Electrodeposition of Hydroxyapatite Coatings for Marble Protection: Preliminary Results





Abstract
1. Introduction
- Possible application of pulsed electric potential. For CaP coating densification, some studies have proposed using pulsed potential, i.e., applying the electric potential by cyclically alternating on/off periods [24,27,29]. According to these studies, during the on-period, OH− ions are produced at the cathode; during the off-period, Ca2+ and PO43− ions have the time to diffuse from the bulk solution towards the cathode, where OH− groups are present so that Reactions (9)–(11) take place [29]. Moreover, pulsed potential reportedly reduces formation of H2 bubbles (cf. Reactions (3–5)), which might otherwise adhere to the substrate and prevent formation of a dense and well adhering coating [24,27].
- Possible addition of ethanol (EtOH) to the electrolyte solution. To reduce the formation of H2 bubbles, the possible addition of ethanol to the aqueous solution has been proposed [23,28]. In fact, ethanol addition reduces the conductivity of the solution, thus reducing H2 bubble formation and promoting coating densification [23,28]. The best results were reported for ethanol additions of about 30–50 vol.% [23,28].
2. Materials and Methods
2.1. Materials
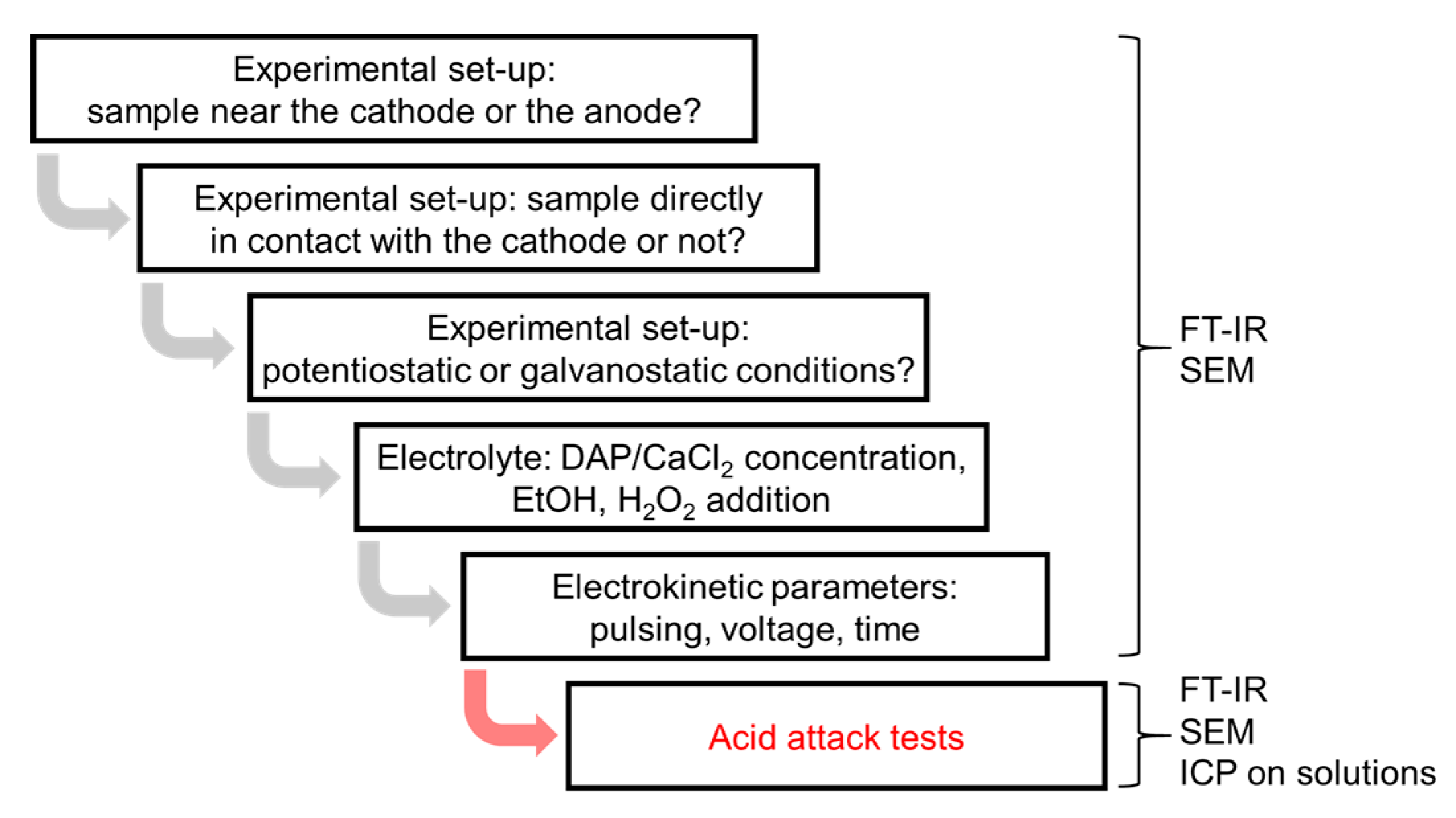
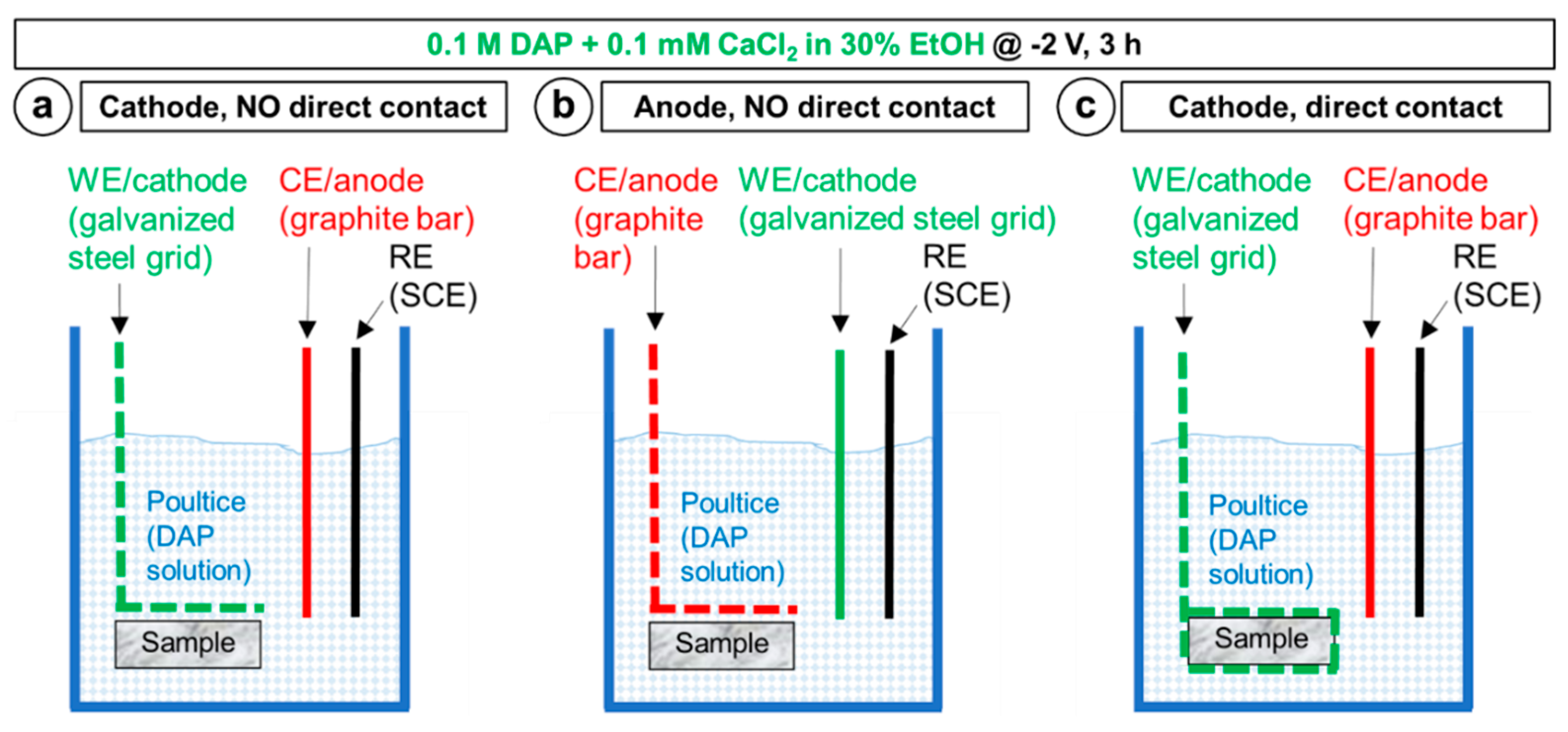
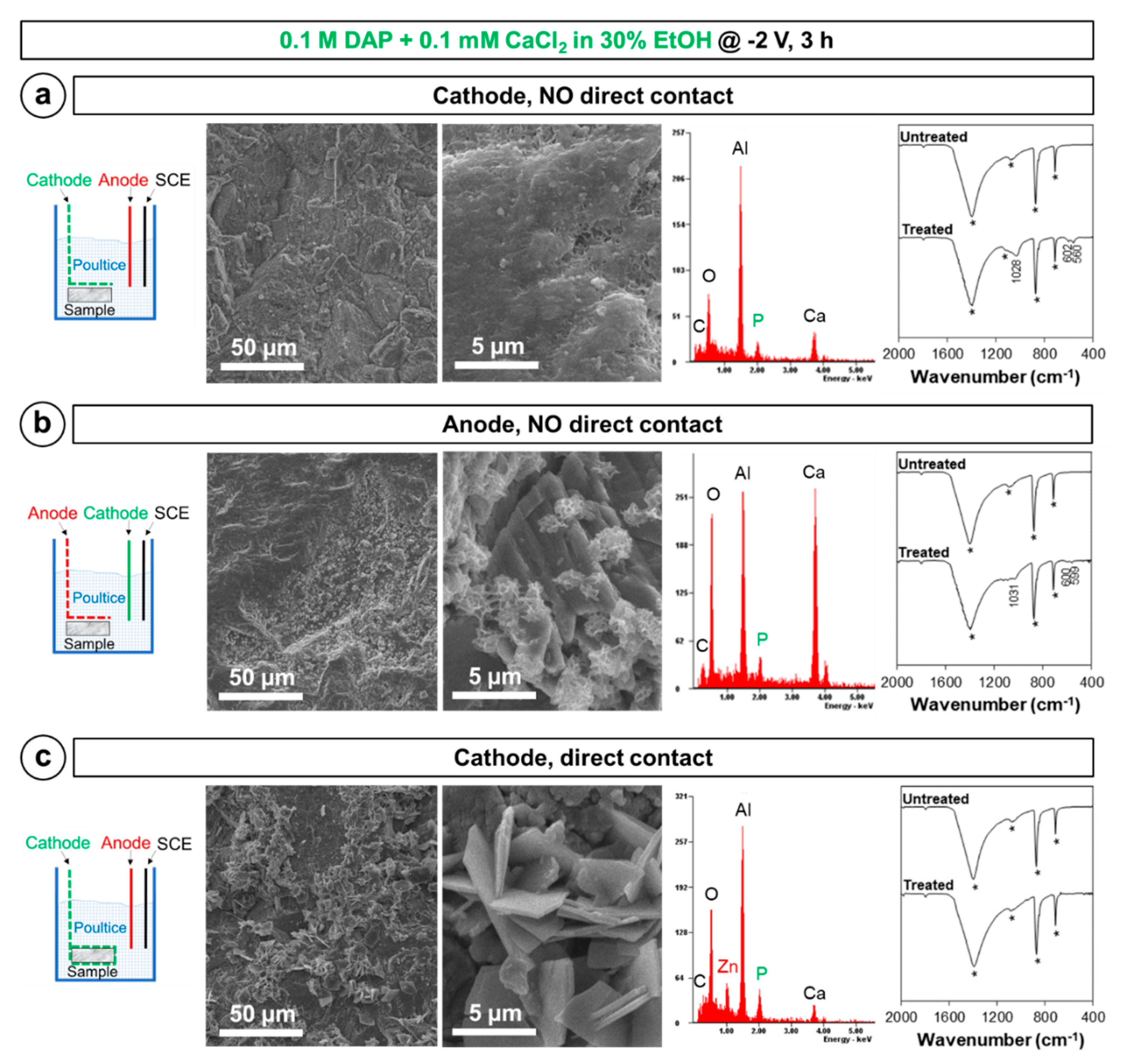
2.2. Influence of the Experimental Setup
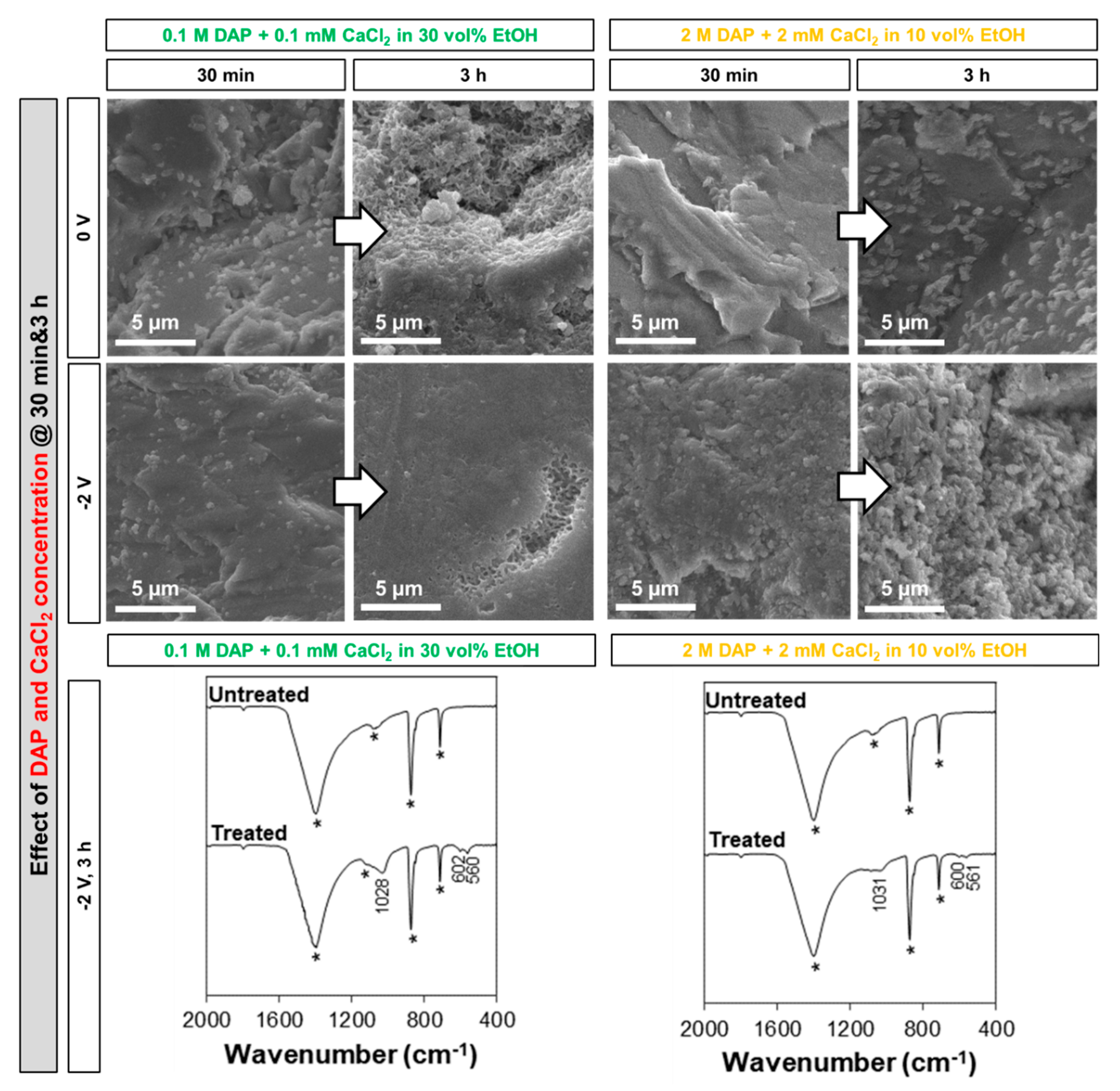
2.3. Influence of the Electrolyte: DAP and CaCl2 Concentration, Ethanol and/or H2O2 Addition
- 30 vol.% EtOH. As mentioned above, this is the maximum concentration not leading to immediate precipitation in the bulk solution at the given DAP and CaCl2 concentrations (only a few isolated crystals were formed in the bulk solution). Even when no current is applied, EtOH promotes HAP nucleation [5,7], so some benefit compared to the reference 0.1 M DAP + 0.1 mM CaCl2 solution is already expected. Moreover, when electrodeposition is adopted, EtOH reportedly reduces H2 bubbling and promotes coating densification [23,28], so an additional benefit is expected.
- 30 vol.% EtOH + 10 vol.% H2O2. To combine the two effects described above, the double addition was also investigated.
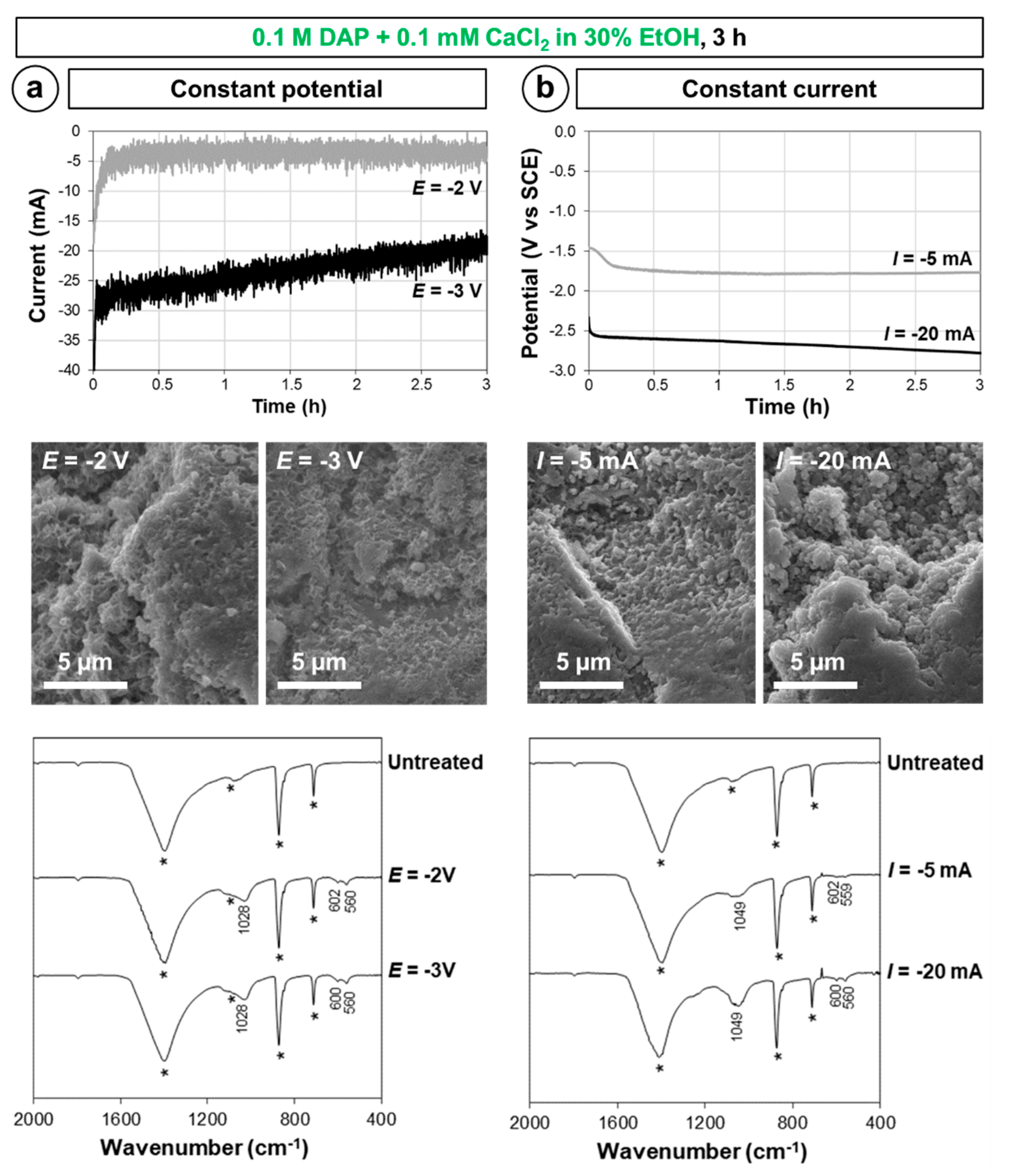
2.4. Influence of Electrokinetic Parameters: Pulsing, Voltage, and Time
2.5. Acid Resistance
2.6. Sample Characterization
3. Results and Discussion
3.1. Influence of the Experimental Setup
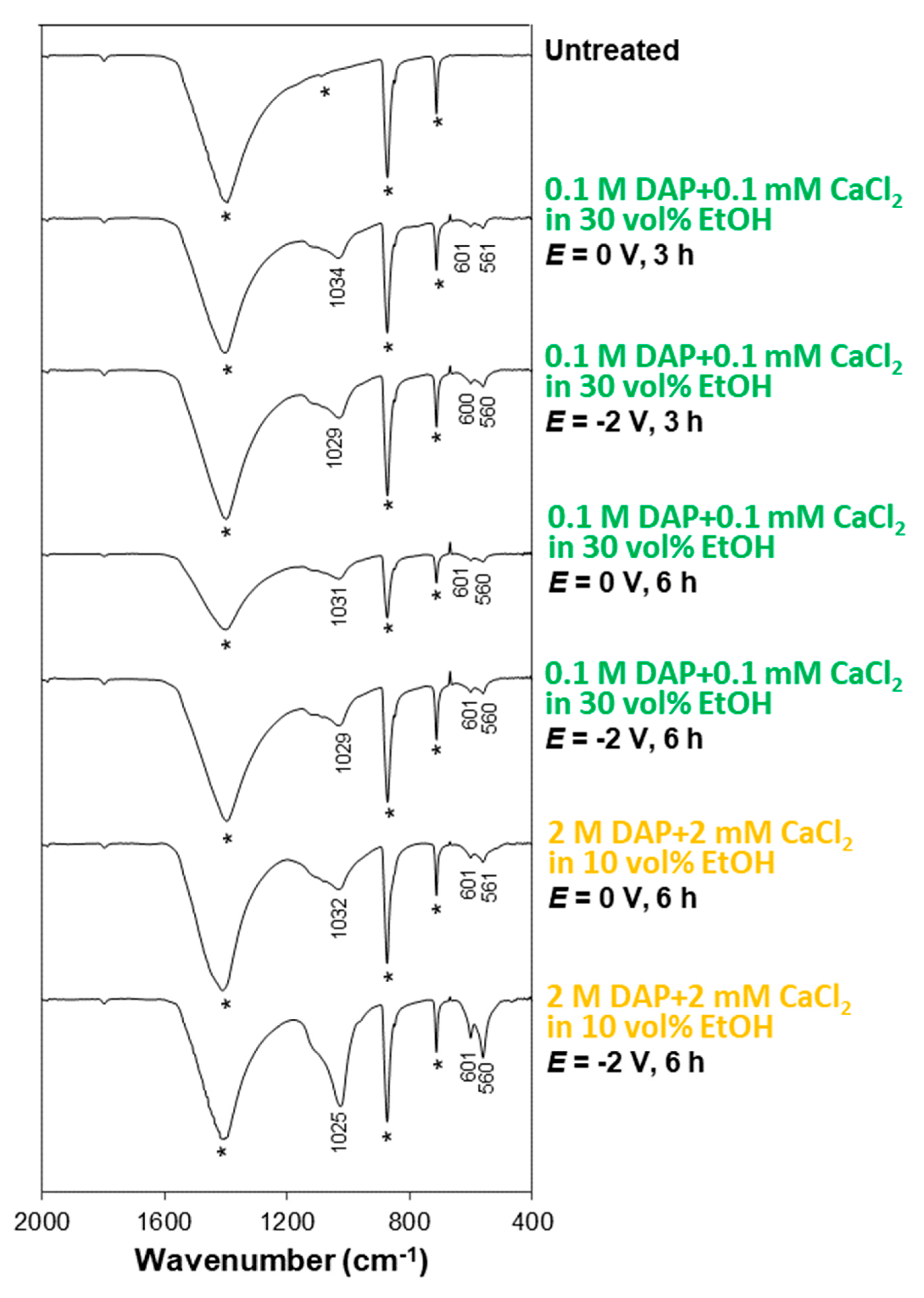
3.2. Influence of the Electrolyte: DAP and CaCl2 Concentration, Ethanol and/or H2O2 Addition
3.3. Influence of Electrokinetic Parameters: Pulsing, Voltage, and Time
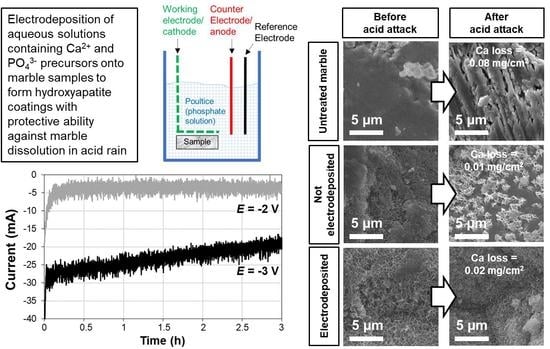
3.4. Acid Resistance
4. Conclusions
- Electrodeposition of HAP over the marble surface can be obtained by placing the marble sample close to the cathode. In these conditions, OH− groups formed near the cathode during electrodeposition favor HAP formation, compared to the situation when no electric potential is applied. Compared to simple increase in pH of the phosphate solution used as HAP precursor, the local increase in OH− near the cathode has the advantage that precipitation of CaP in the bulk solution and excessive film growth are prevented.
- At short times (30 min and 1 h), electrodeposition has a significant effect in accelerating and improving marble coverage by the new HAP coating. At longer times, the effect becomes less evident, because HAP nucleation and growth take place even without electrodeposition.
- Among the two formulations investigated in this study (namely, a solution containing 0.1 M DAP + 0.1 mM CaCl2 in 30 vol.% EtOH and a solution containing 2 M DAP + 2 mM CaCl2 in 10 vol.% EtOH), the less-concentrated one, containing a higher amount of ethanol, leads to formation of more uniform coatings in a shorter time. This is possible thanks to the boosting effect of ethanol, which is visible even without electrodeposition and which is further enhanced when electric potential is applied.
- The HAP coatings formed by electrodeposition in the present most promising conditions (−2 V for 3 and 6 h in the case of the less-concentrated solution and −1 V for 6 h in the case of the more-concentrated one) provide some protective efficacy, even though the substrate dissolution is not completely inhibited. In fact, pores are likely present in the electrodeposited coatings, which reduces their protective efficacy and leads to cracking when the coating thickness is excessive. Moreover, possible damaged parts (e.g., the areas near the edges) proved to have a not negligible influence on the coating protective efficacy, which needs to be taken into account when designing acid resistance tests.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patnaik, P. Handbook of Inorganic Chemical Compounds, 1st ed.; McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- Naidu, S.; Blair, J.; Scherer, G.W. Acid-resistant coatings on marble. J. Am. Ceram. Soc. 2016, 99, 3421–3428. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates: Occurrence, properties, biomineralization, pathological calcification and biomimetic applications. Biomatter 2011, 1, 121–164. [Google Scholar] [CrossRef]
- Naidu, S.; Sassoni, E.; Scherer, G.W. New treatment for corrosion-resistant coatings for marble and consolidation of limestone. In Jardins de Pierres—Conservation of Stone in Parks, Gardens and Cemeteries; Stefanaggi, M., Vergès-Belmin, V., Eds.; XL Print: Paris, France, 2011; pp. 289–294. [Google Scholar]
- Graziani, G.; Sassoni, E.; Franzoni, E.; Scherer, G.W. Hydroxyapatite coatings for marble protection: Optimization of calcite covering and acid resistance. Appl. Surf. Sci. 2016, 368, 241–257. [Google Scholar] [CrossRef]
- Graziani, G.; Sassoni, E.; Scherer, G.W.; Franzoni, E. Resistance to simulated rain of hydroxyapatite- and calcium oxalate-based coatings for protection of marble against corrosion. Corros. Sci. 2017, 127, 168–174. [Google Scholar] [CrossRef]
- Sassoni, E.; Graziani, G.; Franzoni, E.; Scherer, G.W. Calcium phosphate coatings for marble conservation: Influence of ethanol and isopropanol addition to the precipitation medium on the coating microstructure and performance. Corros. Sci. 2018, 136, 255–267. [Google Scholar] [CrossRef]
- Sassoni, E. Hydroxyapatite and other calcium phosphates for the conservation of cultural heritage: A review. Materials 2018, 11, 557. [Google Scholar] [CrossRef] [PubMed]
- Sassoni, E.; Naidu, S.; Scherer, G.W. The use of hydroxyapatite as a new inorganic consolidant for damaged carbonate stones. J. Cult. Herit. 2011, 12, 346–355. [Google Scholar] [CrossRef]
- Possenti, E.; Colombo, C.; Bersani, D.; Bertasa, M.; Botteon, A.; Conti, C.; Lottici, P.P.; Realini, M. New insight on the interaction of diammonium hydrogen phosphate conservation treatment with carbonatic substrates: A multi-analytical approach. Microchem. J. 2016, 127, 79–86. [Google Scholar] [CrossRef]
- Possenti, E.; Colombo, C.; Conti, C.; Gigli, L.; Merlini, M.; Rikkert Plaisier, J.; Realini, M.; Sali, D.; Diego Gatta, G. Diammonium hydrogenphosphate for the consolidation of building materials. Investigation of newly-formed calcium phosphates. Constr. Build. Mater. 2019, 195, 557–563. [Google Scholar] [CrossRef]
- Naidu, S.; Scherer, G.W. Nucleation, growth and evolution of calcium phosphate films on calcite. J. Colloid Interface Sci. 2014, 435, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Redeppening, J.; McIsaac, J.P. Electrocrystallization of brushite coatings on prosthetic alloys. Chem. Mater. 1990, 2, 625–627. [Google Scholar] [CrossRef]
- Shirkhanzadeh, M. Bioactive calcium phosphate coatings prepared by electrodeposition. J. Mater. Sci. Lett. 1991, 10, 1415–1417. [Google Scholar] [CrossRef]
- Monma, H. Electrolytic depositions of calcium phosphates on substrate. J. Mater. Sci. 1994, 29, 949–953. [Google Scholar] [CrossRef]
- Shirkhanzadeh, M. Calcium phosphate coatings prepared by electrocrystallization from aqueous electrolytes. J. Mater. Sci. Mater. Med. 1995, 6, 90–93. [Google Scholar] [CrossRef]
- Redeppening, J.; Schlessinger, T.; Burnham, S.; Lippiello, L.; Miyano, J. Characterization of electrolytically prepared brushite and hydroxyapatite coatings on orthopedic alloys. J. Biomed. Mater. Res. 1996, 30, 287–294. [Google Scholar] [CrossRef]
- Yen, S.K.; Lin, C.M. Cathodic reactions of electrolytic hydroxyapatite coating on pure titanium. Mater. Chem. Phys. 2002, 77, 70–76. [Google Scholar] [CrossRef]
- Kuo, M.C.; Yen, S.K. The process of electrochemical deposited hydroxyapatite coatings on biomedical titanium at room temperature. Mater. Sci. Eng. C 2002, 20, 153–160. [Google Scholar] [CrossRef]
- Eliaz, N.; Sridhar, T.M. Electrocrystallization of hydroxyapatite and its dependence on solution conditions. Cryst. Growth Des. 2008, 8, 3965–3977. [Google Scholar] [CrossRef]
- Hu, R.; Lin, C.; Shi, H.; Wang, H. Electrochemical deposition mechanism of calcium phosphate coating in dilute Ca-P electrolyte system. Mater. Chem. Phys. 2009, 115, 718–723. [Google Scholar] [CrossRef]
- Mokabber, T.; Lu, L.Q.; van Rijn, P.; Vakis, A.I.; Pei, Y.T. Crystal growth mechanism of calcium phosphate coatings on titanium by electrochemical deposition. Surf. Coat. Technol. 2018, 334, 526–535. [Google Scholar] [CrossRef]
- Bobby Kannan, M. Enhancing the performance of calcium phosphate coating on a magnesium alloy for bioimplant applications. Mater. Lett. 2012, 76, 109–112. [Google Scholar] [CrossRef]
- Kannan, M.B.; Wallipa, O. Potentiostatic pulse-deposition of calcium phosphate on magnesium alloy for temporary implant applications—An in vitro corrosion study. Mater. Sci. Eng. C 2013, 33, 675–679. [Google Scholar] [CrossRef]
- Kannan, M.B. Improving the packing density of calcium phosphate coating on a magnesium alloy for enhanced degradation resistance. J. Biomed. Mater. Res. A 2013, 101A, 1248–1254. [Google Scholar] [CrossRef]
- Walter, R.; Kannan, M.B.; He, Y.; Sandham, A. Influence of the cathodic activity of magnesium alloys on the electrochemical deposition of calcium phosphate. Mater. Lett. 2014, 130, 184–187. [Google Scholar] [CrossRef]
- Bobby Kannan, M. Electrochemical deposition of calcium phosphates on magnesium and its alloys for improved biodegradation performance: A review. Surf. Coat. Technol. 2016, 301, 36–41. [Google Scholar] [CrossRef]
- Chen, J.S.; Juang, H.Y.; Hon, M.H. Calcium phosphate coating on titanium substrate by a modified electrocrystallization process. J. Mater. Sci. Mater. Med. 1998, 9, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Gopi, D.; Indira, J.; Kavitha, L. A comparative study on the direct and pulsed current electrodeposition of hydroxyapatite coatings on surgical grade stainless steel. Surf. Coat. Technol. 2012, 206, 2859–2869. [Google Scholar] [CrossRef]
- Monasteiro, N.; Ledesma, J.L.; Aranguiz, I.; Garcia-Romero, A.; Zuza, E. Analysis of electrodeposition processes to obtain calcium phosphate layer on AZ31 alloy. Surf. Coat. Technol. 2017, 319, 12–22. [Google Scholar] [CrossRef]
- Drevet, R.; Benhayoune, H.; Wortham, L.; Potiron, S.; Douglade, J.; Laurent-Maquin, D. Effects of pulsed current and H2O2 amount on the composition on electrodeposited calcium phosphate coatings. Mater. Character 2010, 61, 786–795. [Google Scholar] [CrossRef]
- Blackwood, D.J.; Seah, K.H.W. Electrochemical cathodic deposition of hydroxyapatite: Improvements in adhesion and crystallinity. Mater. Sci. Eng. C 2009, 29, 1233–1238. [Google Scholar] [CrossRef]
- Ottosen, L.M.; Rörig-Dalgård, I. Electrokinetic removal of Ca(NO3)2 from bricks to avoid salt-induced decay. Electrochim. Acta 2007, 52, 3454–3463. [Google Scholar] [CrossRef]
- Ottosen, L.M.; Rörig-Dalgård, I. Desalination of a brick by application of an electric DC field. Mater. Struct. 2009, 42, 961–971. [Google Scholar] [CrossRef]
- Ottosen, L.M.; Christensen, I.V. Electrokinetic desalination of sandstones for NaCl removal—Test of different clay poultices at the electrodes. Electrochim. Acta 2012, 86, 192–202. [Google Scholar] [CrossRef]
- Bernabeu, A.; Expçsito, E.; Montiel, V.; Ordònez, S.; Aldaz, A. A new electrochemical method for consolidation of porous rocks. Electrochem. Commun. 2001, 3, 122–127. [Google Scholar] [CrossRef]
- Feijoo, J.; Ottosen, L.M.; Nòvoa, X.R.; Rivas, T.; de Rosario, I. An improved electrokinetic method to consolidate porous materials. Mater. Struct. 2017, 50, 186. [Google Scholar] [CrossRef]
- Feijoo, J.; Nòvoa, X.R.; Rivas, T. Electrokinetic treatment to increase bearing capacity and durability of granite. Mater. Struct. 2017, 50, 251. [Google Scholar] [CrossRef]
- Meloni, P.; Manca, F.; Carcangiu, G. Marble protection: An inorganic electrokinetic approach. Appl. Surf. Sci. 2013, 273, 377–385. [Google Scholar] [CrossRef]
- Matteini, M.; Moles, A.; Giovannoni, S. Calcium oxalate as a protective mineral system for wall paintings: Methodology and analyses. In Proceedings of the III International Symposium Conservation of Monuments in the Mediterranean Basin, Venice, Italy, 22–25 June 1994. [Google Scholar]
- Sandrolini, F.; Franzoni, E.; Sassoni, E.; Diotallevi, P.P. The contribution of urban-scale environmental monitoring to materials diagnostics: A study on the Cathedral of Modena (Italy). J. Cult. Herit. 2011, 12, 441–450. [Google Scholar] [CrossRef]
- Bonazza, A.; Messina, P.; Sabbioni, C.; Grossi, C.M.; Brimblecombe, P. Mapping the impact of climate change on surface recession of carbonate buildings in Europe. Sci. Total Environ. 2009, 407, 2039–2050. [Google Scholar] [CrossRef]
- Karampas, I.A.; Kontoyannis, C.G. Characterization of calcium phosphates mixtures. Vib. Spectrosc. 2013, 64, 126–133. [Google Scholar] [CrossRef]
- Evans, A.G.; Drory, M.D.; Hu, M.S. The cracking and decohesion of thin films. J. Mater. Res. 1988, 3, 1043–1049. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Voltage, Time | Bare Edges | Coated Edges | ||
|---|---|---|---|---|---|
| Ca (mg/cm2) | P (mg/cm2) | Ca (mg/cm2) | P (mg/cm2) | ||
| Untreated reference | – | 0.09 | – | 0.08 | – |
| 0.1 M DAP + 0.1 mM CaCl2 in 30 vol.% EtOH | 0 V, 3 h | 0.10 | – | 0.03 | – |
| 0.1 M DAP + 0.1 mM CaCl2 in 30 vol.% EtOH | −2 V, 3 h | 0.08 | – | 0.02 | – |
| 0.1 M DAP + 0.1 mM CaCl2 in 30 vol.% EtOH | 0 V, 6 h | 0.09 | – | 0.01 | – |
| 0.1 M DAP + 0.1 mM CaCl2 in 30 vol.% EtOH | −2 V, 6 h | 0.08 | – | 0.02 | – |
| 2 M DAP + 2 mM CaCl2 in 10 vol.% EtOH | 0 V, 6 h | 0.11 | 0.01 | 0.01 | – |
| 2 M DAP + 2 mM CaCl2 in 10 vol.% EtOH | −1 V, 6 h | 0.08 | 0.01 | 0.02 | – |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sassoni, E.; Masi, G.; Bignozzi, M.C.; Franzoni, E. Electrodeposition of Hydroxyapatite Coatings for Marble Protection: Preliminary Results. Coatings 2019, 9, 207. https://doi.org/10.3390/coatings9030207
Sassoni E, Masi G, Bignozzi MC, Franzoni E. Electrodeposition of Hydroxyapatite Coatings for Marble Protection: Preliminary Results. Coatings. 2019; 9(3):207. https://doi.org/10.3390/coatings9030207
Chicago/Turabian StyleSassoni, Enrico, Giulia Masi, Maria Chiara Bignozzi, and Elisa Franzoni. 2019. "Electrodeposition of Hydroxyapatite Coatings for Marble Protection: Preliminary Results" Coatings 9, no. 3: 207. https://doi.org/10.3390/coatings9030207
APA StyleSassoni, E., Masi, G., Bignozzi, M. C., & Franzoni, E. (2019). Electrodeposition of Hydroxyapatite Coatings for Marble Protection: Preliminary Results. Coatings, 9(3), 207. https://doi.org/10.3390/coatings9030207