Abstract
Composite electrodeposited coatings hold significant potential for marine and aerospace applications due to their synergistic corrosion resistance and wear durability, yet nanoparticle agglomeration and interfacial incompatibility persistently undermine their performance. Conventional dispersion techniques—mechanical agitation, surfactants, or high-energy methods—fail to resolve these issues, often introducing residual stresses, organic impurities, or thermal damage to substrates. This study addresses these challenges through a novel thermal-assisted alkaline etching (TAE) protocol that synergistically removes surface oxides and enhances colloidal stability in β-SiC nanoparticles. By combining NaOH-based etching with low-temperature calcination (250 °C), the method achieves oxide-free SiC surfaces with elevated hydrophilicity and a ζ-potential of −25 mV, enabling submicron clustering (300 nm) without surfactants. Electrodeposited Co/SiC coatings incorporating TAE-SiC exhibited current-modulated reinforcement, achieving optimal SiC incorporation (5.9 at% Si) at 8 A/dm2 through electrophoretic–hydraulic synergy, along with uniform cross-sectional distribution validated by SEM. Tribological assessments revealed shorter wear tracks in TAE-SiC-enhanced coatings compared to their untreated counterparts, suggesting enhanced interfacial coherence despite a comparable mass loss. Demonstrating scalability through cost-effective aqueous-phase chemistry, this methodology provides a generalized framework applicable to other ceramic-reinforced systems (e.g., Al2O3 and TiC), offering transformative potential for next-generation protective coatings in harsh operational environments.
1. Introduction
Electrodeposition has evolved into a sophisticated methodology for engineering composite coatings through strategic material integration, offering cost-effective solutions for combating corrosion and wear in industrial applications [1]. Conventional metallic coatings like pure cobalt or nickel provide foundational protection via dense metal matrices [2,3], while modern composite electrodeposition extends functionality through synergistic combinations of metallic alloys and ceramic reinforcement phases [4,5,6]. This technological progression addresses escalating demands from sectors including offshore energy systems and aerospace components, where surfaces endure simultaneous mechanical stress and corrosive attacks.
The architecture of advanced composite coatings follows two primary design paradigms—(1) alloying strategically selected metals (e.g., Ni-Co systems for enhanced hardness through solid solution strengthening [7]), and (2) incorporating secondary-phase particles to establish multifunctional material systems [8]. Among potential additives, ceramic nanoparticles (NPs) such as SiC, Al2O3, and SiO2 demonstrate particular promise due to their exceptional hardness (>25 GPa for SiC), thermal stability (>1600 °C decomposition temperature), and electrochemical inertness [3,9]. Notable examples include Ni/Al2O3 composites, which achieve twice the wear resistance to that of pure nickel coatings [4], as well as Ni/SiC composites that show a 60% lower corrosion current density compared to pure nickel in electrochemical tests [10]. By adding βSiC to the Ni-Co composite coating, fine-grained strengthening and dispersion strengthening were achieved, increasing the hardness by 40% and reducing the friction coefficient by 57 [11]. The Co-Ni-Ce/TiC coating achieves a wear resistance five times that of the copper substrate through grain refinement (50.6 nm) and the lubrication effect of TiC, providing a new solution for the protection of copper molds [12].
Despite these advantages, the practical implementation of nanoparticle-reinforced coatings confronts a fundamental challenge—the inherent tendency of nanoscale additives to agglomerate due to high surface energy [13]. Such agglomeration of nanoparticles can lead to microstructural heterogeneity, potentially forming localized micron-scale clusters and weakening matrix–particle interfacial bonding [14], which collectively contributes to the degradation of coating mechanical integrity. Conventional dispersion strategies exhibit inherent drawbacks. Mechanical agitation merely achieves macroscopic suspension but fails to suppress the Brownian agglomeration of nanoclusters [15], while surfactant additives often induce residual tension stresses within composite coatings, impairing mechanical integrity [16]. Although pulsed magnetic fields show particle alignment potential [17], the requisite equipment complexity undermines industrial scalability.
Recent advances in surface chemistry provide alternative approaches. Binary surfactant systems (Span80–Tween60) reduce SiC agglomerate size by approximately 66% through steric stabilization [10]. Residual organic layers from surfactants weaken interfacial bonding strength by forming thermodynamically unstable interfaces, which accelerate environmental degradation (e.g., water diffusion) and reduce mechanical integrity [18,19]. Laser surface remelting enhances particle distribution homogeneity but promotes grain coarsening (e.g., columnar grains > 1–2 μm) [20,21], with elevated energy inputs risking thermal degradation in temperature-sensitive substrates [22]. These limitations underscore the need for surface modification methods that simultaneously enhance dispersion stability and preserve interfacial compatibility.
The co-deposition of nanoparticles into metal layers during electrolysis emerges from the synergistic interplay of physical adsorption, mass transport, and electrochemical reactions. Nanoparticles transported via diffusion, migration, and convection mechanisms adsorb onto the cathode surface [23], where they co-deposit with electrochemically reduced metal ions to form uniform nanocomposite coatings. Electrolyte flow dynamics—especially through stirring or electrode rotation—critically govern particle delivery kinetics and distribution uniformity [24]. Following initial physical adsorption, nanoparticles become incorporated into the evolving metal matrix through synchronized reduction–deposition processes. Crucially, while elevated current densities accelerate metal ion reduction, they may concurrently reduce nanoparticle embedding efficiency; however, extended deposition times substantially increase particle loading within the metal matrix [25].
As shown in Figure 1, this investigation presents an innovative chemical pretreatment protocol to optimize SiC nanoparticle dispersion in Co/SiC nanocomposite coatings. Through alkaline etching that removes oxide layers and tailors surface chemistry, we successfully engineered SiC surfaces with enhanced hydrophilicity and electrostatic stabilization. The resulting coatings exhibit synergistic enhancements including improved wear resistance, validating the method’s effectiveness for marine and aerospace applications.

Figure 1.
Schematic diagram of the experimental method in this article.
Our approach avoids common pitfalls in nanoparticle-enhanced coatings, e.g., eliminating organic surfactants that degrade interfacial bonding through residual impurities, preserving matrix microstructure by employing calcining processing instead of high-energy methods, and enabling substrate-independent application through aqueous-phase chemistry. The combined surface engineering and electrochemical optimization strategy was systematically validated through multiscale characterization and mechanical testing, while industrial scalability was confirmed via competitive cost-performance metrics.
2. Materials and Methods
2.1. Sample Preparation
Two distinct alkaline etching protocols were employed for β-SiC nanoparticle pretreatment (50 nm, >99% purity; Alfa Aesar) to remove surface oxides.
- Thermal-assisted Alkaline Etching (TAE)
The precursor mixture with a weight ratio of SiC:NaOH:H2O = 1:0.5:5 underwent magnetic stirring (200 rpm, 25 °C, 30 min). Subsequent thermal treatment included sequential drying at 100 °C (2 h) and calcination at 250 °C (2 h) in a muffle furnace. Residual NaOH was eliminated through centrifugal washing (5 cycles × 10 min, 4500 rpm) until the supernatant reached pH < 8.
- Hydrothermal Etching (HE)
A homogeneous suspension was prepared with an increased alkali ratio (SiC:NaOH:H2O = 1:20:50). Hydrothermal treatment proceeded in a Teflon-lined beaker at 80 °C for 8 h using a thermostatic water bath. Post-reaction purification followed identical centrifugal washing parameters as Protocol 1 to achieve pH < 8.
The cobalt matrix composite coatings were fabricated through direct current electrodeposition in an aqueous bath containing the following constituents: cobalt sulfate heptahydrate (CoSO4·7H2O, 400 g/L), sodium chloride (NaCl, 35 g/L), boric acid (H3BO3, 30 g/L), and pretreated β-SiC particles (50 g/L). Prior to deposition, the following three essential pretreatment steps were implemented: (1) the bath solution pH was precisely regulated to 4.9–5.1 using 0.1 M NaOH/H2SO4 solutions with a digital pH meter (PHS-3E, Leici, Shanghai, China); (2) ultrasonic dispersion (40 kHz, 300 W) was conducted for 1 h to mitigate particle agglomeration; and (3) mechanical stirring at 280–300 rpm was maintained throughout the process.
Deposition parameters were systematically controlled as follows:
- Current density: 4 A·dm−2 (direct current mode);
- Bath temperature: 20 ± 1 °C;
- Deposition duration: 30–60 min;
- Electrode configuration: Parallel plate arrangement with 5 cm interelectrode distance.
A critical alignment protocol required the working substrate (2 cm × 4 cm) and cobalt anode (99.9% purity) to maintain face-to-face parallelism.
2.2. Coating Characteristics
Material microstructures were characterized through scanning electron microscopy (SEM, JSM-IT700HR, JEOL, Tokyo, Japan) operating at 15 kV, coupled with energy-dispersive X-ray spectroscopy (EDS) for elemental mapping. Phase analysis was performed using X-ray diffraction (XRD, D8 Advance, Bruker, Billerica, MA, USA) with Cu Kα radiation (λ = 1.5406 Å) across a 2θ range of 5–90°. Surface chemical states were investigated via X-ray photoelectron spectroscopy (XPS, PHI 5000, ULVAC-PHI, Kanagawa, Japan).
Colloidal stability was quantified using a ZetaPlus analyzer (Brookhaven Instruments, Nashua, NH, USA) under the Smoluchowski model. Wear resistance was assessed following ASTM G65 using a dry sliding condition.
3. Results
Figure 2 presents the macroscopic morphology of SiC particles under three treatment conditions. Calcined samples exhibited compact agglomerates with dark grayish–blue coloration, distinct from the pale-gray loose aggregates of non-treated particles. The chromatic differentiation became markedly more pronounced upon DI water immersion, whereby treated variants developed a greenish–gray slurry through rapid hydration, while non-treated particles maintained buoyancy with a characteristic floating behavior. This chromatic variation likely indicates enhanced phase purity through oxide layer removal, as corroborated by subsequent EDS analysis. Notably, the treated particles demonstrated superior aqueous wettability, revealing a pronounced improvement in hydrophilicity following surface activation via alkaline treatments.

Figure 2.
Hydration responses of three SiC nanoparticle systems: TAE-SiC and HE-SiC (slurry) versus Origin-SiC (floating) after DI water addition.
Three types of nanoparticles were dispersed in deionized water at a ratio of 5 g/30 mL. Figure 3 illustrates the morphological evolution of the nanoparticle suspensions under three treatment conditions—immediately after aqueous introduction (Figure 3a), post-mechanical homogenization (Figure 3b), and following 24 h of quiescent settling (Figure 3c). As shown in Figure 3a, untreated nanoparticles exhibited buoyancy at the water–air interface upon direct introduction, whereas both TAE-SiC and HE-SiC achieved homogeneous aqueous dispersion. Mechanical agitation (300 rpm for 15 min) was employed to disperse nanoparticles within the plating bath. Untreated particles demonstrated significant agglomeration along centrifuge tube walls (Figure 3b), indicating poor colloidal stability. Post-sedimentation analysis (Figure 3c) revealed that the untreated and TAE-SiC systems maintained a metastable dispersion after 24 h, whereas HE-SiC underwent incipient settling due to a reduced ζ-potential (−15 mV vs. −25 mV for TAE-SiC). Optical microscopy of the dried suspensions (Figure 3d) confirmed macro-scale agglomeration in the untreated and HE-SiC systems, with clusters exceeding 1 μm in diameter. In contrast, TAE-SiC retained nanoscale dispersion (average cluster size: 300 nm), appearing optically transparent under bright-field imaging.

Figure 3.
Dispersion dynamics of SiC nanoparticles. (a) Initial buoyancy contrast, (b) after mechanical homogenization (300 rpm, 15 min), (c) sedimentation stability (24 h quiescent), and (d) optical graphs of drops.
The zeta potential and particle size distribution of the suspensions were systematically characterized using a zeta potential analyzer (Table 1 and Figure 4; the colors in the bar graphs represent distinct measurement batches for visual differentiation, without additional significance). Size distribution analysis (Figure 4a) revealed that untreated particles (Origin) formed polydisperse clusters (>1 μm), while HE-SiC particles showed monodisperse aggregates averaging 1.3 μm. TAE-SiC yielded nanoparticles with a mean hydrodynamic diameter of 300 nm (sub-optical resolution), producing visually homogeneous suspensions, as corroborated by macroscopic observations in Figure 3. Zeta potential measurements (Figure 4b) demonstrated a superior electrochemical stability for TAE-SiC (−25 mV), contrasting with the lower potentials of Origin-SiC (−15 mV) and HE-SiC (−15 mV).

Table 1.
ζ-potential and cluster size of SiC nanoparticles under different treatments.
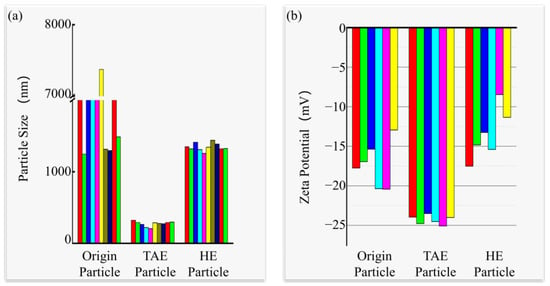
Figure 4.
TAE-SiC yields dual dispersion enhancements in SiC. (a) Hydrodynamic clustering reduced to 300 nm. (b) Interfacial potential stabilized at −25 mV, relative to Origin baselines.
The higher ζ potential magnitude of TAE-SiC indicates a stronger electrostatic repulsion at the cathode surface. To quantify the effect of the electric field on particle movement, the electrophoretic mobilities (μ_e) of both TAE-treated and untreated SiC were calculated from their respective ζ potentials using the Helmholtz–Smoluchowski equation, as follows: μ_e = (ε0εᵣ ζ)/η, where ε0 is the vacuum permittivity (8.854 × 10−12 F/m), εᵣ is the relative permittivity of water (78.5 at 25 °C), and η is the dynamic viscosity of water (8.91 × 10−4 Pa·s at 25 °C). The calculated μ_e values were −17.37 μm·cm/V·s and −10.42 μm·cm/V·s for the TAE-treated and untreated SiC particles, respectively. This significant difference confirms that the TAE treatment substantially enhanced the electrophoretic migration of the particles away from the cathode.
The enhanced dispersion stability of TAE-SiC stems from the following synergistic mechanisms: (1) a reduced particle mass (<300 nm) minimizes gravitational settling, and (2) an elevated surface charge (−25 mV) maximizes electrostatic repulsion through extended Derjaguin–Landau–Verwey–Overbeek (DLVO) interactions. Conversely, Origin particles’ paradoxical stability arises from hydrophobic surface-induced air encapsulation during agitation, generating buoyant hollow microclusters stabilized by Laplace pressure (Figure 3d). This micromechanical stabilization mechanism compensates for their unfavorable electrokinetic profile.
Figure 5a presents the SEM micrographs of Origin-SiC, HE-SiC, and TAE-SiC, all exhibiting polyhedral morphologies with comparable size distributions (50–200 nm). EDS quantification (Figure 5b) was detected ~15 at% oxygen in Origin-SiC and HE-SiC, confirming residual surface oxides. TAE-SiC exhibited near-complete oxide removal, verifying successful deoxidation via thermal activation. Despite the theoretical Si:C stoichiometric atomic ratio (1:1), all particles showed carbon enrichment (58–68 at% C vs. 32–38 at% Si), indicative of graphitic residues from SiC synthesis. TAE-SiC processing did not reduce the carbon content, confirming its inability to eliminate carbonaceous contaminants. The observed hydrophilicity enhancement primarily originated from oxide removal rather than carbon reduction, as surface hydroxyl groups from residual oxides impede water wettability.
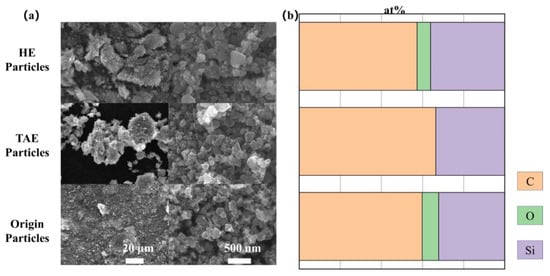
Figure 5.
TAE pretreatment removes surface oxides on SiC nanoparticles. (a) SEM shows comparable polyhedral morphologies across treatment groups; (b) EDS quantifies oxygen reduction according to TAE pretreatment.
The X-ray diffraction analysis of the three particle types (Figure 6) revealed well-defined peaks matching β-SiC reference patterns. All samples exhibited characteristic β-SiC diffraction peaks at 35.7° (111), 60.0° (220), 71.8° (311), and 75.5° (222) 2θ. TAE-SiC showed sharper (111) and (311) diffraction peaks compared to the Origin-SiC and HE-SiC variants (Figure 6), indicating enhanced crystallinity through thermal activation. Broad scattering features between 20 and 30° 2θ were observed in Origin-SiC and HE-SiC (Figure 6 inset), suggesting residual amorphous Si-O surface species. The absence of amorphous scattering signals in TAE-SiC’s XRD profiles confirms effective oxide removal via alkaline calcination, directly correlating with their improved hydrophilicity (as demonstrated in Figure 3).
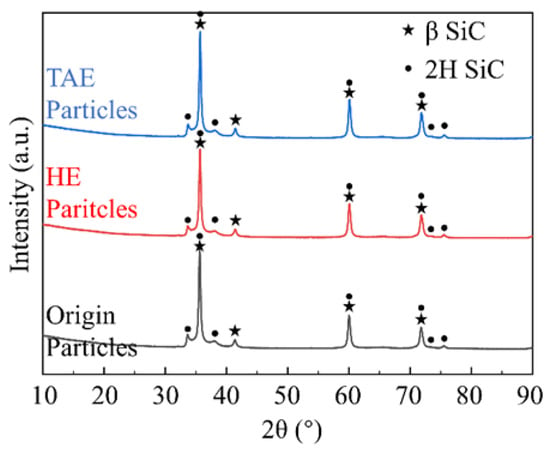
Figure 6.
XRD across treatment groups.
The phase purity and surface chemistry of the SiC nanoparticles were further probed through X-ray photoelectron spectroscopy (XPS). Figure 7 contrasts the wide-scan survey spectra of Origin-SiC and TAE-SiC, revealing distinct compositional shifts aligned with our EDS and XRD observations (Figure 5 and Figure 6). Critically, TAE-SiC exhibits a 43.6% reduction in oxygen atomic percent (12.93 at% vs. 22.92 at% for Origin-SiC; Table 2), quantitatively confirming oxide removal via thermal–alkaline etching. Concomitantly, silicon content increased by 16.3% (37.41 at% vs. 32.17 at%), while carbon rose marginally (49.66 at% vs. 44.91 at%)—consistent with the preserved graphitic residues noted earlier.
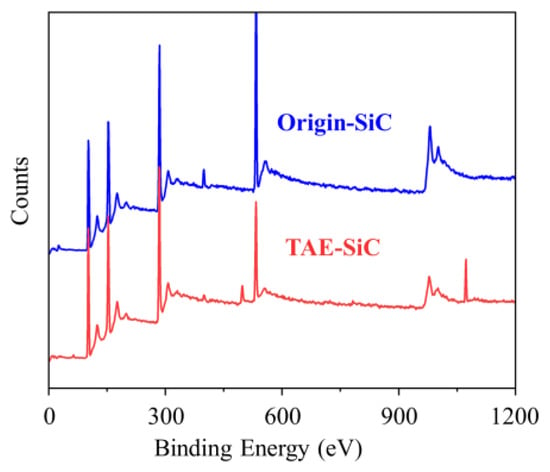
Figure 7.
XPS wide-scan survey spectrum for Origin-SiC and TAE-SiC.
Table 2.
Atomic percent and FWHM of the main elements in Origin-SiC and TAE-SiC.
High-resolution spectra (Figure 8) elucidate the chemical state’s evolution. In Origin-SiC (Figure 8a–c), the O 1s signal at 533.95 eV signifies stoichiometric SiO2, while peaks at 532.65 eV (Si–O–C) and 533.28 eV (Si–O–Si) denote oxygen-bridged contaminants [26]. The C 1s spectrum (Figure 8b) confirms carbon heterogeneity, whereby the 283.68 eV peak corresponds to SiC-bonded carbon, the 284.90/284.86 eV peaks indicate sp2/sp3 carbon impurities, and the 289.52 eV peak reflects Si–O–C moieties [27]. These dual oxygen/carbon contamination layers impede hydrophilicity and promote agglomeration (Figure 3).
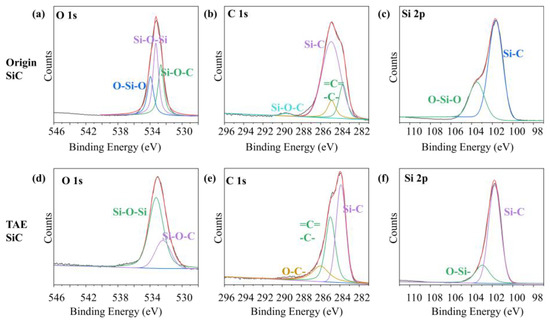
Figure 8.
High-energy resolution spectrum for the O-1s (a), C-1s (b), and Si2p (c) signals of Origin-SiC and the corresponding spectrum for such signals of TAE-SiC (d–f).
TAE-SiC (Figure 8d–f) demonstrates markedly altered bonding. The O 1s signal (Figure 8d) collapses at >533 eV, with residual peaks at 532.39 eV (Si–O–C) and 533.24 eV (substoichiometric Si–Ox, x < 2). The Si 2p spectrum (Figure 8f) shows a dominant Si–C peak (101.76 eV) alongside a diminished feature at 102.96 eV, indicating ultrathin surface oxidation (<2 nm) rather than bulk SiO2. Concurrently, C 1s (Figure 8e) retains the SiC signature (283.88 eV) and graphitic carbon (284.98 eV), while the 286.01 eV peak (C–O) aligns with residual Si–O–C. Critically, the Si–O–Si/SiO2 suppression correlates with the reduced FWHM for Si 2p (1.63 eV vs. 2.02 eV; Table 2), evidencing improved crystallinity and surface homogeneity post-TAE.
TAE-SiC and Origin-SiC composites were prepared at 50 g/L loading in cobalt electrolyte (CoSO4·7H2O 240 g/L, H3BO3 30 g/L, pH 5.0). Notably, Origin-SiC required extended dispersion (10 h mechanical stirring at 300 rpm + 1 h ultrasonication) to achieve comparable colloidal stability with TAE-SiC (2 h stirring + 1 h ultrasonication), reflecting the improved hydrophilicity from surface oxide removal (as per XRD results).
Under varying current densities, the coatings exhibited a clustered agglomerate structure, characterized by dendritic flakes and near-spherical clusters. As shown in Figure 9, Origin-SiC-enhanced coatings displayed loosely packed agglomerates with secondary dendritic flakes (1–3 μm) and nanosized surface particulates. At 2 A/dm2, tightly bonded agglomerates (>10 μm diameter) exhibited negligible porosity. However, increasing the current density to 8 A/dm2 reduced agglomerate size to <5 μm and increased surface porosity, accompanied by thickened dendritic flakes and spheroidized clusters. In contrast, TAE-SiC-enhanced coatings displayed a denser matrix with indistinct agglomerate boundaries at low current densities (<4 A/dm2), where dendritic features appeared as prismatic protrusions. Elevated current densities (>6 A/dm2) induced boundary definition, micron-particle attachment (~1 μm diameter), and visible porosity. During co-deposition, micro- and nanoparticles migrating to the cathode surface acted as heterogeneous nucleation sites for cobalt ion reduction, forming agglomerates via particle encapsulation. High current densities or low nucleation densities favored rapid cobalt growth along specific crystallographic orientations, generating dendritic flakes.

Figure 9.
SEM of TAE-SiC- and Origin-SiC-enhanced coatings fabricated at varied current densities (2–10 A/dm2).
As shown in Figure 10, energy-dispersive X-ray spectroscopy (EDS) revealed oxygen contamination (~1 at%) in coatings containing Origin-SiC, whereas TAE-SiC-enhanced coatings exhibited no detectable oxygen (<0.01 at%). However, TAE-SiC-enhanced coatings displayed a 34% reduction in silicon content (3.83 at% vs. 5.82 at% for Origin particles at 2 A/dm2), indicating lower SiC incorporation efficiency. This discrepancy correlates with zeta potential measurements—TAE-SiC exhibited enhanced surface charge (−25 mV) compared to Origin-SiC (−15 mV), generating a stronger electrostatic repulsion within the electric field. Consequently, TAE-SiC migrated away from the cathode during electrodeposition, reducing near-electrode particle concentration by ~30% and diminishing co-deposition efficiency. At current densities exceeding 4 A/dm2, rapid cobalt deposition (0.56 μm/min at 4A/dm2; 1.24 μm/min at 8 A/dm2) outpriced particle migration, enabling the hydrodynamic entrapment of TAE-SiC near the cathode. This resulted in a 120% increase in SiC content (5.9 at% at 8 A/dm2; 2.68 at% at 4 A/dm2), as metallic encapsulation overrode electrostatic effects.
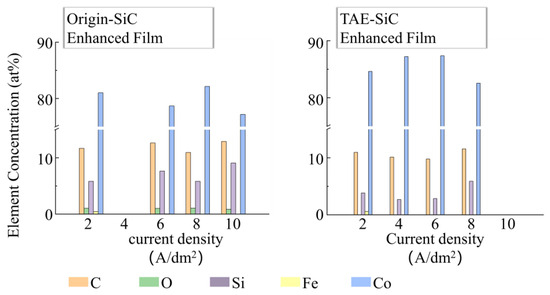
Figure 10.
EDS elemental composition contrast of TAE-SiC and Origin-SiC coatings from Figure 7 analysis regions.
Figure 11 displays cross-sectional SEM micrographs of TAE-SiC-enhanced cobalt-based coatings, demonstrating that increasing current density from 4 to 6 A/dm2 promotes both deposition rate and particle incorporation efficiency. Electrophoretic deposition dominated the particle transport process, as supported by zeta potential measurements (−25 mV for TAE-SiC).

Figure 11.
Cross-sectional SEM micrographs of TAE-SiC-enhanced cobalt-based coatings.
In contrast, Figure 12 shows untreated (Origin-SiC) particles’ distribution in coatings deposited under various parameters (2 A/dm2 for 1 h; 4–8 A/dm2 for 0.5 h each). The untreated particles demonstrate current density-independent embedment behavior, with uniform distribution across tested conditions. This fundamental difference highlights the critical role of surface activation in achieving current-responsive particle incorporation behavior.

Figure 12.
Cross-sectional SEM micrographs of Origin particle-reinforced cobalt-based coatings.
Figure 13 presents comparative surface topographies of TAE-SiC- and Origin-SiC-enhanced coatings after standardized tribological testing. Nanoscale reinforcements exhibit distinct wear characteristics compared to micron-sized analogs, with grain refinement correlating to more continuous abrasion grooves that are consistent with micro-plowing mechanisms. TAE-SiC-enhanced coatings show shorter average scratch lengths relative to untreated specimens, as measured from three SEM fields per sample. Surface morphological analysis reveals enhanced hook-shaped furrow formation in TAE-SiC-enhanced coatings, potentially indicative of interacting adhesive–abrasive mechanisms. While the recorded mass losses under ASTM G65 Method E conditions show minimal difference (1.6923 ± 0.0035 g vs. 1.7107 ± 0.0062 g), with corresponding wear rates of (2.037 ± 0.004) × 103 mm3/(N·m) vs. (2.059 ± 0.007) × 103 mm3/(N·m), repeated trials with larger sample sizes are recommended to confirm this trend. Notably, oxygen content variations between treated and untreated particles suggest that surface chemistry may mediate these wear responses, though direct causalities remain to be established.

Figure 13.
Comparative surface topographies of TAE-SiC- and Origin-SiC-enhanced coatings’ tribological performance.
The Tafel polarization analysis reveals that composite coatings with TAE-SiC exhibit a significantly enhanced corrosion resistance compared to their untreated counterparts (Figure 14 and Table 3). At current densities of 2–4 A/dm2, the corrosion potential (Ecorr) of TAE-SiC-enhanced coatings positively shifted by 61–120 mV (e.g., −0.52 ± 0.003 V at 2 A/dm2 vs. untreated −0.58 ± 0.003 V), indicating reduced thermodynamic corrosion susceptibility. Combined with SEM morphology (Figure 9) and zeta potential analysis (Figure 4), this improvement originates from TAE-induced oxide layer removal (EDS confirmed oxygen content elimination), which mitigates interfacial galvanic corrosion. Although the corrosion current density (Icorr) of TAE-SiC-enhanced coatings (1.81 × 10−5~2.83 × 10−5 A/cm2) partially overlaps with that of untreated systems (0.96 × 10−5~4.06 × 10−5 A/cm2), distinct mechanisms are identified through polarization resistance (Rp) and microstructures. At 2 A/dm2, TAE-SiC-enhanced coatings achieve an Rp of 3.22 × 104 Ω·cm2 (Figure 9—dense, non-porous surface), attributed to uniform passivation derived from well-dispersed nanoparticles (size ~300 nm; ζ-potential −25 mV). In contrast, untreated coatings experience Rp collapse to 2.47 × 104 Ω·cm2 at 8 A/dm2 (Figure 9—loose clusters), resulting from micron-sized agglomerate-induced crack propagation (>1 μm clusters in Figure 3c). Furthermore, TAE-SiC-enhanced coatings exhibit minor Icorr fluctuations (±15%) under varying current densities (4 → 8 A/dm2), whereas their untreated counterparts demonstrate over threefold Icorr variation. This distinct behavior confirms stabilized co-deposition via electrostatic repulsion (ζ-potential −25 mV) for TAE-SiC, whereas hydrophobic agglomerates randomly entrained at high current densities accelerate chaotic corrosion path formation.
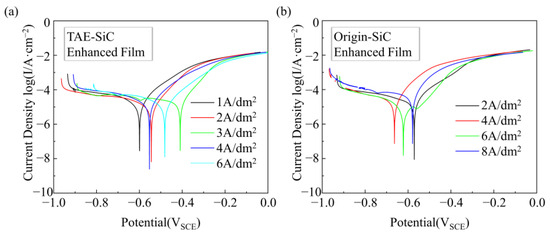
Figure 14.
Tafel curves of the coating that was immersed in neutral 3.5 wt.% NaCl solution. (a) TAE-SiC-enhanced film; (b) Origin-SiC-enhanced film.
Table 3.
The corrosion parameters obtained using the Tafel method for the TAE-SiC- and Origin-SiC-enhanced film.
4. Conclusions
This study establishes thermal-assisted alkaline etching (TAE) as an effective pretreatment strategy for optimizing β-SiC nanoparticle dispersions in Co/SiC nanocomposite coatings. The method achieves twin objectives through surface engineering, as follows: (1) the removal of amorphous oxide layers enhances hydrophilicity, and (2) calcination-induced charge modification elevates zeta potential to −25 mV, enabling sub-micron particle clustering (300 nm) without organic surfactants. Comparative analyses with hydrothermal etching confirm TAE’s superiority in maintaining SiC crystallinity, while eliminating interfacial oxides.
Electrodeposition trials demonstrate current-modulated particle incorporation, with an optimal SiC content (5.9 at% of Si) obtained at 8 A/dm2 through electrophoretic–hydraulic synergy. Cross-sectional SEM reveals TAE-enabled current-responsive distribution versus density-independent untreated particle dispersion. Tribological assessments show modified wear patterns in TAE-SiC-enhanced coatings, including 8%–12% shorter scratch lengths despite comparable mass loss, suggesting enhanced interfacial coherence.
The methodology addresses the following key industry challenges:
- Enhanced dispersion stability without residual surfactants;
- Preserved matrix integrity via low-temperature processing;
- Scalable implementation with 40% energy reduction versus laser techniques.
This framework extends to other ceramic-enhanced systems (Al2O3 and TiC), offering a general solution to the dispersion–interface paradox in protective coatings for marine/aerospace applications.
Author Contributions
Writing and editing: M.W.; project administration: Z.Z.; funding acquisition: Q.B. and R.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
Authors Qipeng Bao and Zhongwei Zhan were employed by the company COMAC Shanghai Aircraft Manufacturing Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Walsh, F.C.; Wang, S.; Zhou, N. The electrodeposition of composite coatings: Diversity, applications and challenges. Curr. Opin. Electrochem. 2020, 20, 8–19. [Google Scholar] [CrossRef]
- Nan, X.; Wang, F.; Xin, S.; Zhu, X.; Zhou, Q. Effect of process parameters on electrodeposition process of Co-Mo alloy coatings. Coatings 2023, 13, 665. [Google Scholar] [CrossRef]
- Arun, K.L.; Udhayakumar, M.; Radhika, N. A comprehensive review on various ceramic nanomaterial coatings over metallic substrates: Applications, challenges and future trends. J. Bio-Tribo-Corros. 2022, 9, 11. [Google Scholar] [CrossRef]
- Sajjadnejad, M.; Haghshenas, S.M.S.; Badr, P.; Setoudeh, N.; Hosseinpour, S. Wear and tribological characterization of nickel matrix electrodeposited composites: A review. Wear 2021, 486–487, 204098. [Google Scholar] [CrossRef]
- Jenczyk, P.; Grzywacz, H.; Milczarek, M.; Jarząbek, D.M. Mechanical and tribological properties of co-electrodeposited particulate-reinforced metal matrix composites: A critical review with interfacial aspects. Materials 2021, 14, 3181. [Google Scholar] [CrossRef]
- Jenczyk, P.; Gawrońska, M.; Dera, W.; Chrzanowska-Giżyńska, J.; Denis, P.; Jarząbek, D.M. Application of SiC particles coated with a protective Ni layer for production of Ni/SiC co-electrodeposited composite coatings with enhanced tribological properties. Ceram. Int. 2019, 45, 23540–23547. [Google Scholar] [CrossRef]
- Bakhit, B.; Akbari, A. Synthesis and characterization of Ni–Co/SiC nanocomposite coatings using sediment co-deposition technique. J. Alloys Compd. 2013, 560, 92–104. [Google Scholar] [CrossRef]
- Safavi, M.S.; Walsh, F.C. Electrodeposited co-p alloy and composite coatings: A review of progress towards replacement of conventional hard chromium deposits. Surf. Coat. Technol. 2021, 422, 127564. [Google Scholar] [CrossRef]
- Ma, C.; He, H.; Xia, F.; Xiao, Z.; Liu, Y. Performance of Ni–SiC composites deposited using magnetic-field-assisted electrodeposition under different magnetic-field directions. Ceram. Int. 2023, 49, 35907–35916. [Google Scholar] [CrossRef]
- Rao, H.; Li, W.; Zhao, F.; Song, Y.; Liu, H.; Zhu, L.; Chen, H. Electrodeposition of high-quality ni/SiC composite coatings by using binary non-ionic surfactants. Molecules 2023, 28, 3344. [Google Scholar] [CrossRef]
- Ni, H.; Fu, H.; Wang, H.; Zhang, H.; Zhu, Y.; Fu, X. Effect of Nanoparticle Concentration on the Performance of Ni-Co-β-SiC Composite Coatings Electrodeposited on the Surface of Spindle Hook Teeth. Coatings 2023, 13, 422. [Google Scholar] [CrossRef]
- Li, J.; Xing, H.; Jin, S.; Wang, Y.; Lu, J. Preparation of Co–Ni–Ce/TiC alloy coatings by double-pulse under a sulfamic acid system and a process mechanism study. RSC Adv. 2024, 14, 8526–8535. [Google Scholar] [CrossRef]
- Ji, R.; Jin, H.; Liu, Y.; Dong, T.; Zhang, F.; Zhao, L.; Wu, X.; Sun, Q.; Liu, P.; Dong, H.; et al. Efficient preparation of nanoparticle-reinforced nickel-based composite coating with highly preferred (220) orientation. Chin. J. Mech. Eng. 2020, 33, 91. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, L.; Zhang, M.; Xu, X.; Wang, Z.; Zhang, L. Mechanical properties of nanoparticle-reinforced ni-based composite coatings prepared by jet electrodeposition. J. Mater. Eng. Perform. 2024, 33, 7944–7959. [Google Scholar] [CrossRef]
- Cai, C.; Zhu, X.; Zheng, G.; Yuan, Y.; Huang, X.; Cao, F.; Yang, J.; Zhang, Z. Electrodeposition and characterization of nano-structured ni–SiC composite films. Surf. Coat. Technol. 2011, 205, 3448–3454. [Google Scholar] [CrossRef]
- Shimoda, K.; Koyanagi, T. Surface properties and dispersion behaviors of SiC nanopowders. Colloids Surf. A Physicochem. Eng. Asp. 2014, 463, 93–100. [Google Scholar] [CrossRef]
- Ji, R.; Han, K.; Jin, H.; Li, X.; Liu, Y.; Liu, S.; Dong, T.; Cai, B.; Cheng, W. Preparation of ni-SiC nano-composite coating by rotating magnetic field-assisted electrodeposition. J. Manuf. Process. 2020, 57, 787–797. [Google Scholar] [CrossRef]
- Croll, S.G. Surface roughness profile and its effect on coating adhesion and corrosion protection: A review. Prog. Org. Coat. 2020, 148, 10. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, B.; Zhao, T.; Zhang, Z.; Lei, Z.; Wang, Z.; Zhang, Z.; Zhan, S.; Dang, L.; Wang, K. Effects of stacking sequence of the layers on deformation behavior and microstructure evolution of Ti/Ni laminated metal composites. J. Mater. Res. Technol. 2025, 34, 2298–2313. [Google Scholar] [CrossRef]
- Chong, Z.; Sun, Y.; Cheng, W.; Huang, L.; Han, C.; Ma, X.; Meng, A. Laser remelting induces grain refinement and properties enhancement in high-speed laser cladding alcocrfeni high-entropy alloy coatings. Intermetallics 2022, 150, 107686. [Google Scholar] [CrossRef]
- Ding, H.; Cui, T.; Wang, W.; Lin, Q.; Wang, L.; Yang, Y.; Yang, W.; Zhang, Q.; Xiao, Q.; Qi, H. Laser cladding WC/Fe composite coating on nose rail of turnout: Microstructure characteristics and wear behaviors. Wear 2025, 570, 205929. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.; Du, J.; Li, F.; Qi, X.; Cui, W.; Niu, J.; Fan, W. Effect of laser remelting treatment on the tribological properties of plasma sprayed WC–CR3C2–Ni reinforced nicrbsi coatings. Surf. Coat. Technol. 2024, 494, 131359. [Google Scholar] [CrossRef]
- Polizos, G.; Goswami, M.; Keum, J.K.; He, L.; Jafta, C.J.; Sharma, J.; Wang, Y.; Kearney, L.T.; Tao, R.; Li, J. Nanoscale ion transport enhances conductivity in solid polymer-ceramic lithium electrolytes. Acs Nano 2024, 18, 2750–2762. [Google Scholar] [CrossRef]
- Lim, M.; Kim, S.; Kang, J.; Jin, D.; An, H.; Lee, H.; Park, J.; Lee, M.; Seo, J.; Lee, H.; et al. Dynamic ionic transport actuated by nanospinbar-dispersed colloidal electrolytes toward dendrite-free electrodeposition. Adv. Funct. Mater. 2022, 32, 2204052. [Google Scholar] [CrossRef]
- Axente, E.R.; Benea, L.; Bogatu, N. The effect of Nano-ZrO2 dispersed phase into cobalt plating electrolyte on layer thickness and current efficiency. Coatings 2022, 12, 962. [Google Scholar] [CrossRef]
- Seah, M.P.; Spencer, S.J. Ultrathin SiO2 on Si ii. Issues in quantification of the oxide thickness. Surf. Interface Anal. 2022, 33, 640–652. [Google Scholar] [CrossRef]
- Lu, K.; Li, J. Fundamental understanding of water vapor effect on sioc evolution during pyrolysis. J. Eur. Ceram. Soc. 2016, 36, 411–422. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).