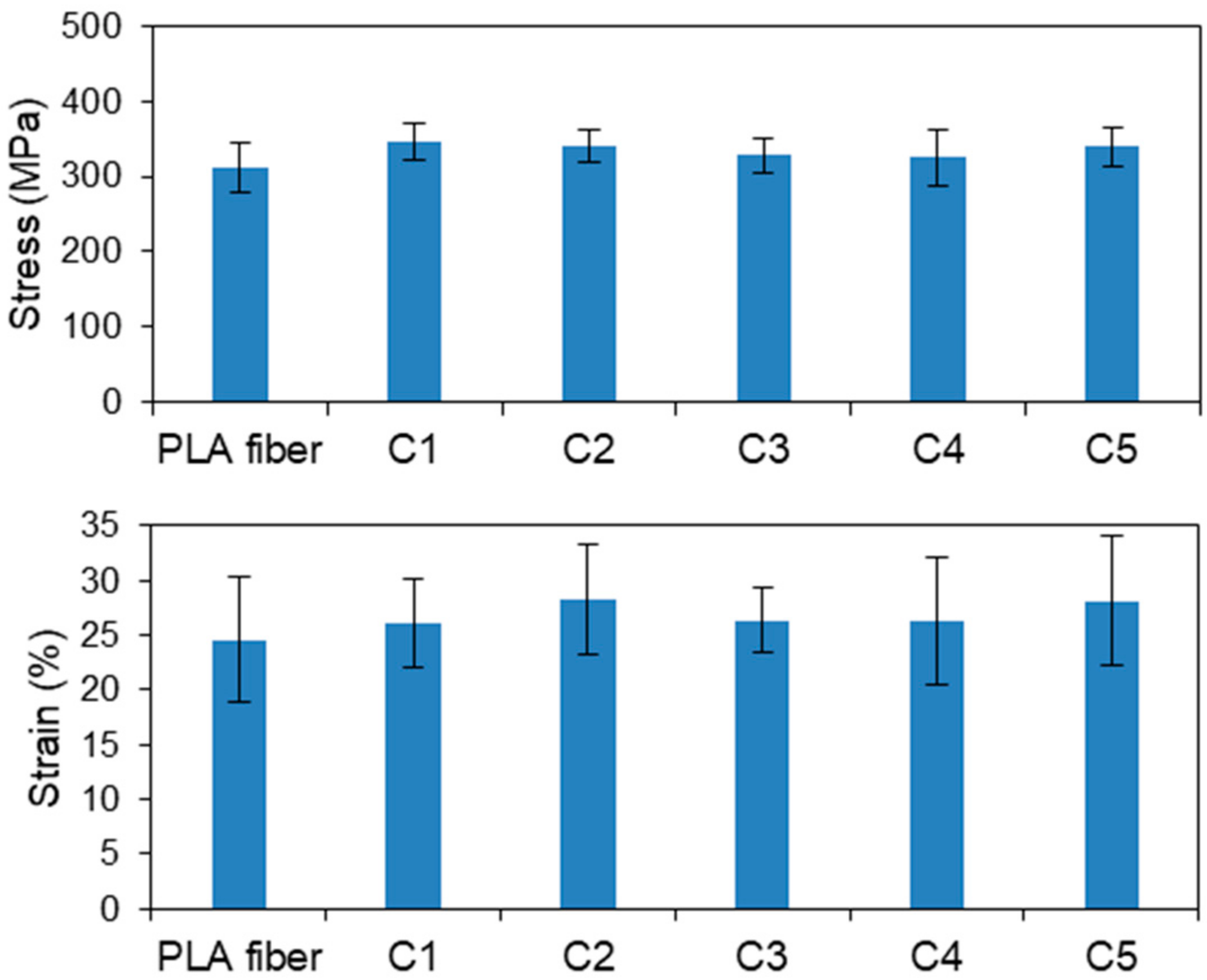
3.1.2. CS-Coated PLA Fibers Prepared by the LBL Method
3.2. Surface Characterization
3.2.1. Plasma-Treated PLA Fibers
3.2.2. CS-Coated PLA Fibers Prepared by the LBL Method
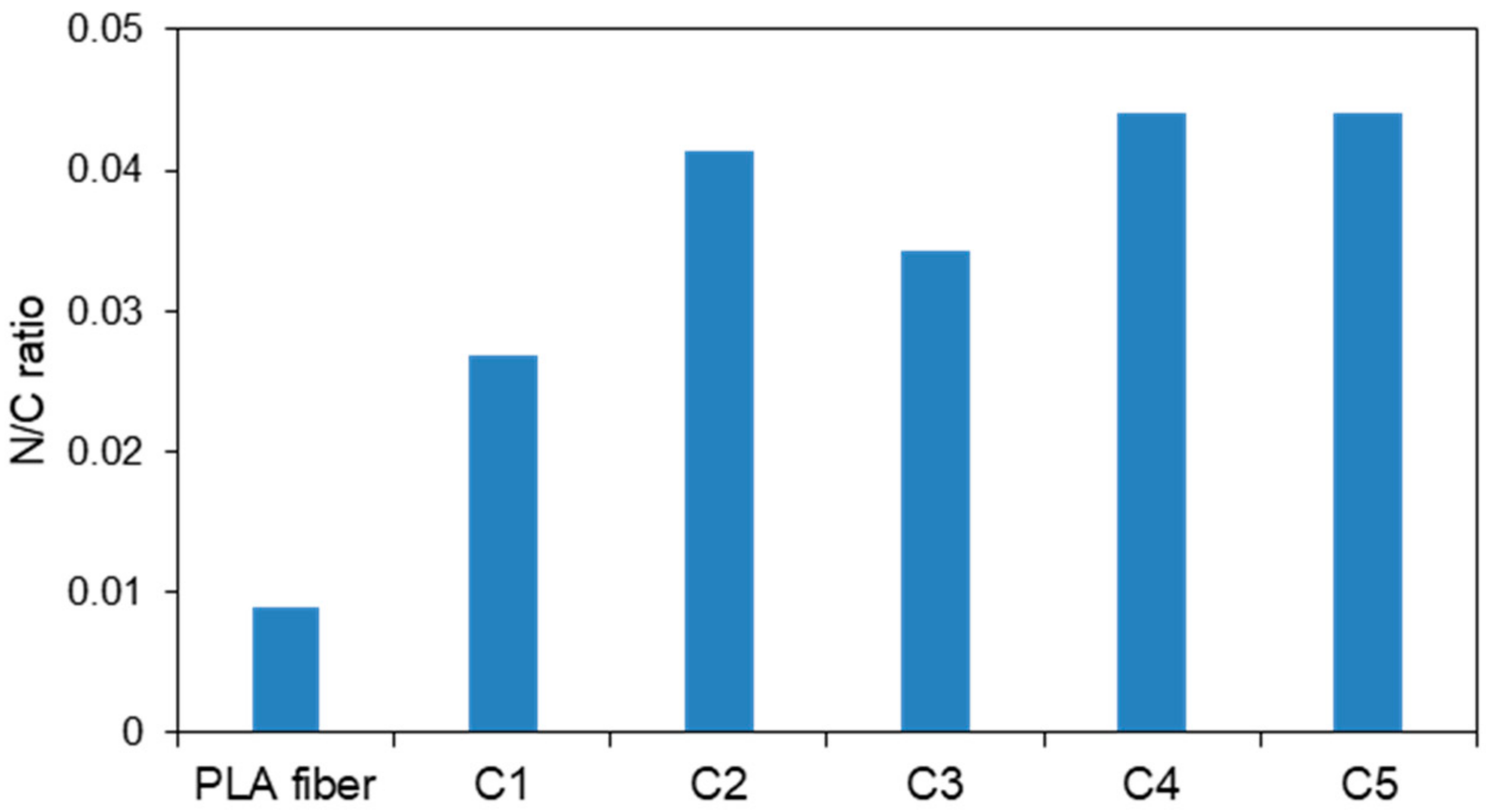
3.3. Nitrogen Analysis
3.4. Degradation Testing
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tomita, K.; Kuroki, Y.; Nagai, K. Isolation of thermophiles degrading poly(l-lactic acid). J. Biosci. Bioeng. 1999, 87, 752–755. [Google Scholar] [CrossRef]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Blends of aliphatic polyesters. II. Hydrolysis of solution-cast blends from poly(l-lactide) and poly(ε-caprolactone) in phosphate-buffered solution. J. Appl. Polym. Sci. 1998, 67, 405–415. [Google Scholar] [CrossRef]
- Lunt, J. Large-scale production, properties and commercial applications of polylactic acid polymers. Polym. Degrad. Stabil. 1998, 59, 145–152. [Google Scholar] [CrossRef]
- Saravanan, M.; Domb, A.J. A contemporary review on—Polymer stereocomplexes and its biomedical application. Eur. J. Nanomed. 2013, 5, 81–96. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Mikos, A.G.; Lyman, M.D.; Freed, L.E.; Langer, R. Wetting of poly(l-lactic acid) and poly(dl-lactic-co-glycolic acid) foams for tissue culture. Biomaterials 1994, 15, 55–58. [Google Scholar] [CrossRef]
- Nagahama, K.; Mori, Y.; Ohya, Y.; Ouchi, T. Biodegradable nanogel formation of polylactide-grafted dextran copolymer in dilute aqueous solution and enhancement of its stability by stereocomplexation. Biomacromolecules 2007, 8, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Rasal, R.M.; Janorkar, A.V.; Hirt, D.E. Poly(lactic acid) modifications. Prog. Polym. Sci. 2010, 35, 338–356. [Google Scholar] [CrossRef]
- Furuike, T.; Nagahama, H.; Chaochai, T.; Tamura, H. Preparation and characterization of chitosan-coated poly(l-lactic acid) fibers and their braided rope. Fibers 2015, 3, 380–393. [Google Scholar] [CrossRef]
- Zhu, A.; Zhang, M.; Wu, J.; Shen, J. Covalent immobilization of chitosan/heparin complex with a photosensitive hetero-bifunctional crosslinking reagent on PLA surface. Biomaterials 2002, 23, 4657–4665. [Google Scholar] [CrossRef]
- Ding, Z.; Chen, J.; Gao, S.; Chang, J.; Zhang, J.; Kang, E.T. Immobilization of chitosan onto poly-L-lactic acid film surface by plasma graft polymerization to control the morphology of fibroblast and liver cells. Biomaterials 2004, 25, 1059–1067. [Google Scholar] [CrossRef]
- Tsuji, H.; Ishida, T. Poly(l-lactide). X. Enhanced surface hydrophilicity and chain-scission mechanisms of poly(l-lactide) film in enzymatic, alkali, and phosphate-buffered solutions. J. Appl. Polym. Sci. 2003, 87, 1628–1633. [Google Scholar] [CrossRef]
- Park, G.E.; Pattison, M.A.; Park, K.; Webster, T.J. Accelerated chondrocyte functions on NaOH-treated PLGA scaffolds. Biomaterials 2005, 26, 3075–3082. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, H.; Nwe, N.; Jayakumar, R.; Koiwa, S.; Furuike, T.; Tamura, H. Novel biodegradable chitin membranes for tissue engineering applications. Carbohyd. Polym. 2008, 73, 295–302. [Google Scholar] [CrossRef]
- Tamura, H.; Nagahama, H.; Tokura, S. Preparation of chitin hydrogel under mild conditions. Cellulose 2006, 13, 357–364. [Google Scholar] [CrossRef]
- Jayakumar, R.; Menon, D.; Manzoor, K.; Nair, S.V.; Tamura, H. Biomedical applications of chitin and chitosan based nanomaterials—A short Review. Carbohyd. Polym. 2010, 82, 227–232. [Google Scholar] [CrossRef]
- Anitha, A.; Rani, V.V.D.; Krishna, R.; Sreeja, V.; Selvamurugan, N.; Nair, S.V.; Tamura, H.; Jayakumar, R. Synthesis, characterization, cytotoxicity and antibacterial studies of chitosan, O-carboxymethyl and N,O-carboxymethyl chitosan nanoparticles. Carbohyd. Polym. 2009, 78, 672–677. [Google Scholar] [CrossRef]
- Carvalho, C.R.; López-Cebral, R.; Silva-Correia, J.; Silva, J.M.; Mano, J.F.; Silva, T.H.; Freier, T.; Reis, R.L.; Oliveira, J.M. Investigation of cell adhesion in chitosan membranes for peripheral nerve regeneration. Mater. Sci. Eng. C 2017, 71, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Dodane, V.; Vilivalam, V.D. Pharmaceutical applications of chitosan. Pharm. Sci. Technol. Today 1998, 1, 246–253. [Google Scholar] [CrossRef]
- Jayakumar, R.; Prabaharan, M.; Kumar, P.T.S.; Nair, S.V.; Tamura, H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol. Adv. 2011, 29, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Javvaji, V.; MacIntire, I.C.; Raghavan, S.R. Gelation of vesicles and nanoparticles using water-soluble hydrophobically modified chitosan. Langmuir 2013, 29, 15302–15308. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.A.; Llanes, F.; Volesky, B.; Diaz-Pulido, G.; McCook, L.; Mucci, A. 1H–NMR study of Na alginates extracted from sargassum spp. in relation to metal biosorption. Appl. Biochem. Biotech. 2003, 110, 75–90. [Google Scholar] [CrossRef]
- Gomez, C.G.; Lambrecht, M.V.P.; Lozano, J.E.; Rinaudo, M.; Villar, M.A. Influence of the extraction-purification conditions on final properties of alginates obtained from brown algae (Macrocystis pyrifera). Int. J. Biol. Macromol. 2009, 44, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Tsuruta, Y.; Tokura, S. Preparation of chitosan-coated alginate filament. Mater. Sci. Eng. C 2002, 20, 143–147. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lin, H.; Shen, L.; Liao, B.-Q.; Wu, X.; Li, R. Effect of calcium ions on fouling properties of alginate solution and its mechanisms. J. Membr. Sci. 2017, 525, 320–329. [Google Scholar] [CrossRef]
- Ariga, K.; Hill, J.P.; Ji, Q. Layer-by-layer assembly as a versatile bottom-up nanofabrication technique for exploratory research and realistic application. Phys. Chem. Chem. Phys. 2007, 9, 2319–2340. [Google Scholar] [CrossRef] [PubMed]
- Antipov, A.A.; Sukhorukov, G.B.; Donath, E.; Mohwald, H. Sustained release properties of polyelectrolyte multilayer capsules. J. Phys. Chem. B 2001, 105, 2281–2284. [Google Scholar] [CrossRef]
- Kyung, W.K.-H.; Kim, S.-H.; Siratori, S. Preparation and characterization of antithrombogenic chitosan/alginate films with enhanced physical stability by cross-linking using layer-bylayer. MATEC Web Conf. 2013, 4, 05008. [Google Scholar] [CrossRef]
- Wan, Y.; Tu, C.; Yang, J.; Bei, J.; Wang, S. Influences of ammonia plasma treatment on modifying depth and degradation of poly(l-lactide) scaffolds. Biomaterials 2006, 27, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.V.; Merino, E.G.; Mano, J.F.; Alves, N.M. Crosslink Effect and Albumin Adsorption onto Chitosan/Alginate Multilayered Systems: An in situ QCM-D Study. Macromol. Biosci. 2010, 10, 1444–1455. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Power (W) | Time (s) |
|---|---|---|
| P1 | 100 | 60 |
| P2 | 100 | 300 |
| P3 | 100 | 1800 |
| P4 | 200 | 60 |
| P5 | 200 | 300 |
| P6 | 200 | 1800 |
| Sample | Number of CS Layers | CS Content (%) | Thickness of the CS Layer (nm) |
|---|---|---|---|
| C1 | 1 | 0.331 | 31.5 |
| C2 | 3 | 0.377 | 35.9 |
| C3 | 5 | 0.585 | 55.8 |
| C4 | 10 | 0.730 | 69.7 |
| C5 | 15 | 1.083 | 103.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoto, D.; Ikeda, R.; Furuike, T.; Tamura, H. Preparation of Chitosan-Coated Poly(L-Lactic Acid) Fibers for Suture Threads. Fibers 2018, 6, 84. https://doi.org/10.3390/fib6040084
Komoto D, Ikeda R, Furuike T, Tamura H. Preparation of Chitosan-Coated Poly(L-Lactic Acid) Fibers for Suture Threads. Fibers. 2018; 6(4):84. https://doi.org/10.3390/fib6040084
Chicago/Turabian StyleKomoto, Daiki, Ryoka Ikeda, Tetsuya Furuike, and Hiroshi Tamura. 2018. "Preparation of Chitosan-Coated Poly(L-Lactic Acid) Fibers for Suture Threads" Fibers 6, no. 4: 84. https://doi.org/10.3390/fib6040084
APA StyleKomoto, D., Ikeda, R., Furuike, T., & Tamura, H. (2018). Preparation of Chitosan-Coated Poly(L-Lactic Acid) Fibers for Suture Threads. Fibers, 6(4), 84. https://doi.org/10.3390/fib6040084