Metal Fluorides as Lithium-Ion Battery Materials: An Atomic Layer Deposition Perspective



Abstract
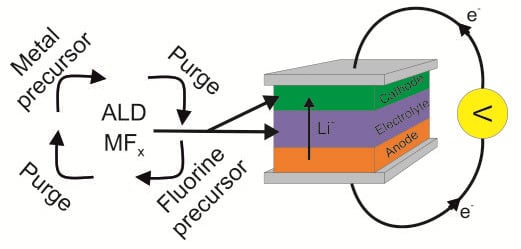
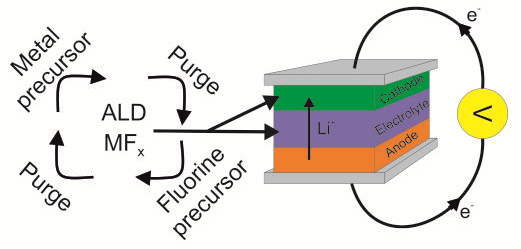
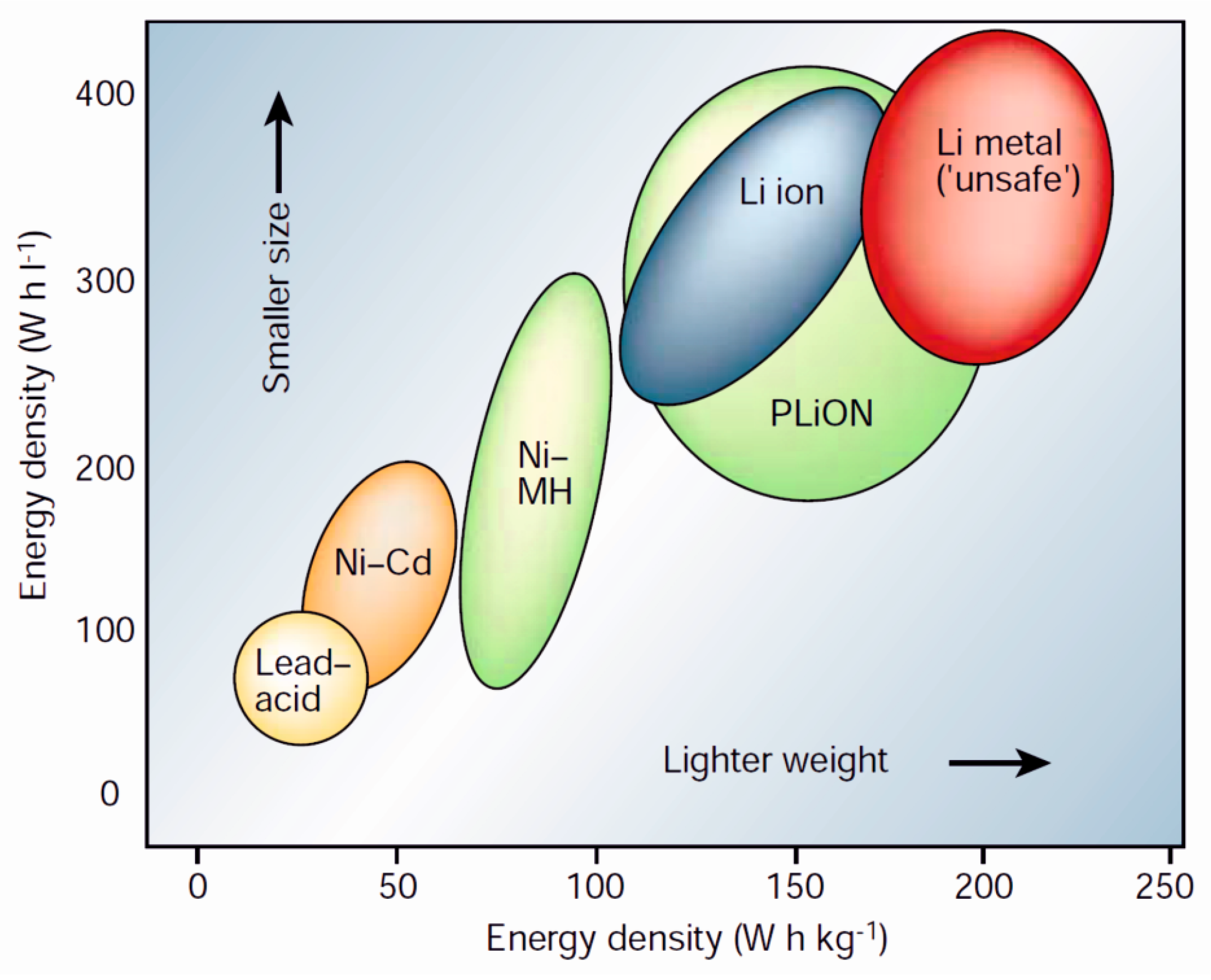
1. Introduction
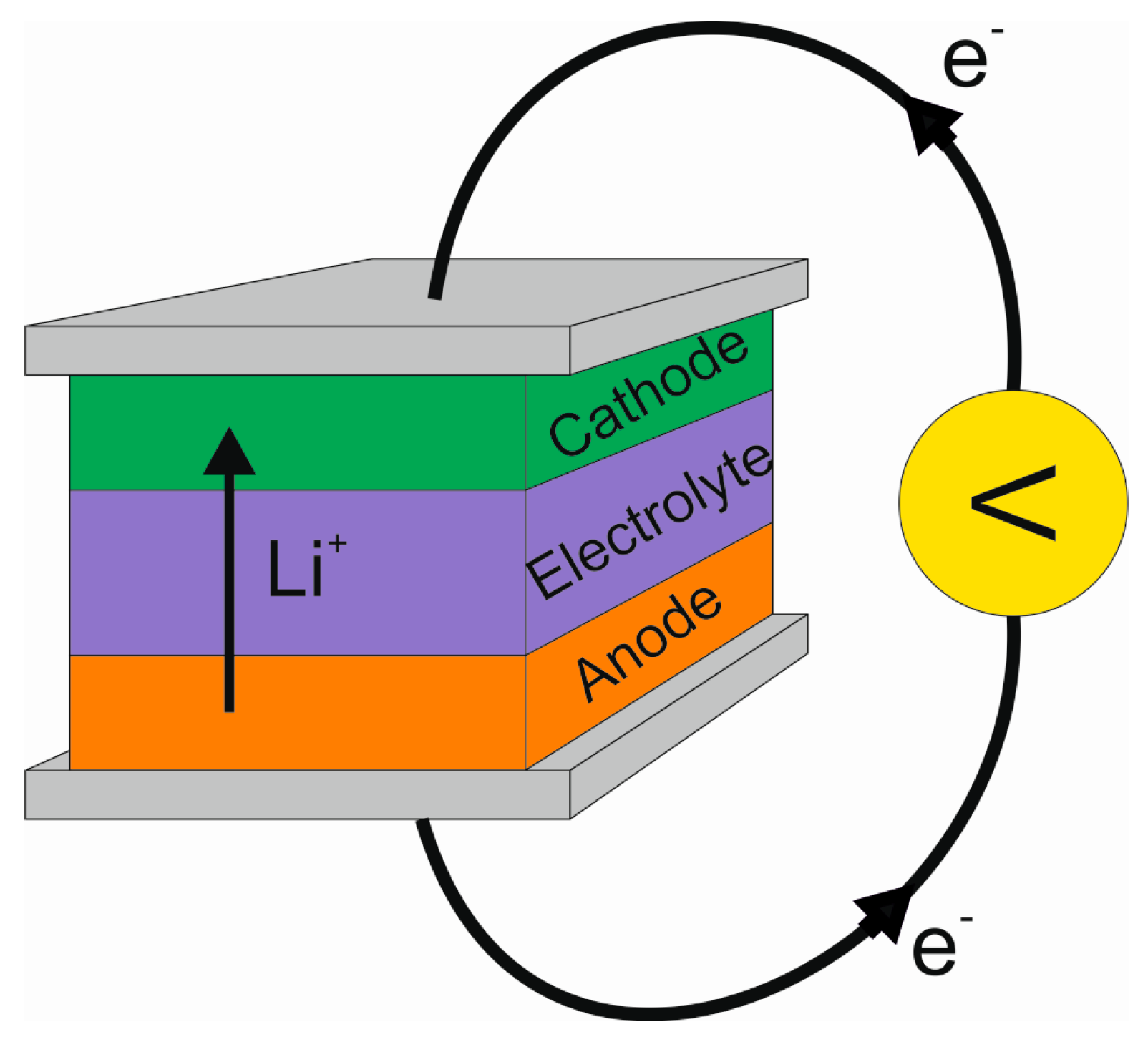
2. The Lithium-Ion Battery
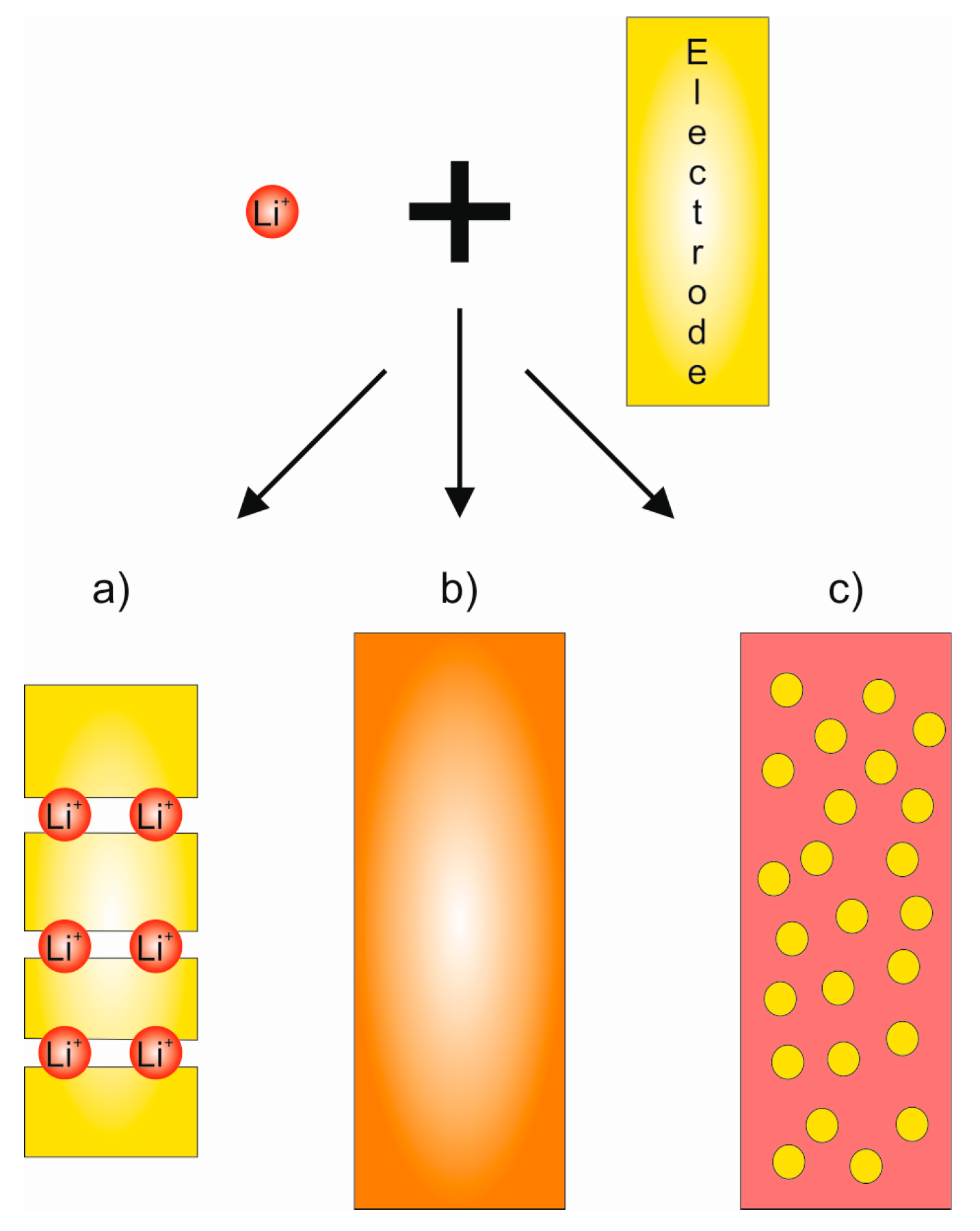
2.1. Basic Principle
2.2. Conventional Electrode Materials
2.2.1. Cathodes
2.2.2. Anodes
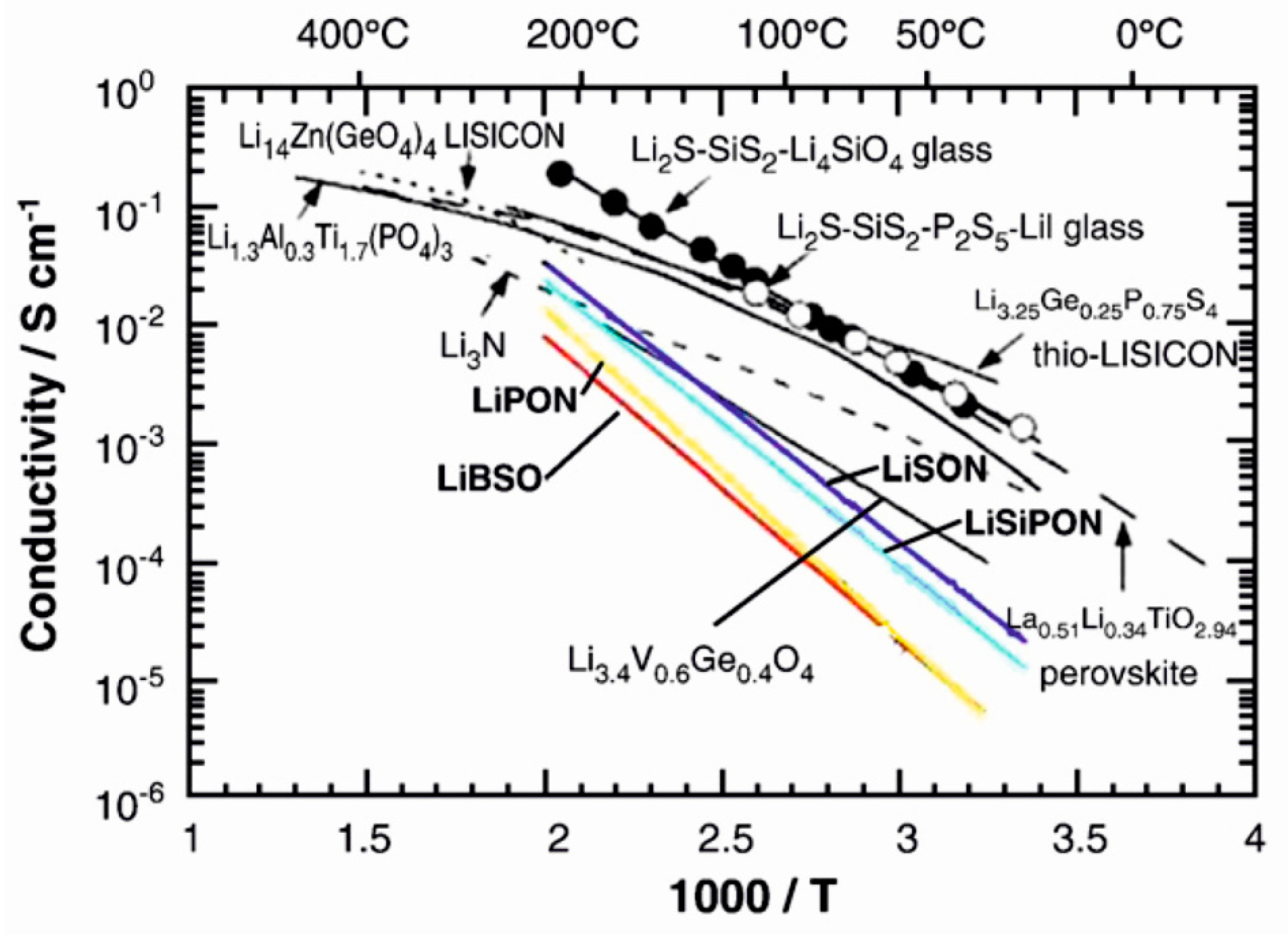
2.3. Conventional Solid Electrolyte Materials
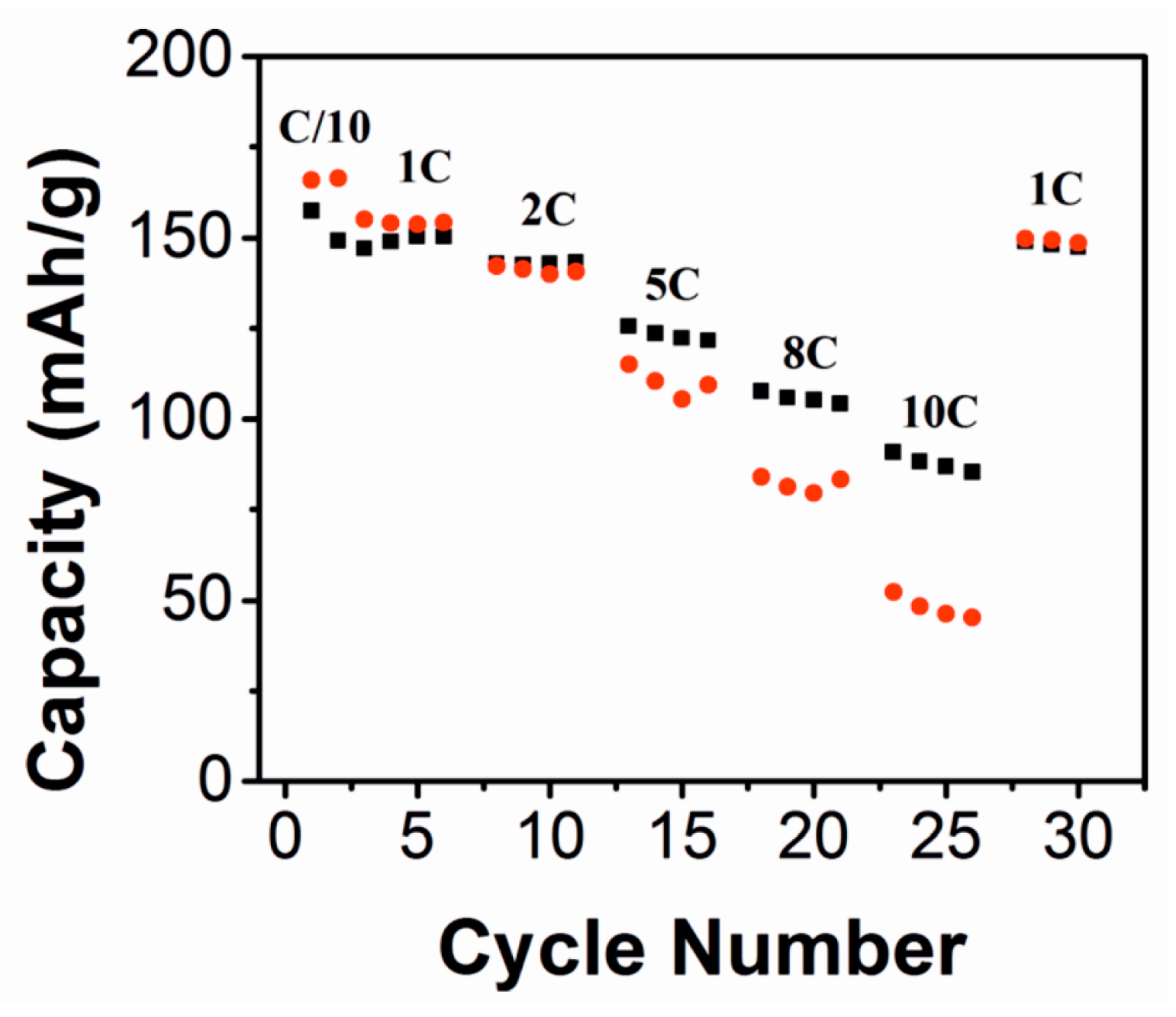
3. Metal Fluorides as Lithium-Ion Battery Materials
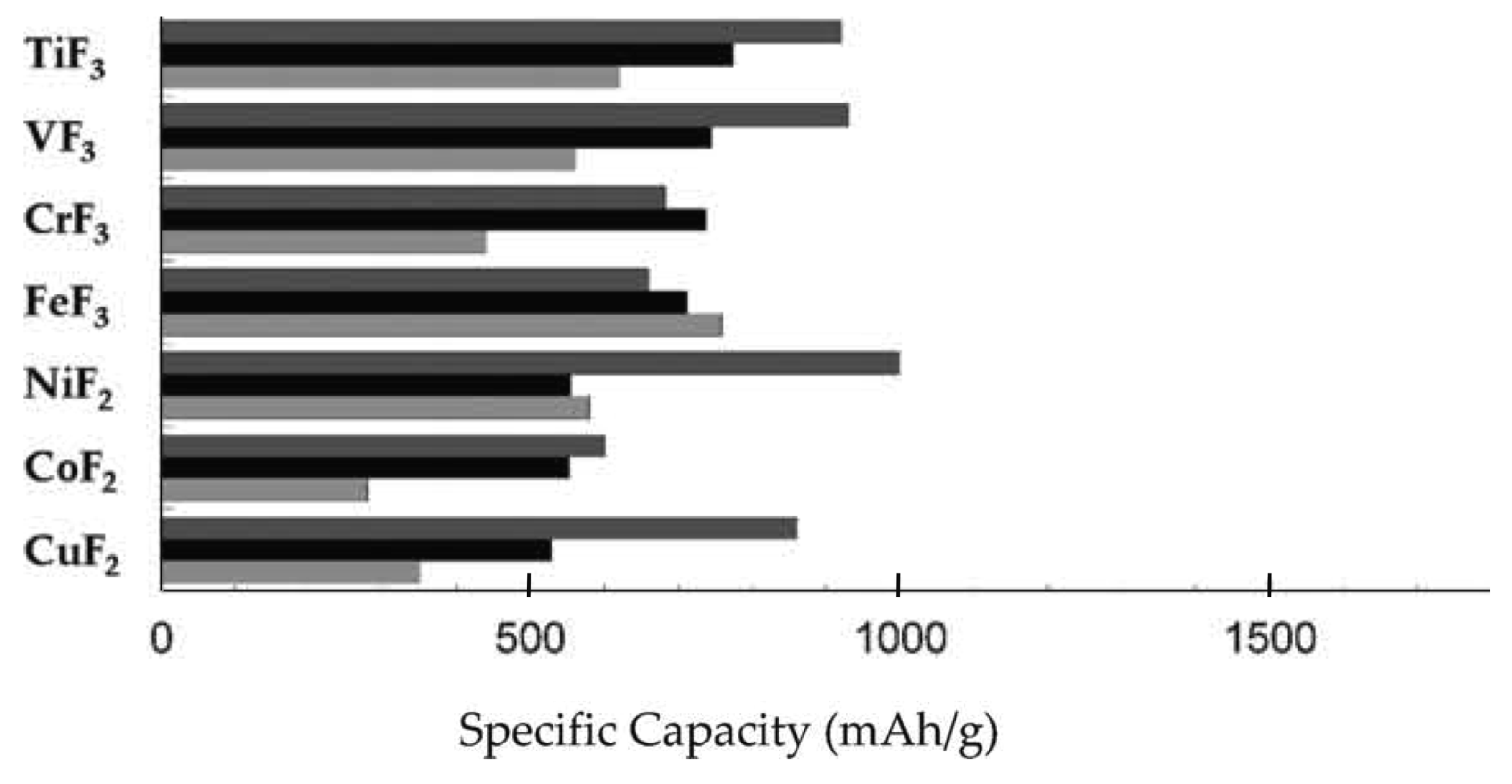
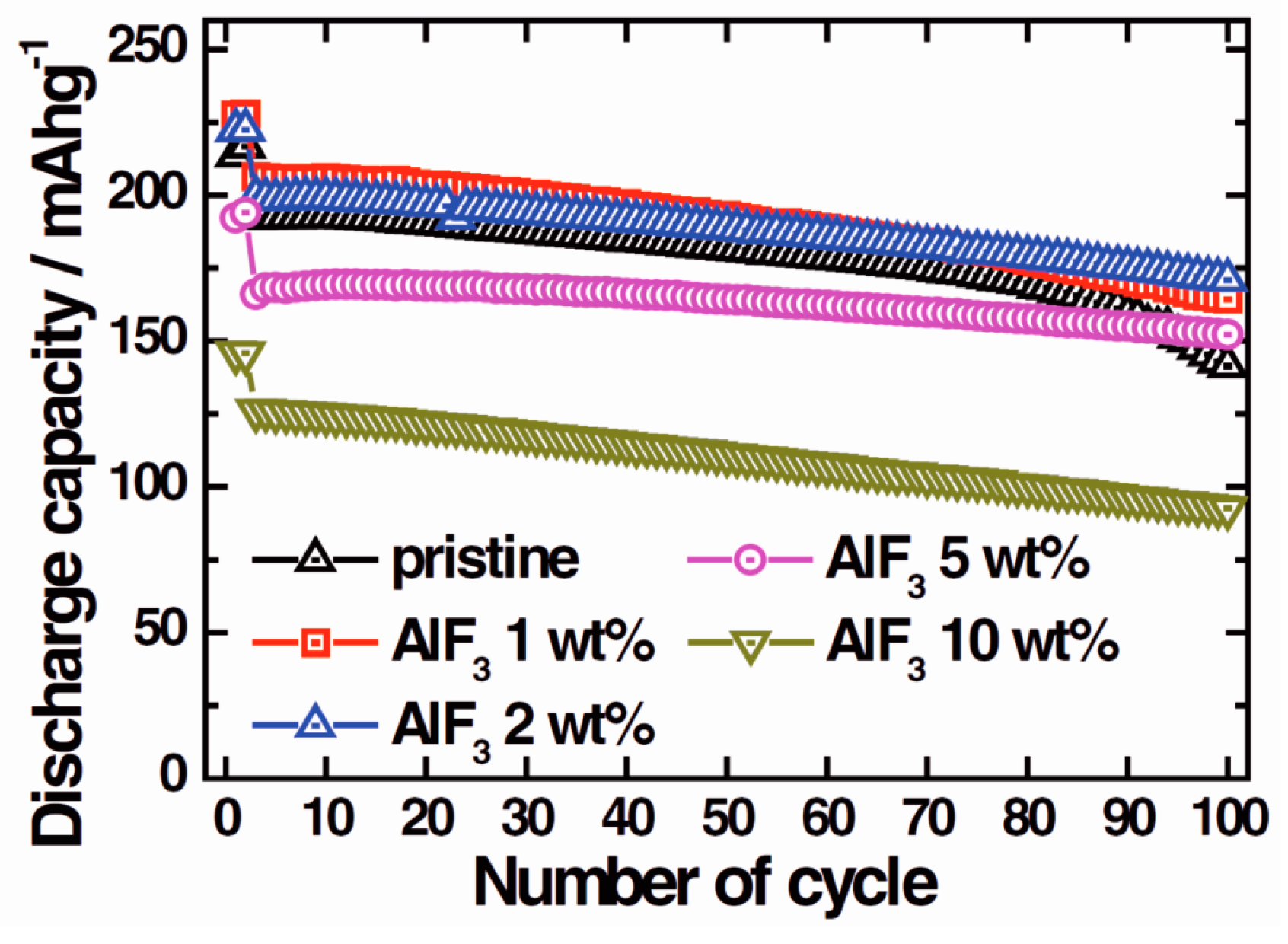
3.1. Electrode Materials
3.2. Solid Electrolyte Materials
4. Atomic Layer Deposition
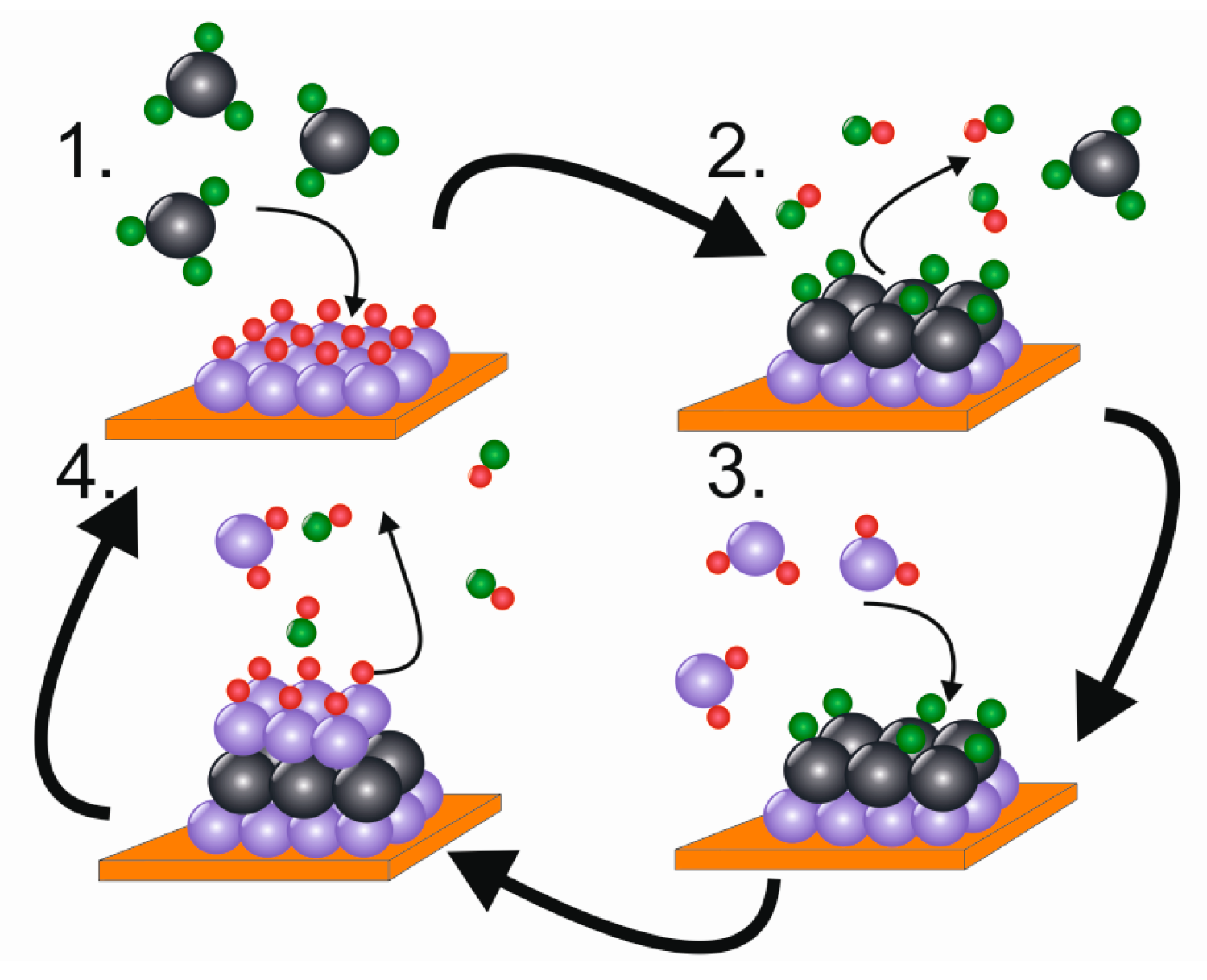
4.1. Basic Principle
4.2. Atomic Layer Deposition of Conventional Lithium-Ion Battery Materials
4.2.1. Cathodes
4.2.2. Anodes
4.2.3. Solid Electrolytes
5. Atomic Layer Deposition of Metal Fluorides
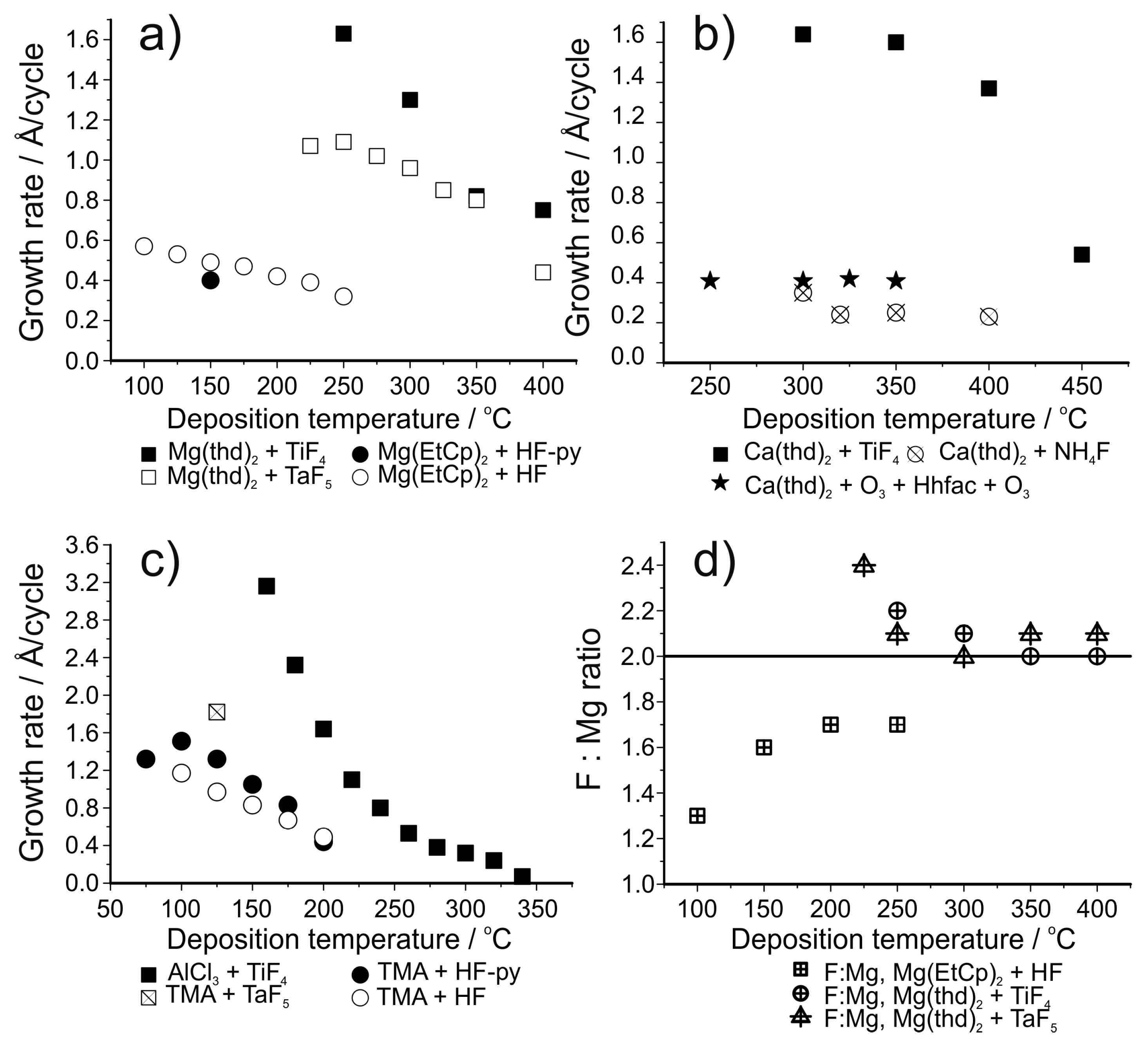
5.1. ALD of Metal Fluorides Using HF as the Fluorine Source
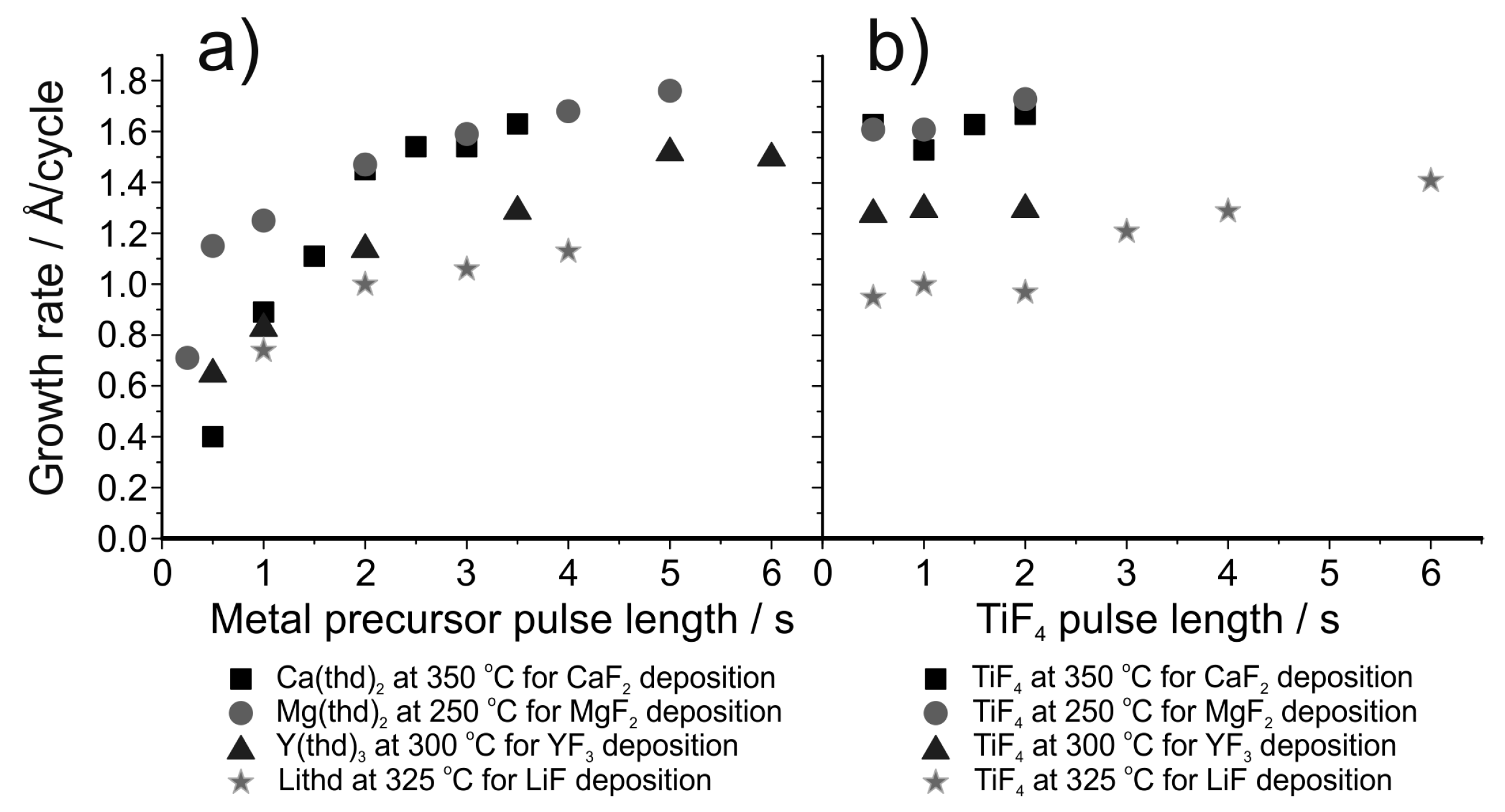
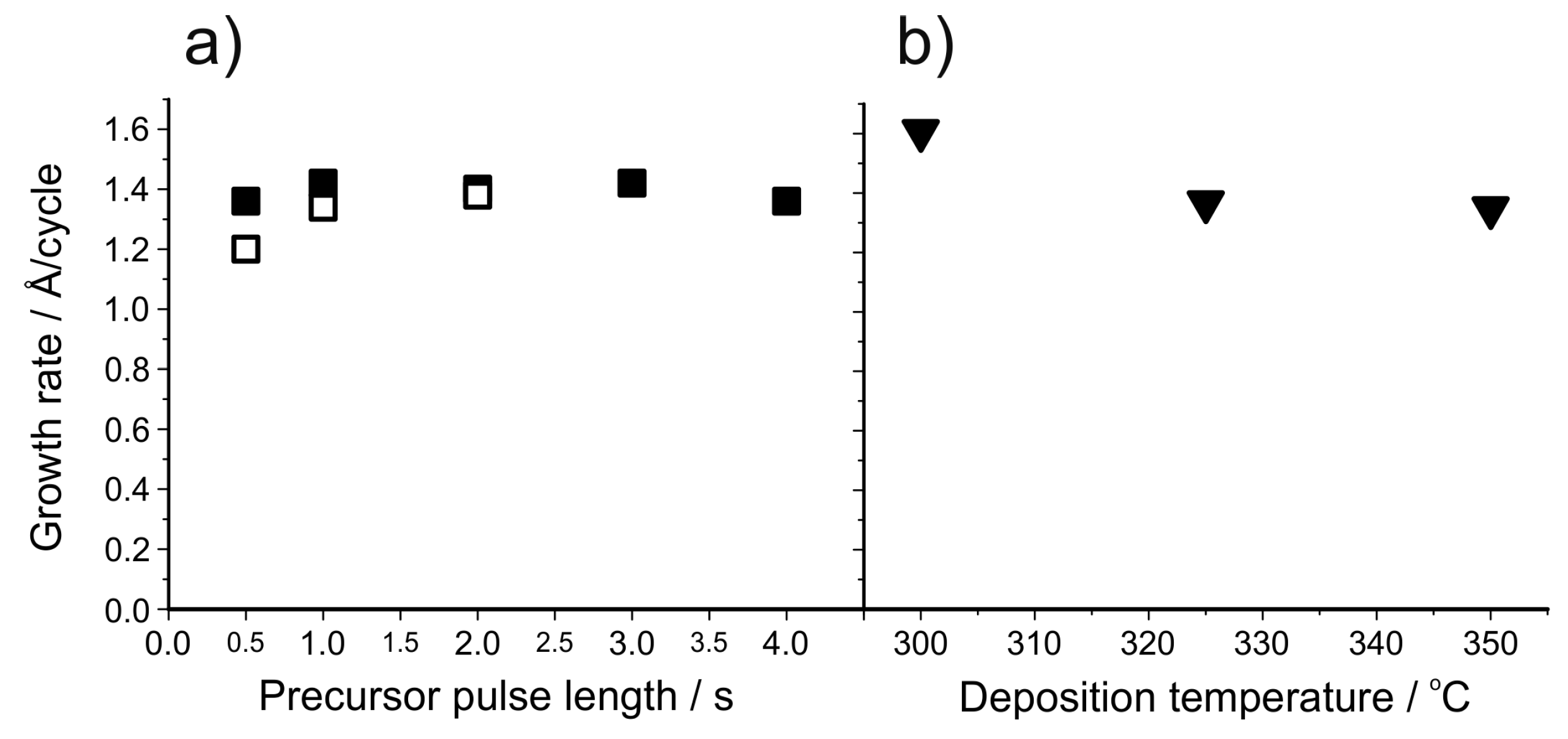
5.2. ALD of Metal Fluorides Using Metal Fluorides as the Fluorine Source
5.3. Other Approaches to ALD of Metal Fluorides
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tesla Solar Roof. Available online: https://www.tesla.com/solarroof (accessed on 12 May 2018).
- Pillot, C. The worldwide battery market 2011–2025. In Proceedings of the Batteries 2012, Nice, France, 24–26 October 2012. [Google Scholar]
- Batteries storing power seen as big as rooftop solar in 12 years. Available online: https://www.bloomberg.com/news/articles/2016-06-13/batteries-storing-power-seen-as-big-as-rooftop-solar-in-12-years (accessed on 12 May 2018).
- Oudenhoven, J.F.M.; Baggetto, L.; Notten, P.H.L. All-solid-state lithium-ion microbatteries: A review of various three-dimensional concepts. Adv. Energy Mater. 2011, 1, 10–33. [Google Scholar] [CrossRef]
- Hayner, C.M.; Zhao, X.; Kung, H.H. Materials for Rechargeable Lithium-Ion Batteries. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 445–471. [Google Scholar] [CrossRef] [PubMed]
- Knoops, H.C.M.; Donders, M.E.; van de Sanden, M.C.M.; Notten, P.H.L.; Kessels, W.M.M. Atomic layer deposition for nanostructured Li-ion batteries. J. Vac. Sci. Technol. A 2012, 30, 010801. [Google Scholar] [CrossRef]
- Marichy, C.; Bechelany, M.; Pinna, N. Atomic Layer Deposition of Nanostructured Materials for Energy and Environmental Applications. Adv. Mater. 2012, 24, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Long, J.W.; Dunn, B.; Rolison, D.R.; White, H.S. Three-Dimensional Battery Architectures. Chem. Rev. 2004, 104, 4463–4492. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Yushin, G. High-capacity anode materials for lithium-ion batteries: Choice of elements and structures for active particles. Part. Part. Syst. Charact. 2014, 31, 317–336. [Google Scholar] [CrossRef]
- Zhu, C.; Han, K.; Geng, D.; Ye, H.; Meng, X. Achieving high-performance silicon anodes of lithium-ion batteries via atomic and molecular layer deposited surface coatings: An overview. Electrochim. Acta 2017, 251, 710–728. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Pereira, N. Fluoride based electrode materials for advanced energy storage devices. J. Fluor. Chem. 2007, 128, 243–262. [Google Scholar] [CrossRef]
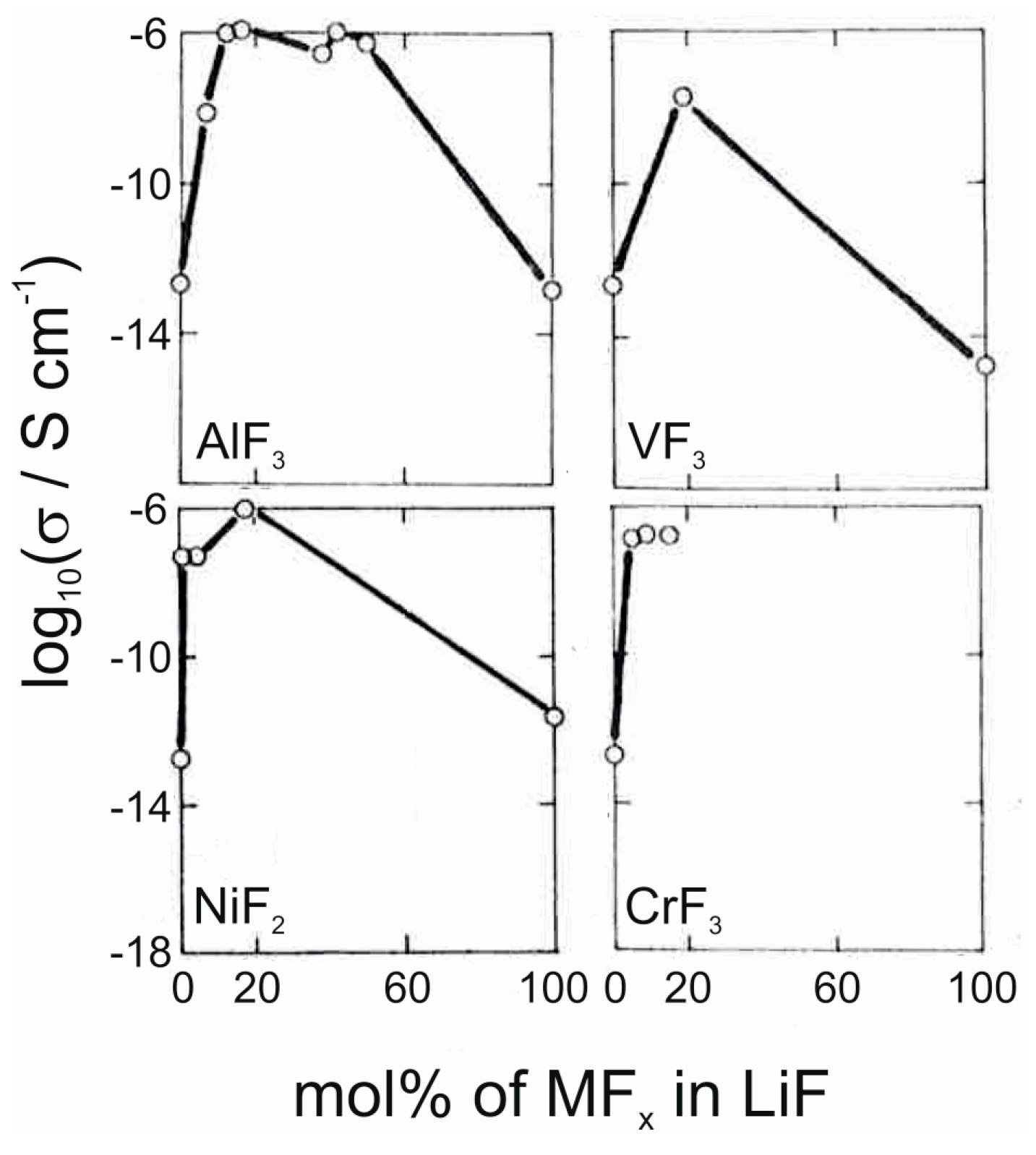
- Oi, T. Ionic conductivity of amorphous mLiFnMF3 thin films (M = Al, Cr, Sc or Al + Sc). Mat. Res. Bull. 1984, 19, 1343–1348. [Google Scholar] [CrossRef]
- Oi, T. Ionic conductivity of LiF thin films containing Di- or trivalent metal fluorides. Mat. Res. Bull. 1984, 19, 451–457. [Google Scholar] [CrossRef]
- Oi, T. Ionic conductivity of amorphous ternary fluoride thin films of composition Li2MIIMIIIF7 (MII = Mg, Fe; MIII = Al, Sc. Mat. Res. Bull. 1984, 19, 1077–1082. [Google Scholar] [CrossRef]
- Huggins, R.A. Advanced Batteries: Materials Science Aspects; Springer: New York, NY, USA, 2009; ISBN 978-0-387-76424-5. [Google Scholar]
- Tarascon, J.-M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Baggetto, L.; Niessen, R.A.H.; Roozeboom, F.; Notten, P.H.L. High energy density all-solid-state batteries: A challenging concept towards 3D integration. Adv. Funct. Mater. 2008, 18, 1057–1066. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium batteries and cathode materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef] [PubMed]
- Ellis, B.L.; Lee, K.T.; Nazar, L.F. Positive electrode materials for Li-Ion and Li-batteries. Chem. Mater. 2010, 22, 691–714. [Google Scholar] [CrossRef]
- Chen, Z.; Qin, Y.; Amine, K.; Sun, Y.-K. Role of surface coating on cathode materials for lithium-ion batteries. J. Mater. Chem. 2010, 20, 7606–7612. [Google Scholar] [CrossRef]
- Park, J.S.; Meng, X.; Elam, J.W.; Hao, S.; Wolverton, C.; Kim, C.; Cabana, J. Ultrathin lithium-ion conducting coatings for increased interfacial stability in high voltage lithium-ion batteries. Chem. Mater. 2014, 26, 3128–3134. [Google Scholar] [CrossRef]
- Guan, D.; Jeevarajan, J.A.; Wang, Y. Enhanced cycleability of LiMn2O4 cathodes by atomic layer deposition of nanosized-thin Al2O3 coatings. Nanoscale 2011, 3, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Cavanagh, A.S.; Riley, L.A.; Kang, S.-H.; Dillon, A.C.; Groner, M.D.; George, S.M.; Lee, S.-H. Ultrathin direct atomic layer deposition on composite electrodes for highly durable and safe Li-Ion batteries. Adv. Mater. 2010, 22, 2172–2176. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Cavanagh, A.S.; Dillon, A.C.; Groner, M.D.; George, S.M.; Lee, S.-H. Enhanced stability of LiCoO2 cathodes in lithium-ion batteries using surface modification by atomic layer deposition. J. Electrochem. Soc. 2010, 157, A75–A81. [Google Scholar] [CrossRef]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Xu, W.; Wang, J.; Ding, F.; Chen, X.; Nasybulin, E.; Zhang, Y.; Zhang, J.-G. Lithium metal anodes for rechargeable batteries. Energy Environ. Sci. 2014, 7, 513–537. [Google Scholar] [CrossRef]
- Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.-M. Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries. Nature 2000, 407, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-M.; Kim, J.-H.; Kim, H.; Sohn, H.-J. Li-alloy based anode materials for Li secondary batteries. Chem. Soc. Rev. 2010, 39, 3115–3141. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Balaya, P.; Maier, J. Li-Storage via heterogeneous reaction in selected binary metal fluorides and oxides. J. Electrochem. Soc. 2004, 151, A1878–A1885. [Google Scholar] [CrossRef]
- Thangadurai, V.; Weppner, W. Solid state lithium ion conductors: Design considerations by thermodynamic approach. Ionics 2002, 8, 281–292. [Google Scholar] [CrossRef]
- Thangadurai, V.; Weppner, W. Recent progress in solid oxide and lithium ion conducting electrolytes research. Ionics 2006, 12, 81–92. [Google Scholar] [CrossRef]
- Xia, H.; Wang, H.L.; Xiao, W.; Lai, M.O.; Lu, L. Thin film Li electrolytes for all-solid-state micro-batteries. Int. J. Surf. Sci. Eng. 2009, 3, 23–43. [Google Scholar] [CrossRef]
- Knauth, P. Inorganic solid Li ion conductors: An overview. Solid State Ion. 2009, 180, 911–916. [Google Scholar] [CrossRef]
- Fergus, J.W. Ceramic and polymeric solid electrolytes for lithium-ion batteries. J. Power Sources 2010, 195, 4554–4569. [Google Scholar] [CrossRef]
- Hu, M.; Pang, X.; Zhou, Z. Recent progress in high-voltage lithium ion batteries. J. Power Sources 2013, 237, 229–242. [Google Scholar] [CrossRef]
- Cabana, J.; Monconduit, L.; Larcher, D.; Palacín, M.R. Beyond intercalation-based Li-ion batteries: The state of the art and challenges of electrode materials reacting through conversion reactions. Adv. Mater. 2010, 22, E170–E192. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Richter, G.; Maier, J. Reversible formation and decomposition of LiF clusters using transition metal fluorides as precursors and their application in rechargeable Li batteries. Adv. Mater. 2003, 15, 736–739. [Google Scholar] [CrossRef]
- Badway, F.; Cosandey, F.; Pereira, N.; Amatucci, G.G. Carbon metal fluoride nanocomposites: High-capacity reversible metal fluoride conversion materials as rechargeable positive electrodes for Li batteries. J. Electrochem. Soc. 2003, 150, A1318–A1327. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.-N.; Sun, Q.; Fu, Z.-W. Nanostructured nickel fluoride thin film as a new Li storage material. Solid State Sci. 2008, 10, 1166–1172. [Google Scholar] [CrossRef]
- Bervas, M.; Badway, F.; Klein, L.C.; Amatucci, G.G. Bismuth fluoride nanocomposite as a positive electrode material for rechargeable lithium batteries. Electrochem. Solid-State Lett. 2005, 8, A179–A183. [Google Scholar] [CrossRef]
- Gonzalo, E.; Kuhn, A.; García-Alvarado, F. On the room temperature synthesis of monoclinic Li3FeF6: A new cathode material for rechargeable lithium batteries. J. Power Sources 2010, 195, 4990–4996. [Google Scholar] [CrossRef]
- Basa, A.; Gonzalo, E.; Kuhn, A.; García-Alvarado, F. Reaching the full capacity of the electrode material Li3FeF6 by decreasing the particle size to nanoscale. J. Power Sources 2012, 197, 260–266. [Google Scholar] [CrossRef]
- Basa, A.; Gonzalo, E.; Kuhn, A.; García-Alvarado, F. Facile synthesis of β-Li3VF6: A new electrochemically active lithium insertion material. J. Power Sources 2012, 207, 160–165. [Google Scholar] [CrossRef]
- Koyama, Y.; Tanaka, I.; Adachi, H. New fluoride cathodes for rechargeable lithium batteries. J. Electrochem. Soc. 2000, 147, 3633–3636. [Google Scholar] [CrossRef]
- Amalraj, F.; Talianker, M.; Markovsky, B.; Burlaka, L.; Leifer, N.; Goobes, G.; Erickson, E.M.; Haik, O.; Grinblat, J.; Zinigrad, E.; et al. Studies of Li and Mn-rich Lix[MnNiCo]O2 electrodes: electrochemical performance, structure, and the effect of the aluminum fluoride coating. J. Electrochem. Soc. 2013, 160, A2220–A2233. [Google Scholar] [CrossRef]
- Lee, S.-H.; Yoon, C.S.; Amine, K.; Sun, Y.-K. Improvement of long-term cycling performance of Li[Ni0.8Co0.15Al0.05]O2 by AlF3 coating. J. Power Sources 2013, 234, 201–207. [Google Scholar] [CrossRef]
- Sun, Y.-K.; Lee, M.-J.; Yoon, C.S.; Hassoun, J.; Amine, K.; Scrosati, B. The role of AlF3 coatings in improving electrochemical cycling of Li-enriched nickel-manganese oxide electrodes for Li-ion batteries. Adv. Mater. 2012, 24, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.; Park, Y.D.; Mun, J. AlF3-coated LiMn2O4 as cathode material for aqueous rechargeable lithium battery with improved cycling stability. J. Power Sources 2016, 325, 360–364. [Google Scholar] [CrossRef]
- Sun, Y.-K.; Cho, S.-W.; Myung, S.-T.; Amine, K.; Prakash, J. Effect of AlF3 coating amount on high voltage cycling performance of LiCoO2. Electrochim. Acta 2007, 53, 1013–1019. [Google Scholar] [CrossRef]
- Ding, F.; Xu, W.; Choi, D.; Wang, W.; Li, X.; Engelhard, M.H.; Chen, X.; Yang, Z.; Zhang, J.-G. Enhanced performance of graphite anode materials by AlF3 coating for lithium-ion batteries. J. Mater. Chem. 2012, 22, 12745–12751. [Google Scholar] [CrossRef]
- Lee, Y.; Piper, D.M.; Cavanagh, A.S.; Young, M.J.; Lee, S.-H.; George, S.M. Atomic layer deposition of lithium ion conducting (AlF3)(LiF)x alloys using trimethylaluminum, lithium hexamethyldisilazide and hydrogen fluoride-pyridine. In Proceedings of the 14th International Conference on Atomic Layer Deposition, Kyoto, Japan, 15–18 June 2014. [Google Scholar]
- Miyazaki, R.; Maekawa, H. Li+-ion conduction of Li3AlF6 mechanically milled with LiCl. ECS Electrochem. Lett. 2012, 1, A87–A89. [Google Scholar] [CrossRef]
- Dance, J.-M.; Oi, T. Ionic conductivity of amorphous lithium fluoride-nickel fluoride (mLiF-nNiF2) thin films. Thin Solid Films 1983, 104, L71–L73. [Google Scholar]
- Kawamoto, Y.; Fujiwara, J.; Ichimura, C. Ionic conduction in xMF·(95 − x)ZrF4·5LaF3 (M: Alkali metals) glasses: I. Lithium ion conduction in xLiF·(95 − x)ZrF4·5LaF3 glasses. J. Non-Cryst. Solids 1989, 111, 245–251. [Google Scholar] [CrossRef]
- Reau, J.M.; Kahnt, H.; Poulain, M. Ionic transport studies in mixed alkali-fluorine conductor glasses of composition ZrF4-BaF2-LaF3-AF (A = Li, Na) and ZrF4-BaF2-ThF4-LiF. J. Non-Cryst. Solids 1990, 119, 347–350. [Google Scholar] [CrossRef]
- Senegas, J.; Reau, J.M.; Aomi, H.; Hagenmuller, P.; Poulain, M. Ionic conductvity and NMR investigation of quaternary glasses in the ZrF4-BaF2-ThF4-LiF system. J. Non-Cryst. Solids 1986, 85, 315–334. [Google Scholar] [CrossRef]
- Trnovcová, V.; Fedorov, P.P.; Bárta, Č.; Labaš, V.; Meleshina, V.A.; Sobolev, B.P. Microstructure and physical properties of superionic eutectic composites of the LiF-RF3 (R = rare earth element) system. Solid State Ion. 1999, 119, 173–180. [Google Scholar] [CrossRef]
- Trnovcová, V.; Fedorov, P.P.; Furár, I. Fluoride solid electrolytes containing rare earth elements. J. Rare Earths 2008, 26, 225–232. [Google Scholar] [CrossRef]
- Dieudonné, B.; Chable, J.; Mauvy, F.; Fourcade, S.; Durand, E.; Lebraud, E.; Leblanc, M.; Legein, C.; Body, M.; Maisonneuve, V.; et al. Exploring the Sm1–xCaxF3–x tysonite solid solution as a solid-state electrolyte: Relationships between structural features and F– ionic conductivity. J. Phys. Chem. C 2015, 119, 25170–25179. [Google Scholar] [CrossRef]
- Sorokin, N.I.; Sobolev, B.P. Nonstoichiometric fluorides-solid electrolytes for electrochemical devices: A review. Crystallogr. Rep. 2007, 52, 842–863. [Google Scholar] [CrossRef]
- Reddy, M.A.; Fichtner, M. Batteries based on fluoride shuttle. J. Mater. Chem. 2011, 21, 17059–17062. [Google Scholar] [CrossRef]
- Leskelä, M.; Ritala, M. Atomic layer deposition chemistry: Recent developments and future challenge. Angew. Chem. Int. Ed. 2003, 42, 5548–5554. [Google Scholar] [CrossRef] [PubMed]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Ritala, M.; Leskelä, M. Atomic layer deposition. In Handbook of Thin Film Materials; Nalwa, H.S., Ed.; Academic Press: San Diego, CA, USA, 2002; Volume 1, pp. 103–159. ISBN 9780125129084. [Google Scholar]
- Profijt, H.B.; Potts, S.E.; van de Sanden, M.C.M.; Kessels, W.M.M. Plasma-assisted atomic layer deposition: Basics, opportunities, and challenges. J. Vac. Sci. Technol. A 2011, 29, 050801. [Google Scholar] [CrossRef]
- Chalker, P.R. Photochemical atomic layer deposition and etching. Surf. Coat. Technol. 2016, 291, 258–263. [Google Scholar] [CrossRef]
- Miikkulainen, V.; Väyrynen, K.; Kilpi, V.; Han, Z.; Vehkamäki, M.; Mizohata, K.; Räisänen, J.; Ritala, M. Photo-assisted ALD: Process development and application perspectives. ECS Trans. 2017, 80, 49–60. [Google Scholar] [CrossRef]
- Puurunen, R.L. Growth per cycle in atomic layer deposition: A theoretical model. Chem. Vap. Depos. 2003, 9, 249–257. [Google Scholar] [CrossRef]
- Dunn, B.; Long, J.W.; Rolison, D.R. Rethinking multifunction in three dimensions for miniaturizing electrical energy storage. Electrochem. Soc. Interface 2008, 17, 49–53. [Google Scholar]
- Putkonen, M.; Aaltonen, T.; Alnes, M.; Sajavaara, T.; Nilsen, O.; Fjellvåg, H. Atomic layer deposition of lithium containing thin films. J. Mater. Chem. 2009, 19, 8767–8771. [Google Scholar] [CrossRef]
- Nilsen, O.; Miikkulainen, V.; Gandrud, K.B.; Østreng, E.; Ruud, A.; Fjellvåg, H. Atomic layer deposition of functional films for Li-ion microbatteries. Phys. Status Solidi A 2014, 211, 357–367. [Google Scholar] [CrossRef]
- Aaltonen, T.; Miikkulainen, V.; Gandrud, K.B.; Pettersen, A.; Nilsen, O.; Fjellvåg, H. ALD of Thin Films for Lithium-Ion Batteries. ECS Trans. 2011, 41, 331–339. [Google Scholar] [CrossRef]
- Meng, X.; Wang, X.; Geng, D.; Ozgit-Akgun, C.; Schneider, N.; Elam, J.W. Atomic layer deposition for nanomaterial synthesis and functionalization in energy technology. Mater. Horiz. 2017, 4, 133–154. [Google Scholar] [CrossRef]
- Meng, X.; Elam, J.W. Atomic layer deposition of nanophase materials for electrical energy storage. ECS Trans. 2015, 69, 39–57. [Google Scholar] [CrossRef]
- Meng, X.; Yang, X.-Q.; Sun, X. Emerging applications of atomic layer deposition for lithium-ion battery studies. Adv. Mater. 2012, 24, 3589–3615. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Wang, J. Recent development of advanced electrode materials by atomic layer deposition for electrochemical energy storage. Adv. Sci. 2016, 3, 1500405. [Google Scholar] [CrossRef] [PubMed]
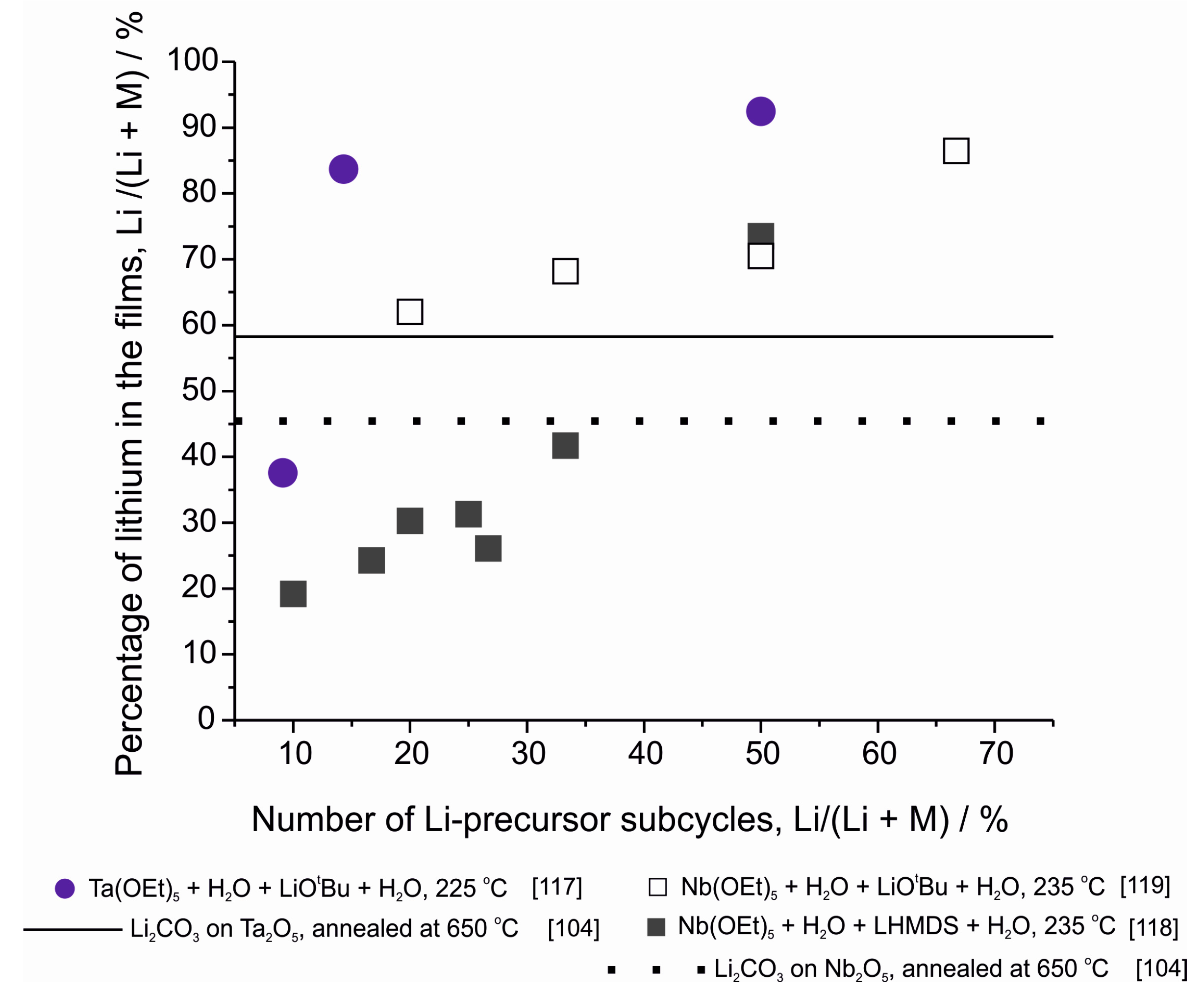
- Ma, L.; Nuwayhid, R.B.; Wu, T.; Lei, Y.; Amine, K.; Lu, J. Atomic layer deposition for lithium-based batteries. Adv. Mater. Interfaces 2016, 3, 1600564. [Google Scholar] [CrossRef]
- Nilsen, O.; Gandrud, K.B.; Ruud, A.; Fjellvåg, H. Atomic layer deposition for thin-film lithium-ion batteries. In Atomic Layer Deposition in Energy Conversion Applications; Bachmann, J., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; pp. 183–207. ISBN 978-3-527-33912-9. [Google Scholar]
- Mäntymäki, M. Atomic Layer Deposition and Lithium-ion Batteries: Studies on New Materials and Reactions for Battery Development. Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 9 June 2017. [Google Scholar]
- Liu, J.; Sun, X. Elegant design of electrode and electrode/electrolyte interface in lithium-ion batteries by atomic layer deposition. Nanotechnology 2015, 26, 024001. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yushin, G. Chemical vapor deposition and atomic layer deposition for advanced lithium ion batteries and supercapacitors. Energy Environ. Sci. 2015, 8, 1889–1904. [Google Scholar] [CrossRef]
- Noked, M.; Liu, C.; Hu, J.; Gregorczyk, K.; Rubloff, G.W.; Lee, S.B. Electrochemical thin layers in nanostructures for energy storage. Acc. Chem. Res. 2016, 49, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Share, K.; Westover, A.; Li, M.; Pint, C.L. Surface engineering of nanomaterials for improved energy storage —A review. Chem. Eng. Sci. 2016, 154, 3–19. [Google Scholar] [CrossRef]
- Sun, Q.; Lau, K.C.; Geng, D.; Meng, X. Atomic and molecular layer deposition for superior lithium-sulfur batteries: Strategies, performance, and mechanisms. Batter. Supercaps 2018. [Google Scholar] [CrossRef]
- Le Van, K.; Groult, H.; Mantoux, A.; Perrigaud, L.; Lantelme, F.; Lindström, R.; Badour-Hadjean, R.; Zanna, S.; Lincot, D. Amorphous vanadium oxide films synthesised by ALCVD for lithium rechargeable batteries. J. Power Sources 2006, 160, 592–601. [Google Scholar] [CrossRef]
- Chen, X.; Pomerantseva, E.; Gregorczyk, K.; Ghodssi, R.; Rubloff, G. Cathodic ALD V2O5 thin films for high-rate electrochemical energy storage. RSC Adv. 2013, 3, 4294–4302. [Google Scholar] [CrossRef]
- Donders, M.E.; Knoops, H.C.M.; Kessels, W.M.M.; Notten, P.H.L. Remote plasma atomic layer deposition of thin films of electrochemically active LiCoO2. ECS Trans. 2011, 41, 321–330. [Google Scholar] [CrossRef]
- Donders, M.E.; Arnoldbik, W.M.; Knoops, H.C.M.; Kessels, W.M.M.; Notten, P.H.L. Atomic layer deposition of LiCoO2 thin-film electrodes for all-solid-state Li-ion micro-batteries. J. Electrochem. Soc. 2013, 160, A3066–A3071. [Google Scholar] [CrossRef]
- Liu, J.; Banis, M.N.; Sun, Q.; Lushington, A.; Li, R.; Sham, T.-K.; Sun, X. Rational design of atomic-layer-deposited LiFePO4 as a high-performance cathode for lithium-ion batteries. Adv. Mater. 2014, 26, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Miikkulainen, V.; Ruud, A.; Østreng, E.; Nilsen, O.; Laitinen, M.; Sajavaara, T.; Fjellvåg, H. Atomic layer deposition of spinel lithium manganese oxide by film-body-controlled lithium incorporation for thin-film lithium-ion batteries. J. Phys. Chem. C 2014, 118, 1258–1268. [Google Scholar] [CrossRef]
- Meng, X.; Comstock, D.J.; Fisher, T.T.; Elam, J.W. Vapor-phase atomic-controllable growth of amorphous Li2S for high-performance lithium–sulfur batteries. ACS Nano 2014, 8, 10963–10972. [Google Scholar] [CrossRef] [PubMed]
- Lantelme, F.; Mantoux, A.; Groult, H.; Lincot, D. Electrochemical study of phase transition processes in lithium insertion in V2O5 electrodes. J. Electrochem. Soc. 2003, 150, A1202–A1208. [Google Scholar] [CrossRef]
- Gandrud, K.B.; Pettersen, A.; Nilsen, O.; Fjellvåg, H. Growth of LiFePO4 cathode material by ALD. In Proceedings of the 10th International Conference on Atomic Layer Deposition, Seoul, Korea, 20–23 June 2010. [Google Scholar]
- Gandrud, K.B.; Pettersen, A.; Nilsen, O.; Fjellvåg, H. High-performing iron phosphate for enhanced lithium ion solid state batteries as grown by atomic layer deposition. J. Mater. Chem. A 2013, 1, 9054–9059. [Google Scholar] [CrossRef]
- Kozen, A.C.; Pearse, A.J.; Lin, C.-F.; Schoeder, M.A.; Noked, M.; Lee, S.B.; Rubloff, G.W. Atomic layer deposition and in situ characterization of ultraclean lithium oxide and lithium hydroxide. J. Phys. Chem. C 2014, 118, 27749–27753. [Google Scholar] [CrossRef]
- Wang, W.; Tian, M.; Abdulagatov, A.; George, S.M.; Lee, Y.-C.; Yang, R. Three-dimensional Ni/TiO2 nanowire network for high areal capacity lithium ion microbattery applications. Nano Lett. 2012, 12, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Cheah, S.K.; Perre, E.; Rooth, M.; Fondell, M.; Hårsta, A.; Nyholm, L.; Boman, M.; Gustafsson, T.; Lu, J.; Simon, P.; et al. Self-supported three-dimensional nanoelectrodes for microbattery applications. Nano Lett. 2009, 9, 3230–3233. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Yoon, Y.; Jung, H.S.; Yoon, W.-S.; Shin, H. Nanoscale size effect of titania (anatase) nanotubes with uniform wall thickness as high performance anode for lithium-ion secondary battery. J. Power Sources 2012, 204, 162–167. [Google Scholar] [CrossRef]
- Miikkulainen, V.; Nilsen, O.; Laitinen, M.; Sajavaara, T.; Fjellvåg, H. Atomic layer deposition of LixTiyOz thin films. RSC Adv. 2013, 3, 7537–7542. [Google Scholar] [CrossRef]
- Meng, X.; Liu, J.; Li, X.; Banis, M.N.; Yang, J.; Li, R.; Sun, X. Atomic layer deposited Li4Ti5O12 on nitrogen-doped carbon nanotubes. RSC Adv. 2013, 3, 7285–7288. [Google Scholar] [CrossRef]
- Miikkulainen, V.; Nilsen, O.; Laitinen, M.; Sajavaara, T.; Fjellvåg, H. Atomic layer deposition of LixTiyOz films. In Proceedings of the 12th International Conference on Atomic Layer Deposition, Dresden, Germany, 17–20 June 2012. [Google Scholar]
- Nisula, M.; Karppinen, M. Atomic/molecular layer deposition of lithium terephthalate thin films as high rate capability Li-Ion battery anodes. Nano Lett. 2016, 16, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Søndergaard, M.; Christensen, M.; Birgisson, S.; Iversen, B.B. Solid state formation mechanism of Li4Ti5O12 from an anatase TiO2 source. Chem. Mater. 2014, 26, 3679–3686. [Google Scholar] [CrossRef]
- Atosuo, E.; Mäntymäki, M.; Mizohata, K.; Heikkilä, M.J.; Räisänen, J.; Ritala, M.; Leskelä, M. Preparation of lithium containing oxides by the solid state reaction of atomic layer deposited thin films. Chem. Mater. 2017, 29, 998–1005. [Google Scholar] [CrossRef]
- Sundberg, P.; Karppinen, M. Organic and inorganic–organic thin film structures by molecular layer deposition: A review. Beilstein J. Nanotechnol. 2014, 5, 1104–1136. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.; Grugeon, S.; Vezin, H.; Laruelle, S.; Ribière, P.; Poizot, P.; Tarascon, J.-M. Conjugated dicarboxylate anodes for Li-ion batteries. Nat. Mater. 2009, 8, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Nisula, M.; Karppinen, M. In situ lithiated quinone cathode for ALD/MLD-fabricated high-power thin-film battery. J. Mater. Chem. A 2018, 6, 7027–7033. [Google Scholar] [CrossRef]
- Nisula, M.; Shindo, Y.; Koga, H.; Karppinen, M. Atomic layer deposition of lithium phosphorus oxynitride. Chem. Mater. 2015, 27, 6987–6993. [Google Scholar] [CrossRef]
- Kozen, A.C.; Pearse, A.J.; Lin, C.-F.; Noked, M.; Rubloff, G.W. Atomic layer deposition of the solid electrolyte LiPON. Chem. Mater. 2015, 27, 5324–5331. [Google Scholar] [CrossRef]
- Hämäläinen, J.; Munnik, F.; Hatanpää, T.; Holopainen, J.; Ritala, M.; Leskelä, M. Study of amorphous lithium silicate thin films grown by atomic layer deposition. J. Vac. Sci. Technol. A 2012, 30, 01A106. [Google Scholar] [CrossRef]
- Tomczak, Y.; Knapas, K.; Sundberg, M.; Leskelä, M.; Ritala, M. In situ reaction mechanism studies on lithium hexadimethyldisilazide and ozone atomic layer deposition process for lithium silicate. J. Phys. Chem. C 2013, 117, 14241–14246. [Google Scholar] [CrossRef]
- Ruud, A.; Miikkulainen, V.; Mizohata, K.; Fjellvåg, H.; Nilsen, O. Enhanced process and composition control for atomic layer deposition with lithium trimethylsilanolate. J. Vac. Sci. Technol. A 2017, 35, 01B133-1–01B133-8. [Google Scholar] [CrossRef]
- Wang, B.; Liu, J.; Banis, M.N.; Sun, Q.; Zhao, Y.; Li, R.; Sham, T.-K.; Sun, X. Atomic layer deposited lithium silicates as solid-state electrolytes for all-solid-state batteries. ACS Appl. Mater. Interfaces 2017, 9, 31786–31793. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, J.; Holopainen, J.; Munnik, F.; Hatanpää, T.; Heikkilä, M.; Ritala, M.; Leskelä, M. Lithium phosphate thin films grown by atomic layer deposition. J. Electrochem. Soc. 2012, 159, A259–A263. [Google Scholar] [CrossRef]
- Wang, B.; Liu, J.; Sun, Q.; Li, R.; Sham, T.-K.; Sun, X. Atomic layer deposition of lithium phosphates as solid-state electrolytes for all-solid-state microbatteries. Nanotechnology 2014, 25, 504007. [Google Scholar] [CrossRef] [PubMed]
- Létiche, M.; Eustache, E.; Freixas, J.; Demortière, A.; De Andrade, V.; Morgenroth, L.; Tilmant, P.; Vaurette, F.; Troadec, D.; Roussel, P.; et al. Atomic layer deposition of functional layers for on chip 3D Li-ion all solid state microbattery. Adv. Energy Mater. 2017, 7, 1601402. [Google Scholar] [CrossRef]
- Liu, J.; Banis, M.N.; Li, X.; Lushington, A.; Cai, M.; Li, R.; Sham, T.-K.; Sun, X. Atomic layer deposition of lithium tantalate solid-state electrolytes. J. Phys. Chem. C 2013, 117, 20260–20267. [Google Scholar] [CrossRef]
- Østreng, E.; Sønsteby, H.H.; Sajavaara, T.; Nilsen, O.; Fjellvåg, H. Atomic layer deposition of ferroelectric LiNbO3. J. Mater. Chem. C 2013, 1, 4283–4290. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, Y.; Banis, M.N.; Sun, Q.; Adair, K.R.; Li, R.; Sham, T.-K.; Sun, Q. Atomic layer deposition of lithium niobium oxides as potential solid-state electrolytes for lithium-ion batteries. ACS Appl. Mater. Interfaces 2018, 10, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, T.; Alnes, M.; Nilsen, O.; Costelle, L.; Fjellvåg, H. Lanthanum titanate and lithium lanthanum titanate thin films grown by atomic layer deposition. J. Mater. Chem. 2010, 20, 2877–2881. [Google Scholar] [CrossRef]
- Perng, Y.-C.; Cho, J.; Sun, S.Y.; Membreno, D.; Cirigliano, N.; Dunn, B.; Chang, J.P. Synthesis of ion conducting LixAlySizO thin films by atomic layer deposition. J. Mater. Chem. A 2014, 2, 9566–9573. [Google Scholar] [CrossRef]
- Kazyak, E.; Chen, K.-H.; Wood, K.N.; Davis, A.L.; Thompson, T.; Bielinski, A.R.; Sanchez, A.J.; Wang, X.; Wang, C.; Sakamoto, J.; et al. Atomic layer deposition of the solid electrolyte garnet Li7La3Zr2O12. Chem. Mater. 2017, 29, 3785–3792. [Google Scholar] [CrossRef]
- Cao, Y.; Meng, X.; Elam, J.W. Atomic Layer Deposition of LixAlyS Solid-state electrolytes for stabilizing lithium-metal anodes. ChemElectroChem 2016, 3, 858–863. [Google Scholar] [CrossRef]
- Xie, J.; Sendek, A.D.; Cubuk, E.D.; Zhang, X.; Lu, Z.; Gong, Y.; Wu, T.; Shi, F.; Liu, W.; Reed, E.J.; et al. Atomic layer deposition of stable LiAlF4 lithium ion conductive interfacial layer for stable cathode cycling. ACS Nano 2017, 11, 7019–7027. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Kuwata, N.; Matsuda, Y.; Kawamura, J. Characterization of stable solid electrolyte lithium silicate for thin film lithium battery. J. Phys. Soc. Jpn. Suppl. A 2010, 79, 98–101. [Google Scholar] [CrossRef]
- Furusawa, S.; Kamiyama, A.; Tsurui, T. Fabrication and ionic conductivity of amorphous lithium meta-silicate thin film. Solid State Ion. 2008, 179, 536–542. [Google Scholar] [CrossRef]
- Furusawa, S.; Kasahara, T.; Kamiyama, A. Fabrication and ionic conductivity of Li2SiO3 thin film. Solid State Ion. 2009, 180, 649–653. [Google Scholar] [CrossRef]
- Liu, J.; Wang, B.; Sun, Q.; Li, R.; Sham, T.-K.; Sun, X. Atomic layer deposition of hierarchical CNTs@FePO4 architecture as a 3D electrode for lithium-ion and sodium-ion batteries. Adv. Mater. Interfaces 2016, 3, 1600468. [Google Scholar] [CrossRef]
- Wang, B.; Liu, J.; Sun, Q.; Xiao, B.; Li, R.; Sham, T.-K.; Sun, X. Titanium dioxide/lithium phosphate nanocomposite derived from atomic layer deposition as a high-performance anode for lithium ion batteries. Adv. Mater. Interfaces 2016, 3, 1600369. [Google Scholar] [CrossRef]
- Shibata, S. Thermal atomic layer deposition of lithium phosphorus oxynitride as a thin-film solid electrolyte. J. Electrochem. Soc. 2016, 163, A2555–A2562. [Google Scholar] [CrossRef]
- Lin, C.-F.; Noked, M.; Kozen, A.C.; Liu, C.; Zhao, O.; Gregorczyk, K.; Hu, L.; Lee, S.B.; Rubloff, G.W. Solid Electrolyte Lithium Phosphous Oxynitride as a Protective Nanocladding Layer for 3D high-capacity conversion electrodes. ACS Nano 2016, 10, 2693–2701. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.T. Elastic, piezoelectric, and dielectric properties of lithium tantalate. Appl. Phys. Lett. 1967, 11, 146–148. [Google Scholar] [CrossRef]
- Tomeno, I.; Matsumura, S. Dielectric properties of LiTaO3. Phys. Rev. B 1988, 38, 606–614. [Google Scholar] [CrossRef]
- Glass, A.M.; Nassau, K.; Negran, T.J. Ionic conductivity of quenched alkali niobate and tantalate glasses. J. Appl. Phys. 1978, 49, 4808–4811. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Banis, M.N.; Lushington, A.; Li, R.; Cai, M.; Sun, X. Atomic layer deposition of solid-state electrolyte coated cathode materials with superior high-voltage cycling behavior for lithium ion battery application. Energy Environ. Sci. 2014, 7, 768–778. [Google Scholar] [CrossRef]
- Comstock, D.; Elam, J.W. Mechanistic study of lithium aluminum oxide atomic layer deposition. J. Phys. Chem. C 2013, 117, 1677–1683. [Google Scholar] [CrossRef]
- Murugan, R.; Thangadurai, V.; Weppner, W. Fast lithium ion conduction in garnet-type Li7La3Zr2O12. Angew. Chem. Int. Ed. 2007, 46, 7778–7781. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Cao, Y.; Libera, J.A.; Elam, J.W. Atomic layer deposition of aluminum sulfide: growth mechanism and electrochemical evaluation in lithium-ion batteries. Chem. Mater. 2017, 29, 9043–9052. [Google Scholar] [CrossRef]
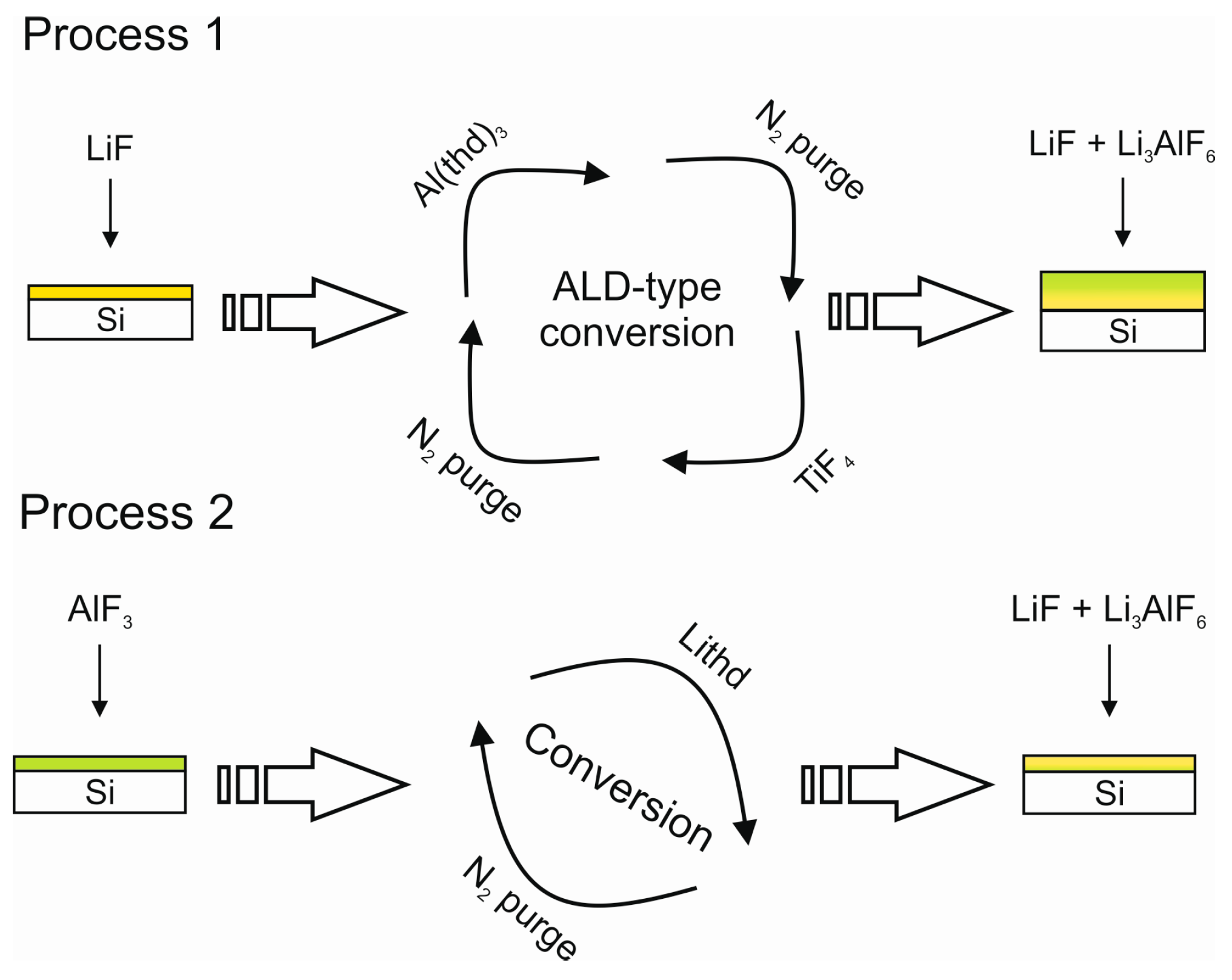
- Mäntymäki, M.; Mizohata, K.; Heikkilä, M.J.; Räisänen, J.; Ritala, M.; Leskelä, M. Studies on Li3AlF6 thin film deposition utilizing conversion reactions of thin films. Thin Solid Films 2017, 636, 26–33. [Google Scholar] [CrossRef]
- Nykänen, E.; Soininen, P.; Niinistö, L.; Leskelä, M.; Rauhala, E. Electroluminescent SrS:Ce,F thin films deposited by the atomic layer epitaxy process. In Proceedings of the 1994 International Workshop on Electroluminescence, Beijing, China, 10–12 October 1994; pp. 437–444. [Google Scholar]
- Ylilammi, M.; Ranta-aho, T. Metal fluoride thin films prepared by atomic layer deposition. J. Electrochem. Soc. 1994, 141, 1278–1284. [Google Scholar] [CrossRef]
- Pilvi, T. Atomic Layer Deposition for Optical Applications: Metal fluoride thin films and novel devices. Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 5 December 2008. [Google Scholar]
- Lee, Y.; DuMont, J.W.; Cavanagh, A.S.; George, S.M. Atomic Layer Deposition of AlF3 using trimethylaluminum and hydrogen fluoride. J. Phys. Chem. C 2015, 119, 14185–14194. [Google Scholar] [CrossRef]
- Park, J.S.; Mane, A.U.; Elam, J.W.; Croy, J.R. Amorphous metal fluoride passivation coatings prepared by atomic layer deposition on LiCoO2 for Li-ion batteries. Chem. Mater. 2015, 27, 1917–1920. [Google Scholar] [CrossRef]
- Jackson, D.H.K.; Laskar, M.R.; Fang, S.; Xu, S.; Ellis, R.G.; Li, X.; Dreibelbis, M.; Babcock, S.E.; Mahanthappa, M.K.; Morgan, D.; et al. Optimizing AlF3 atomic layer deposition using trimethylaluminum and TaF5: Application to high voltage Li-ion battery cathodes. J. Vac. Sci. Technol. A 2016, 34, 031503-1–031503-8. [Google Scholar] [CrossRef]
- Lee, Y. Atomic layer etching of metal oxides and atomic layer deposition of metal fluorides. Ph.D. Thesis, University of Colorado, Boulder, CO, USA, January 2015. [Google Scholar]
- Lee, Y.; Sun, H.; Young, M.J.; George, S.M. Atomic layer deposition of metal fluorides using HF–pyridine as the fluorine precursor. Chem. Mater. 2016, 28, 2022–2032. [Google Scholar] [CrossRef]
- Hennessy, J.; Jewell, A.P.; Greer, F.; Lee, M.C.; Nikzad, S. Atomic layer deposition of magnesium fluoride via bis(ethylcyclopentadienyl)magnesium and anhydrous hydrogen fluoride. J. Vac. Sci. Technol. A 2015, 33, 01A125. [Google Scholar] [CrossRef]
- Hennessy, J.; Jewell, A.D.; Balasubramanian, K.; Nikzad, S. Ultraviolet optical properties of aluminum fluoride thin films deposited by atomic layer deposition. J. Vac. Sci. Technol. A 2016, 34, 01A120. [Google Scholar] [CrossRef]
- Mäntymäki, M.; Hämäläinen, J.; Puukilainen, E.; Munnik, F.; Ritala, M.; Leskelä, M. Atomic layer deposition of LiF thin films from lithd and TiF4 precursors. Chem. Vap. Depos. 2013, 19, 111–116. [Google Scholar] [CrossRef]
- Mäntymäki, M.; Hämäläinen, J.; Puukilainen, E.; Sajavaara, T.; Ritala, M.; Leskelä, M. Atomic layer deposition of LiF thin films from Lithd, Mg(thd)2, and TiF4 precursors. Chem. Mater. 2013, 25, 1656–1663. [Google Scholar] [CrossRef]
- Mane, A.; Libera, J.; Elam, J. Atomic layer deposition of LiF thin films using lithium tert-butoxide and metal fluoride precursors. In Proceedings of the 16th International Conference on Atomic Layer Deposition, Dublin, Ireland, 24–27 July 2016. [Google Scholar]
- Pilvi, T.; Hatanpää, T.; Puukilainen, E.; Arstila, K.; Bischoff, M.; Kaiser, U.; Kaiser, N.; Leskelä, M.; Ritala, M. Study of a novel ALD process for depositing MgF2 thin films. J. Mater. Chem. 2007, 17, 5077–5083. [Google Scholar] [CrossRef]
- Pilvi, T.; Puukilainen, E.; Kreissig, U.; Leskelä, M.; Ritala, M. Atomic layer deposition of MgF2 thin films using TaF5 as a novel fluorine source. Chem. Mater. 2008, 20, 5023–5028. [Google Scholar] [CrossRef]
- Pilvi, T.; Arstila, K.; Leskelä, M.; Ritala, M. Novel ALD process for depositing CaF2 thin films. Chem. Mater. 2007, 19, 3387–3392. [Google Scholar] [CrossRef]
- Mäntymäki, M.; Heikkilä, M.J.; Puukilainen, E.; Mizohata, K.; Marchand, B.; Räisänen, J.; Ritala, M.; Leskelä, M. Atomic layer deposition of AlF3 thin films using halide precursors. Chem. Mater. 2015, 27, 604–611. [Google Scholar] [CrossRef]
- Pilvi, T.; Puukilainen, E.; Munnik, F.; Leskelä, M.; Ritala, M. ALD of YF3 thin films from TiF4 and Y(thd)3 precursors. Chem. Vap. Depos. 2009, 15, 27–32. [Google Scholar] [CrossRef]
- Pilvi, T.; Puukilainen, E.; Arstila, K.; Leskelä, M.; Ritala, M. Atomic layer deposition of LaF3 thin films using La(thd)3 and TiF4 as precursors. Chem. Vap. Depos. 2008, 14, 85–91. [Google Scholar] [CrossRef]
- Park, J.S.; Mane, A.U.; Elam, J.W.; Croy, J.R. Atomic layer deposition of Al–W–Fluoride on LiCoO2 cathodes: Comparison of particle- and electrode-level coatings. ACS Omega 2017, 2, 3724–3729. [Google Scholar] [CrossRef]
- Putkonen, M.; Szeghalmi, A.; Pippel, E.; Knez, M. Atomic layer deposition of metal fluorides through oxide chemistry. J. Mater. Chem. 2011, 21, 14461–14465. [Google Scholar] [CrossRef]
- Vos, M.F.J.; Knoops, H.C.M.; Synowicki, R.A.; Kessels, W.M.M.; Mackus, A.J.M. Atomic layer deposition of aluminum fluoride using Al(CH3)3 and SF6 plasma. Appl. Phys. Lett. 2017, 111, 113105-1–113105-5. [Google Scholar] [CrossRef]
- Bridou, F.; Cuniot-Ponsard, M.; Desvignes, J.-M.; Richter, M.; Kroth, U.; Gottwald, A. Experimental determination of optical constants of MgF2 and AlF3 thin films in the vacuum ultra-violet wavelength region (60–124 nm), and its application to optical designs. Opt. Commun. 2010, 283, 1351–1358. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Zhang, W.; Yi, K.; Shao, J. Effects of substrate temperatures and deposition rates on properties of aluminum fluoride thin films in deep-ultraviolet region. Appl. Opt. 2012, 51, 8481–8489. [Google Scholar] [CrossRef] [PubMed]
- König, D.; Scholz, R.; Zahn, D.R.T.; Ebest, G. Band diagram of the AlF3/SiO2/Si system. J. Appl. Phys. 2005, 97, 093707-1–093707-9. [Google Scholar] [CrossRef]
- Song, G.-M.; Wu, Y.; Liu, G.; Xu, Q. Influence of AlF3 coating on the electrochemical properties of LiFePO4/graphite Li-ion batteries. J. Alloy. Compd. 2009, 487, 214–217. [Google Scholar] [CrossRef]
- Lee, D.-J.; Lee, K.-S.; Myung, S.-T.; Yashiro, H.; Sun, Y.-K. Improvement of electrochemical properties of Li1.1Al0.05Mn1.85O4 achieved by an AlF3 coating. J. Power Sources 2011, 196, 1353–1357. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, S.B.; Park, Y.J. Enhanced electrochemical properties of fluoride-coated LiCoO2 thin films. Nanoscale Res. Lett. 2012, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Lapiano-Smith, D.A.; Eklund, E.A.; Himpsel, F.J.; Terminello, L.J. Epitaxy of LiF on Ge(100). Appl. Phys. Lett. 1991, 59, 2174–2176. [Google Scholar] [CrossRef]
- Li, H.H. Refractive index of alkali halides and its wavelength and temperature derivatives. J. Phys. Chem. Ref. Data 1976, 5, 329–528. [Google Scholar] [CrossRef]
- Lee, Y.; Huffman, C.; George, S.M. Selectivity in thermal atomic layer etching using sequential, self-limiting fluorination and ligand-exchange reactions. Chem. Mater. 2016, 28, 7657–7665. [Google Scholar] [CrossRef]
- Zywotko, D.R.; George, S.M. Thermal atomic layer etching of ZnO by a “Conversion-Etch” mechanism using sequential exposures of hydrogen fluoride and trimethylaluminum. Chem. Mater. 2017, 29, 1183–1191. [Google Scholar] [CrossRef]
- DuMont, J.W.; George, S.M. Competition between Al2O3 atomic layer etching and AlF3 atomic layer deposition using sequential exposures of trimethylaluminum and hydrogen fluoride. J. Chem. Phys. 2017, 146, 052819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lee, Y.; Sun, H.; Wallas, J.M.; George, S.M.; Xie, M. Coating solution for high-voltage cathode: AlF3 atomic layer deposition for freestanding LiCoO2 electrodes with high energy density and excellent flexibility. ACS Appl. Mater. Interfaces 2017, 9, 9614–9619. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Jain, A.; Kodas, T.T.; Hampden-Smith, M.; Farr, J.D.; Muenchausen, R. Low-temperature dry etching of metal oxides and ZnS via formation of volatile metal β-diketonate complexes. J. Mater. Chem. 1992, 2, 893–894. [Google Scholar] [CrossRef]
- Ritala, M.; Leskelä, M.; Nykänen, E.; Soininen, P.; Niinistö, L. Growth of titanium dioxide thin films by atomic layer epitaxy. Thin Solid Films 1993, 225, 288–295. [Google Scholar] [CrossRef]
- Miikkulainen, V.; Leskelä, M.; Ritala, M.; Puurunen, R.L. Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends. J. Appl. Phys. 2013, 113, 021301-1–021301-101. [Google Scholar] [CrossRef]
- Krause, R.F., Jr.; Douglas, T.B. Heats of formation of AlClF2 and AlCl2F from subliming aluminum fluoride in the presence of aluminum chloride vapor. J. Phys. Chem. 1968, 72, 3444–3451. [Google Scholar] [CrossRef]
- Haukka, S. ALD technology—Present and future challenges. ECS Trans. 2007, 3, 15–26. [Google Scholar] [CrossRef]
- Klug, J.A.; Proslier, T.; Elam, J.W.; Cook, R.E.; Hiller, J.M.; Claus, H.; Becker, N.G.; Pellin, M.J. Atomic layer deposition of amorphous niobium carbide-based thin film superconductors. J. Phys. Chem. C 2011, 115, 25063–25071. [Google Scholar] [CrossRef]
- Kaipio, M.; Kemell, M.; Vehkamäki, M.; Mattinen, M.; Mizohata, K.; Ritala, M.; Leskelä, M. Atomic layer deposition of metal carbides—The TiCl4/TMA process as an example. In Proceedings of the 14th Baltic Conference on Atomic Layer Deposition, St. Petersburg, Russia, 2–4 October 2016. [Google Scholar]
- Chen, Y.; Ould-Chikh, S.; Abou-Hamad, E.; Callens, E.; Mohandas, J.C.; Khalid, S.; Basset, J.-M. Facile and efficient synthesis of the surface tantalum hydride (≡SiO)2TaIIIH and tris-siloxy tantalum (≡SiO)3TaIII starting from novel tantalum surface species (≡SiO)TaMe4 and (≡SiO)2TaMe3. Organometallics 2014, 33, 1205–1211. [Google Scholar] [CrossRef]
- Schrock, R.R.; Meakin, P. Pentamethyl complexes of niobium and tantalum. J. Am. Chem. Soc. 1974, 96, 5288–5290. [Google Scholar] [CrossRef]
- Nilsen, O.; Fjellvåg, H.; Kjekshus, A. Growth of calcium carbonate by the atomic layer chemical vapour deposition technique. Thin Solid Films 2004, 450, 240–247. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Type | Potential (V) (vs. Li/Li+) | Specific Capacity (mAh/g) | Ref. |
|---|---|---|---|---|
| LiCoO2 | Intercalation | ~3.9 | 140 | [4] |
| LiNiO2 | Intercalation | 2.7–4.1 | 140–200 | [4,19] |
| LiMn2O4 | Intercalation | 3.5–4.5 | 150 | [4] |
| LiFePO4 | Intercalation | 3.4 | 170 | [4,5,19] |
| V2O5 | Intercalation | 3.2–3.4 | 120 | [4] |
| Material | Type | Potential (V) (vs. Li+/Li) | Specific Capacity (mAh/g) | Ref. |
|---|---|---|---|---|
| C6 | Intercalation | <0.6 | 370 | [4,5] |
| Li4Ti5O12 | Intercalation | 1.5 | 175 | [4] |
| Li | Alloying | 0 | 3800 | [26] |
| Si | Alloying | 0.1–0.3 | 3580 | [4,5] |
| Sn | Alloying | 0.6–0.8 | 990 | [4,5,9] |
| MO (M = Co2+, Fe2+, Cu2+, Ni2+) | Conversion | ~1.8–2.0 | 670–750 | [27,28,29] |
| Material | Structure | Ionic Conductivity at RT (S/cm) | Ref. |
|---|---|---|---|
| LiPON | Amorphous | 10−8–10−6 | [31,32] |
| Li2O–SiO2–V2O5 (LVSO) | Crystalline/Amorphous | 10−7–10−5 | [32] |
| Li2S–GeS2–Ga2S3 | Amorphous | 10−4 | [32] |
| (Li,La)TiO3 (LLT) | Perovskite | 10−3 | [31,33] |
| LiTi2(PO4)3 | NASICON | 10−5 | [31,32] |
| Li14ZnGe4O16 | LISICON | 10−6 | [33] |
| Li6BaLa2Ta2O12 | Garnet | 10−5 | [31] |
| Material | Precursors | TDep (°C) | Growth Rate (Å)/Binary Cycle | Capacity (mAh/g) | Ref. |
|---|---|---|---|---|---|
| V2O5 | VO(OiPr)3 + H2O | 105 | 0.15–0.2 | 455 | [85,86] |
| V2O5 | VO(OiPr)3 + O3 | 170–185 | 0.25 | 440 | [86] |
| LixCoyO | LiOtBu + CoCp2 + O2 plasma | 325 | 1.0 with Li:Co pulsing ratio 1:1 | 96 for Li:Co pulsing 1:4 | [87,88] |
| LixFeyPO4 | LiOtBu + FeCp2 + O3 + TMPO + H2O | 300 | 0.85 with Li:Fe pulsing ratio 1:5 | 150 at 0.1 C | [89] |
| LixMnyO | Lithd + Mn(thd)3 + O3 | 225 | 0.2 with Li:Mn pulsing ratio 1:19 | – | [90] |
| LixMnyO | LiOtBu + H2O or Lithd + O3 on MnO2 | 225 | – | 230 | [90] |
| Li2S | LiOtBu + H2S | 150–300 | 1.1 at 150–300 °C | 500 | [91] |
| Material | Precursors | TDep (°C) | Growth Rate (Å)/Binary Cycle | Capacity (mAh/g) | Ref. |
|---|---|---|---|---|---|
| TiO2 | Ti(OiPr)4 + H2O | 160 | 0.33 | 330 | [98] |
| LixTiyO | LiOtBu + Ti(OiPr)4 + H2O | 225, 250 | 0.7 with Li:Ti pulsing ratio 1:1 | 40 | [99,100,101] |
| LiTP | Lithd + TPA | 200–280 | 3.0 at 200 °C, decreases with TDep | 350 | [102] |
| Material | Precursors | TDep (°C) | Growth Rate (Å)/Binary Cycle | Ionic Conductivity (S/cm) | Ref. |
|---|---|---|---|---|---|
| LixSiyO | LiHMDS + O3 | 150–400 | Varies with temperature | Not measured | [110,111] |
| LiTMSO + O3 + H2O | 175–300 | 1.5 at 200–300 °C | Not measured | [112] | |
| LiOtBu + TEOS + H2O | 225–300 | 0.5 at 250 °C | 10−10–10−9 at 30 °C | [113] | |
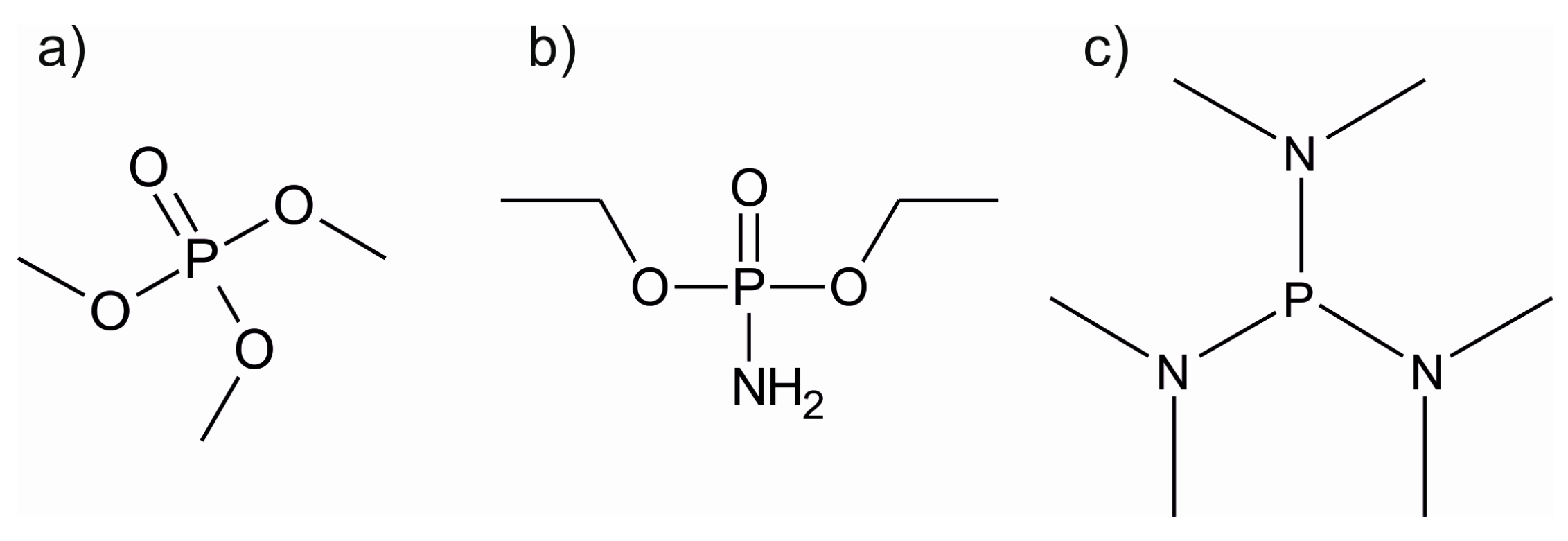
| Li3PO4 | LiOtBu + TMPO | 225–300 [114] 250–350 [115] | 0.7 at 225–275 °C [114], 0.69 at 300 °C [115] | 10−8–10−7 at RT [115,116] | [114,115,116] |
| LiPON | LiHMDS + DEPA | 250–350 | 0.7 at 270–310 °C | 6.6 × 10−7 at RT (9.7 at.% nitrogen) | [108] |
| LiOtBu + H2O + TMPO + N2 plasma | 250 | 1.05 | 1.45 × 10−7 (5.5 at.% nitrogen, increases with nitrogen contents) | [109] | |
| LixTayO | LiOtBu + Ta(OEt)5 + H2O | 225 | 0.7 with Li:Ta pulsing ratio 1:6. | 1.2 × 10−8 at RT | [117] |
| LixNbyO | LiHMDS + Nb(OEt)5 + H2O | 235 | ~0.64 with Li:Nb pulsing ratio 1:2 | Not measured | [118] |
| LiOtBu + Nb(OEt)5 + H2O | 235 | ~0.68 with Li:Nb pulsing ratio 1:2 | 6.4 × 10−8 at 30 °C | [119] | |
| LixLayTizO | LiOtBu + La(thd)3 + TiCl4 + O3 + H2O | 225 | Varies with pulsing ratio | Not measured. | [120] |
| LixAlySizO | LiOtBu + TMA + TEOS + H2O | 290 | 1.0 with Li:Al pulsing ratio 6:10 | 10−9–10−7 at RT, depends on Li contents | [121] |
| LixLayZrzO:Al | LiOtBu + La(FMAD)3 + TDMAZ + TMA + O3 | 225 | 1.0 with Li:La:Zr:Al ratio 8:28:12:1 | 1 × 10−8 at RT for amorphous film | [122] |
| LixAlyS | LiOtBu + TDMA-Al + H2S | 150 | 0.5 with Li:Al ratio 1:1 | 2.5 × 10−7 at RT | [123] |
| LixAlyF | LiHMDS + TMA + HF-py | 150 | 0.45 with Li:Al pulsing ratio 1:1 | 7.5 × 10−6 at RT | [51] |
| LiOtBu + AlCl3 + TiF4 | 250 | 1 with Li:Al pulsing ratio 1:1 | (3.5 ± 0.5) × 10−8 at RT | [124] |
| Material | Precursors | TDep (°C) | Growth Rate (Å)/Cycle | Ref. |
|---|---|---|---|---|
| Fluorides Using HF as the Fluorine Precursor | ||||
| LiF | LiHMDS + HF-py | 150 | 0.5 | [147] |
| MgF2 | Mg(EtCp)2 + HF-py | 150 | 0.4 | [147] |
| MgF2 | Mg(EtCp)2 + HF | 100–250 | Varies, 0.6 at 100 °C | [148] |
| CaF2 | Ca(thd)2 + HF/NH4F | 300–400 | 0.2 at 320–400 °C | [141] |
| SrF2 | Sr(thd)2 + HF/NH4F | 260–320 | Varies, 0.6 at 300 °C | [141] |
| AlF3 | TMA + HF-py | 75–300 | Varies, 1.0 at 150 °C | [143] |
| AlF3 | TMA + HF | 100–200 | Varies, 1.2 at 100 °C | [149] |
| MnF2 | Mn(EtCp)2 + HF-py | 150 | 0.4 | [147] |
| ZnF2 | Zn(Ac)2·2H2O + HF/NH4F | 260–320 | 0.7 at 260–300 °C | [141] |
| ZnF2 | DEZ + HF-py | 150 | 0.7 | [147] |
| ZrF4 | TEMAZ + HF-py | 150 | 0.9 | [147] |
| ZrF4 | Zr(OtBu)4 + HF-py | 150 | 0.6 | [147] |
| HfF4 | TDMAH + HF-py | 150 | 0.8 | [147] |
| LixAlyF | LiHMDS + TMA + HF-py | 150 | 0.45 with Li:Al pulsing ratio 1:1 | [51,146] |
| Fluorides Using Metal Fluorides as the Fluorine Precursor | ||||
| LiF | Lithd + TiF4 | 250–350 | 1.0 at 325 °C | [150] |
| LiF | Mg(thd)2 + Lithd + TiF4 | 300–350 | 1.4 at 325 °C | [151] |
| LiF | LiOtBu + WF6 | 150–300 | – | [152] |
| LiF | LiOtBu + MoF6 | 150–300 | 2.6 | [152] |
| LiF | LiOtBu + TiF4 | 200–300 | 0.5 at 250 °C | [124] |
| MgF2 | Mg(thd)2 + TiF4 | 250–400 | Varies, 1.6 at 250 °C | [153] |
| MgF2 | Mg(thd)2 + TaF5 | 225–400 | Varies, 1.1 at 225–250 °C | [154] |
| CaF2 | Ca(thd)2 + TiF4 | 300–450 | Varies, 1.6 at 300–350 °C | [155] |
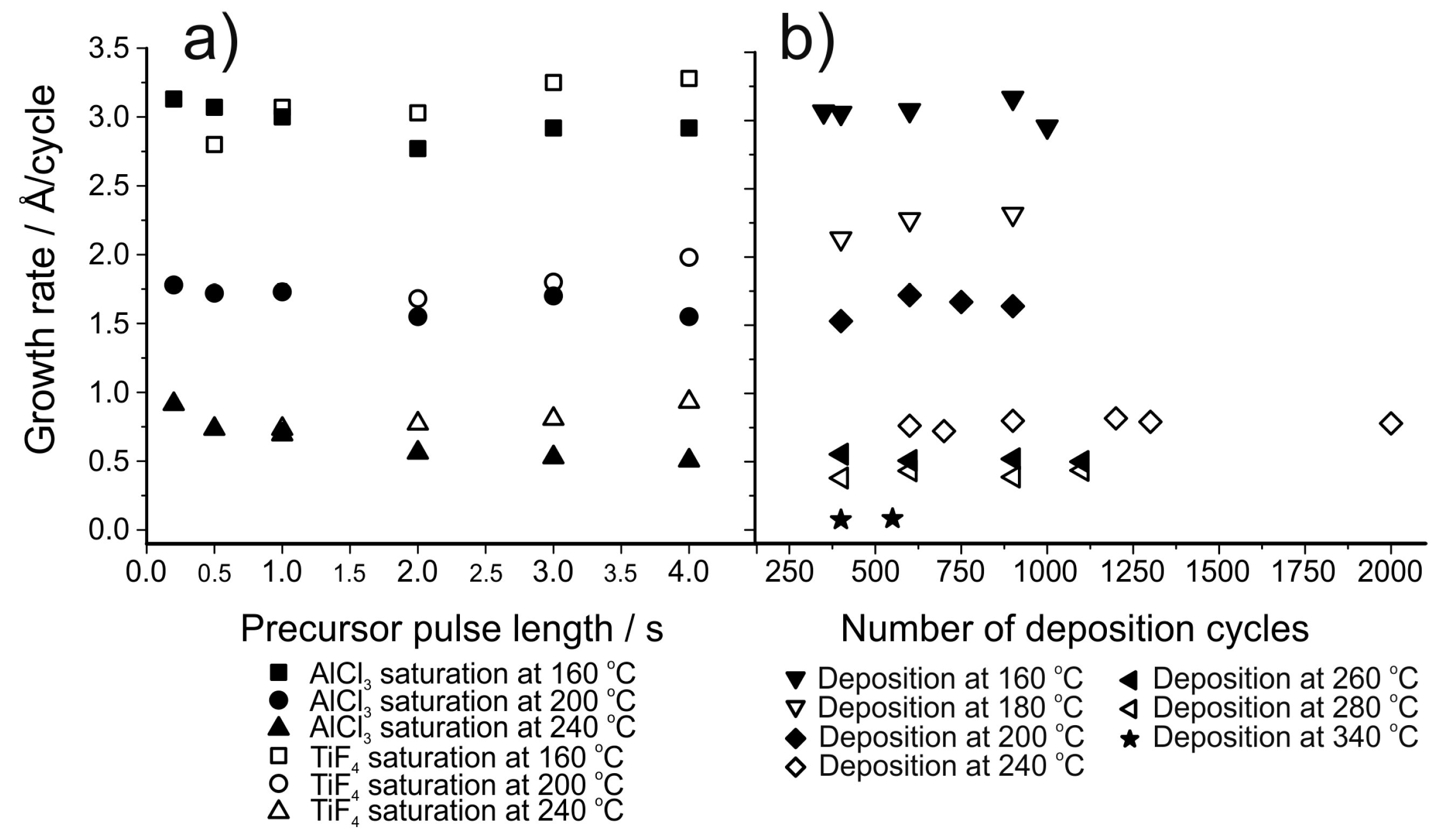
| AlF3 | AlCl3 + TiF4 | 160–340 | Varies, 0.75 at 240 °C | [156] |
| AlF3 | TMA + TaF5 | 125–350 | Varies, 1.9 at 125 °C | [145] |
| YF3 | Y(thd)3 + TiF4 | 175–325 | Varies, 1.3–1.5 at 200–300 °C | [157] |
| LaF3 | La(thd)3 + TiF4 | 225–350 | Varies, 5.2 at 225–250 °C | [158] |
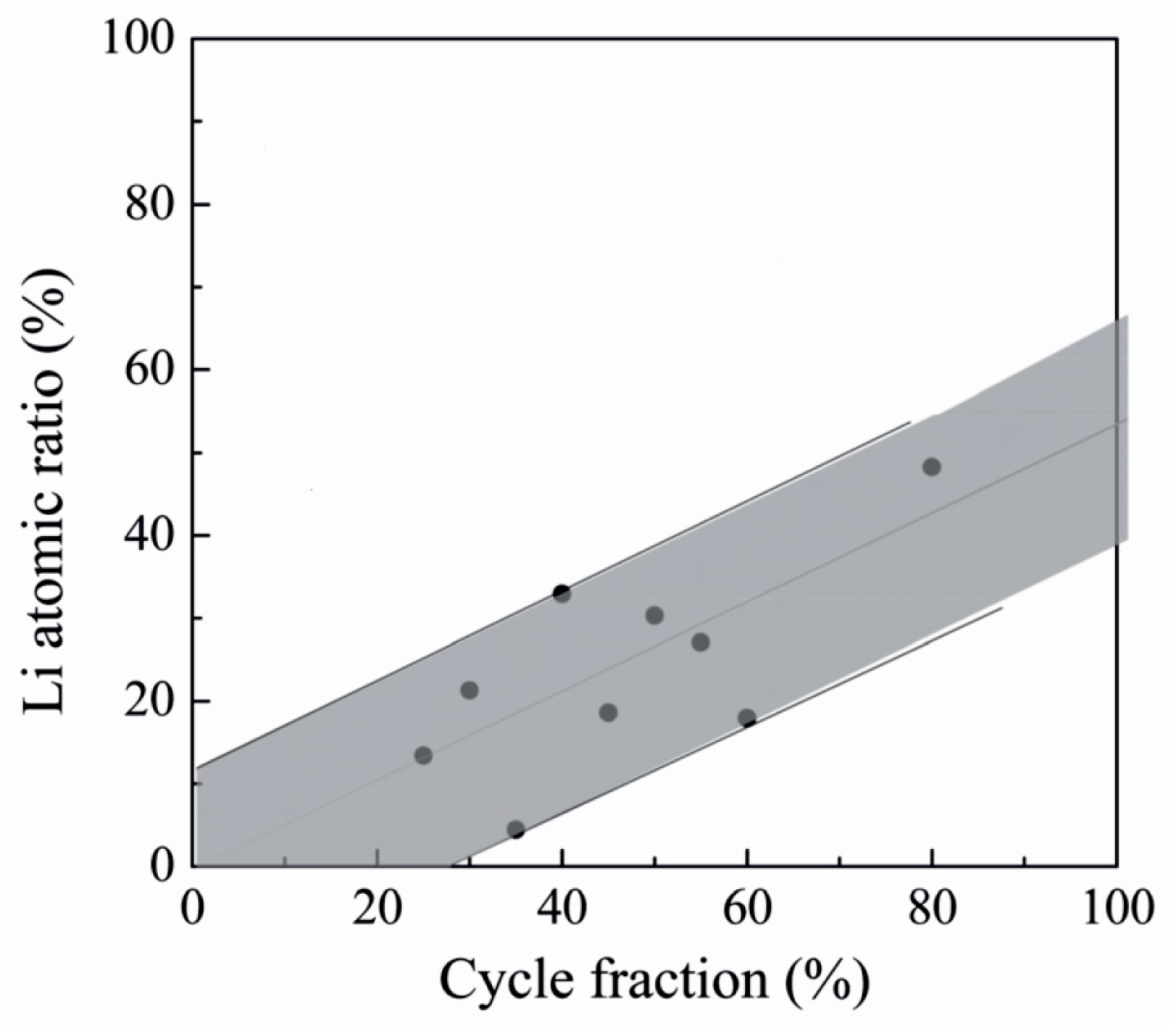
| LixAlyF | LiF + Al(thd)3 + TiF4 | 250–350 | – | [139] |
| LixAlyF | LiOtBu + AlCl3 + TiF4 | 250 | 1 | [124] |
| AlWxFy | TMA + WF6 | 200 | 1–1.5 | [144,159] |
| Fluorides Using Other Processes | ||||
| LiF | MgF2 + Lithd | 275–325 | – | [151] |
| MgF2 | Mg(thd)2 + Hhfac + O3 | – | 0.38 | [160] |
| CaF2 | Ca(hfac)2 + O3 | 300 | 0.3 | [160] |
| CaF2 | Ca(thd)2 + Hhfac + O3 | 250–350 | 0.4 | [160] |
| AlF3 | TMA + SF6 plasma | 50–300 | Varies, 0.85 at 200 °C | [161] |
| LaF3 | La(thd)3 + Hhfac + O3 | – | 0.49 | [160] |
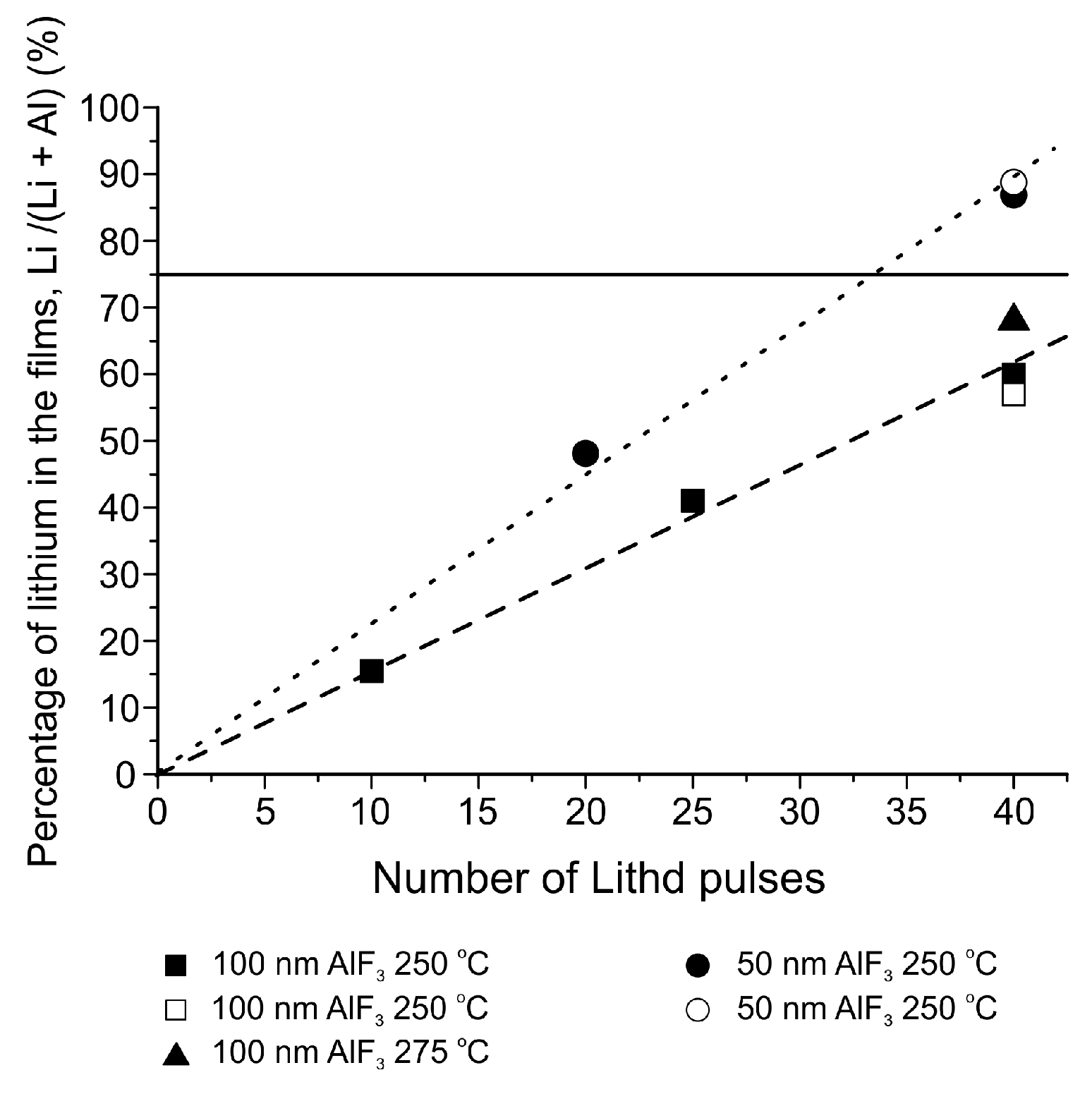
| LixAlyF | AlF3 + Lithd | 250–300 | – | [139] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mäntymäki, M.; Ritala, M.; Leskelä, M. Metal Fluorides as Lithium-Ion Battery Materials: An Atomic Layer Deposition Perspective. Coatings 2018, 8, 277. https://doi.org/10.3390/coatings8080277
Mäntymäki M, Ritala M, Leskelä M. Metal Fluorides as Lithium-Ion Battery Materials: An Atomic Layer Deposition Perspective. Coatings. 2018; 8(8):277. https://doi.org/10.3390/coatings8080277
Chicago/Turabian StyleMäntymäki, Miia, Mikko Ritala, and Markku Leskelä. 2018. "Metal Fluorides as Lithium-Ion Battery Materials: An Atomic Layer Deposition Perspective" Coatings 8, no. 8: 277. https://doi.org/10.3390/coatings8080277
APA StyleMäntymäki, M., Ritala, M., & Leskelä, M. (2018). Metal Fluorides as Lithium-Ion Battery Materials: An Atomic Layer Deposition Perspective. Coatings, 8(8), 277. https://doi.org/10.3390/coatings8080277