
Modeling the Deposition of Thin Films of Transition Metal Nitrides

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
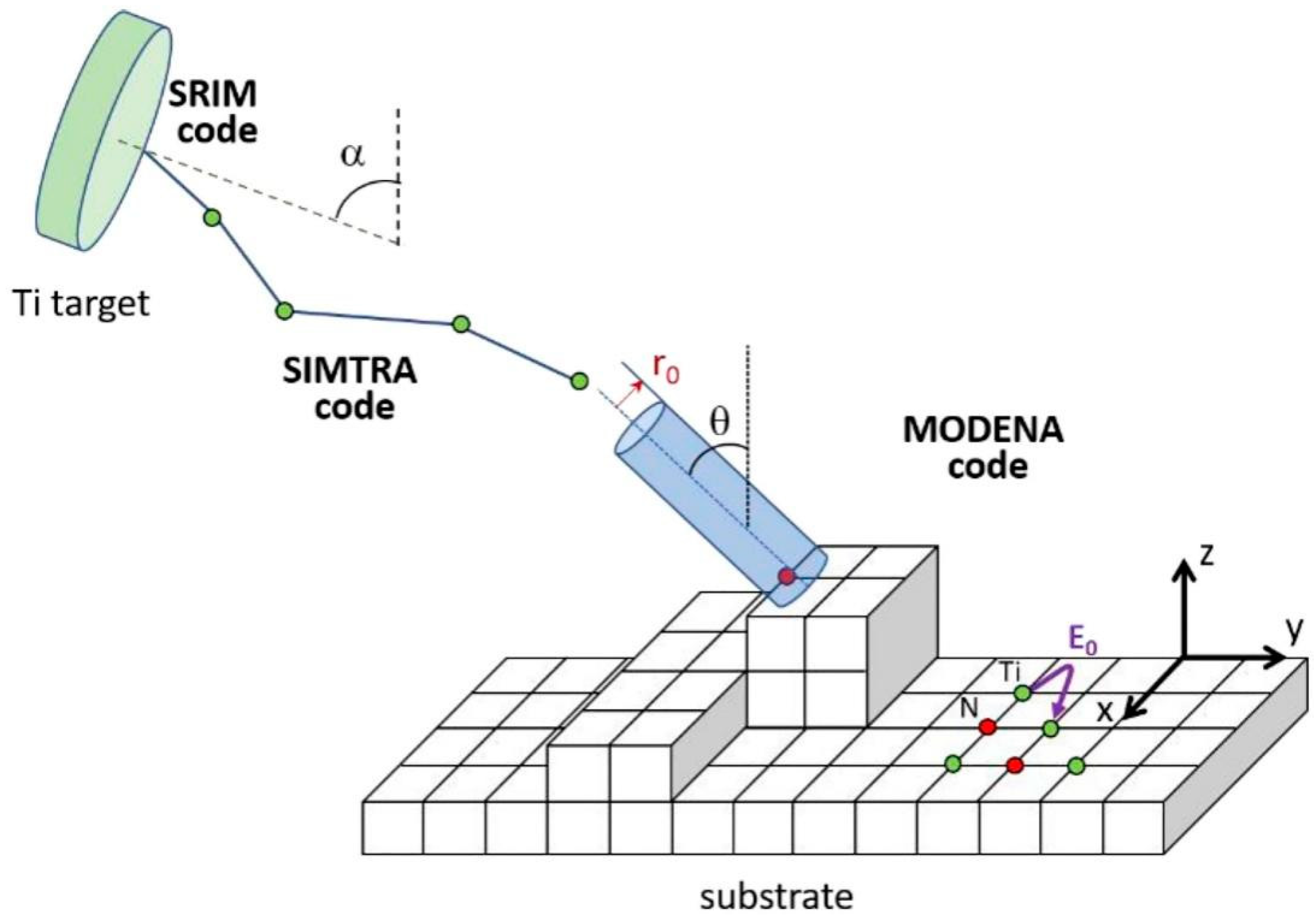{kind=link}
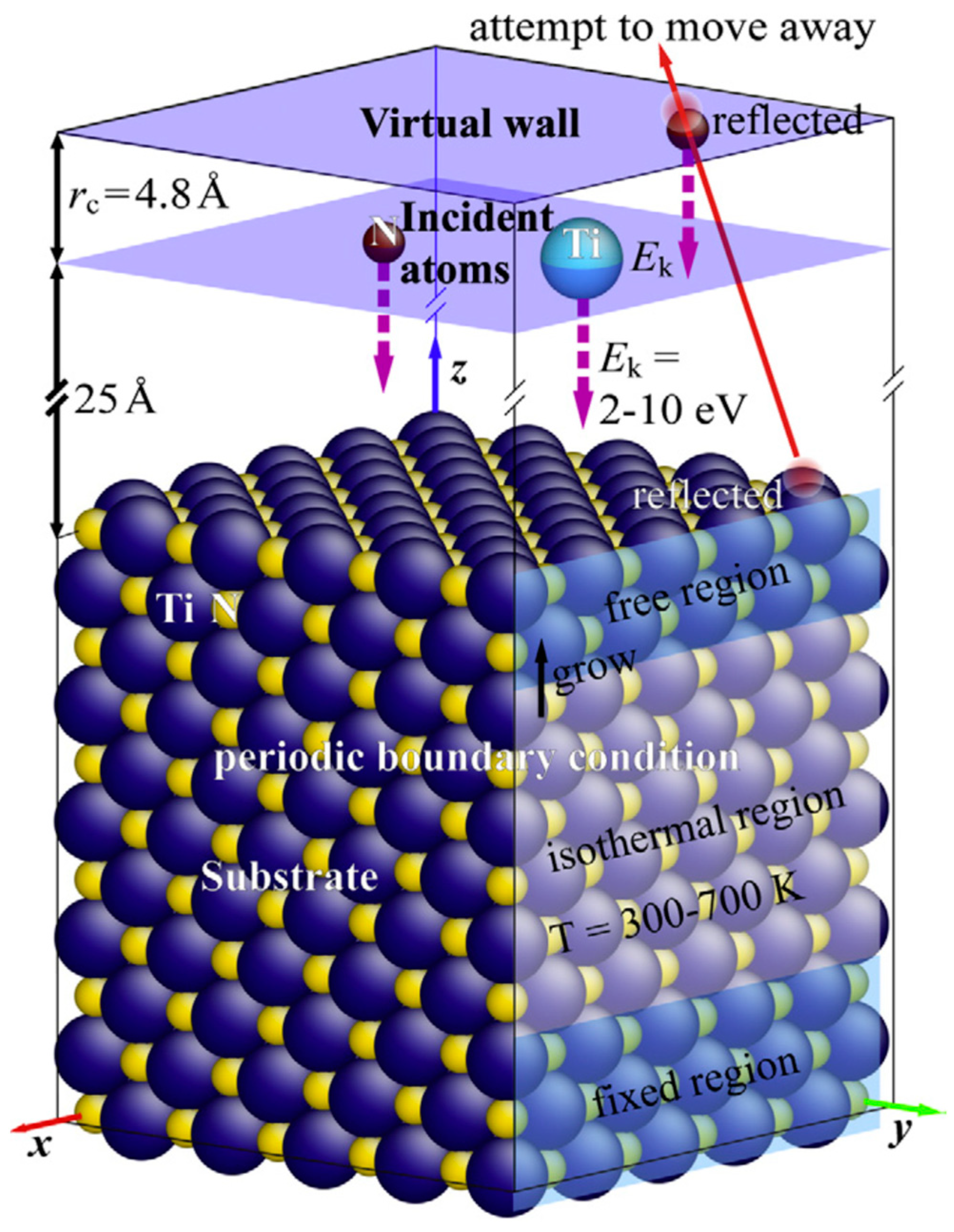{kind=link}
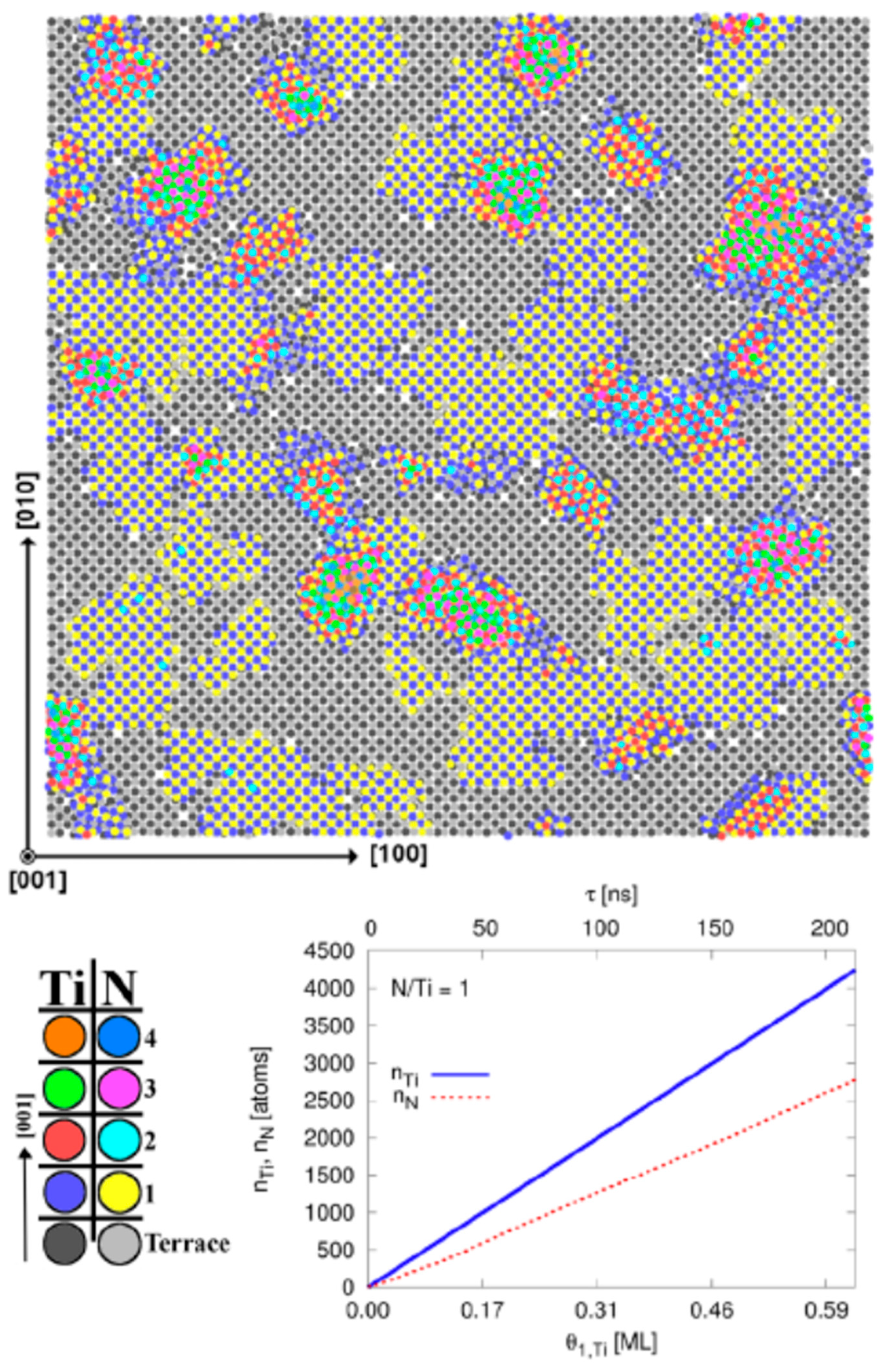{kind=link}
Abstract
:1. Introduction
2. Methods for Modeling Multiatomic Systems
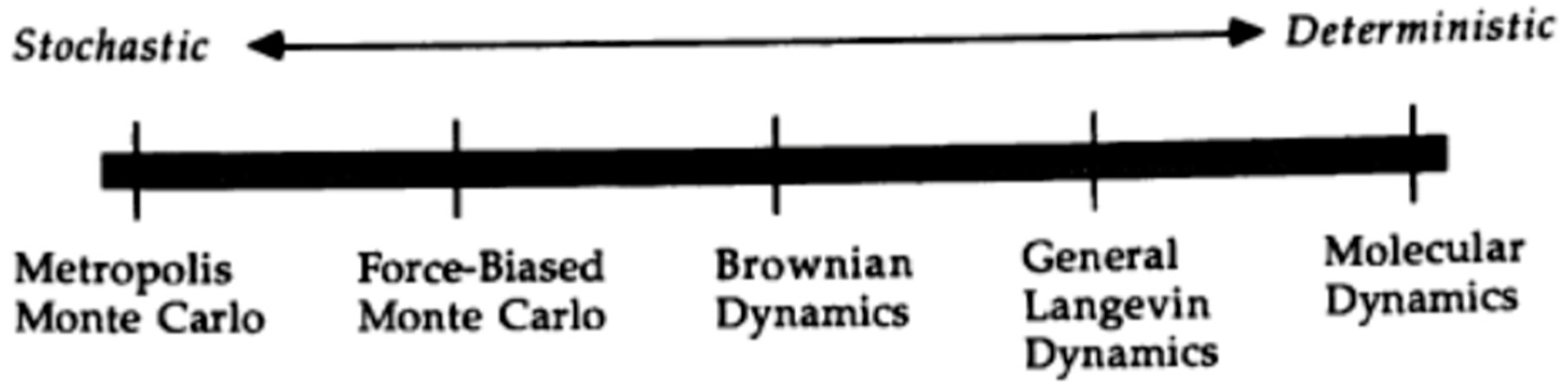
2.1. Monte Carlo Simulation
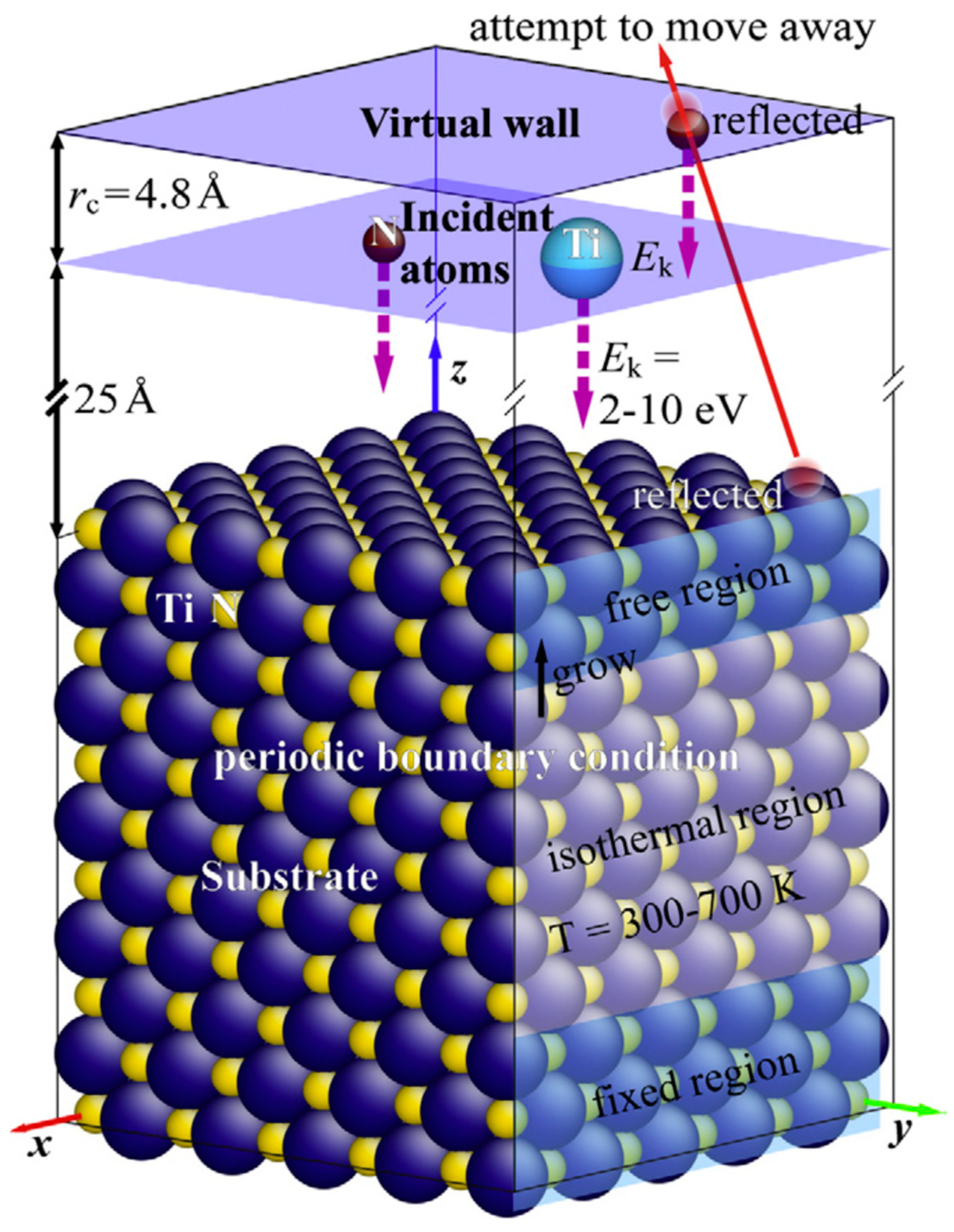
2.2. Molecular Dynamics Simulation
2.2.1. VN, TiN
2.2.2. TaN
2.2.3. ZrN
2.2.4. FeN
2.2.5. CrN
2.2.6. Ternary Alloys
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pogrebnjak, O.; Goncharov, O. Structural Features of Formed Coatings and Films of Refractory Compounds. Metallofiz. Noveishie Tekhnol. 2016, 38, 1145–1166. [Google Scholar] [CrossRef]
- Ahmed, S.; Bakar, S.A. Synthesis of Thin Film and Its Application. In Contemporary Nanomaterials in Material Engineering Applications; Mubarak, N.M., Khalid, M., Walvekar, R., Numan, A., Eds.; Engineering Materials; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Makhlouf, A.S.H. Current and advanced coating technologies for industrial applications. In Nanocoatings and Ultra-Thin Films; Woodhead Publishing: Sawston, UK, 2011; pp. 3–23. [Google Scholar] [CrossRef]
- Pogrebnjak, A.D.; Goncharov, A.; Yunda, A.; Shelest, I.; Swic, A.; Lebedynskyi, I. Structural features of the formation of multicomponent and high-entropy transition metal nitride films. High Temp. Mater. Process. Int. Q. High-Technol. Plasma Process. 2018, 22, 7–15. [Google Scholar] [CrossRef]
- Goncharov, A.; Yunda, A.; Kolinko, I.; Maksakova, O.V. Structural regularities of the formation of nitride and boride coatings based on transition metals. High Temp. Mater. Process. Int. Q. High-Technol. Plasma Process. 2023, 27, 31–52. [Google Scholar] [CrossRef]
- Movchan, B.A.; Demchishin, A.V. Structure and properties of thick condensates of nickel, titanium, tungsten, aluminum oxides, and zirconium dioxide in vacuum. Fiz. Metal. Metalloved. 1969, 28, 653–660. [Google Scholar]
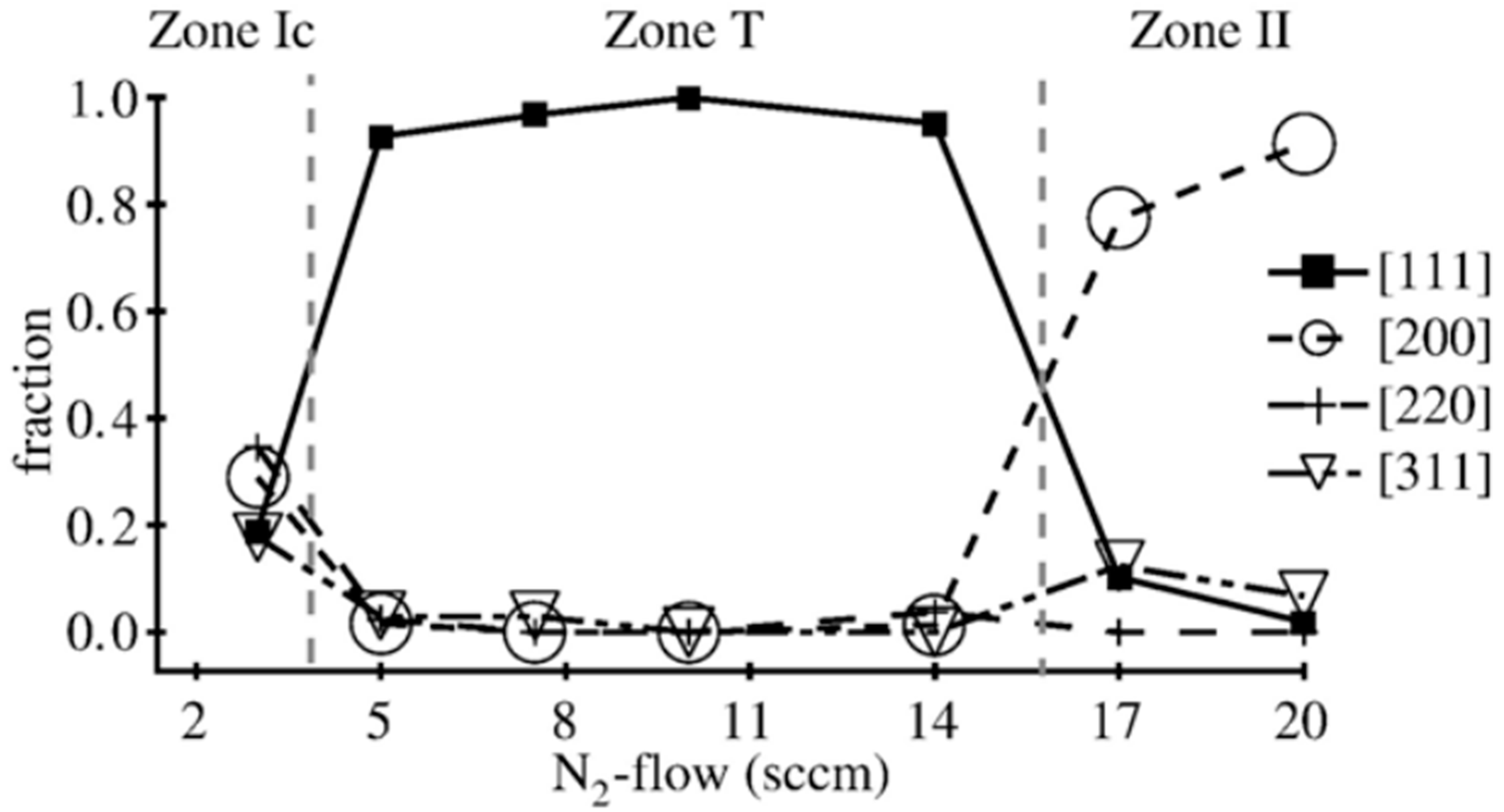
- Thornton, J.A. Influence of apparatus geometry and deposition conditions on the structure and topography of thick sputtered coatings. J. Vac. Sci. Technol. 1974, 11, 666–670. [Google Scholar] [CrossRef]
- Messier, R.; Giri, A.P.; Roy, R.A. Revised structure zone model for thin film physical structure. J. Vac. Sci. Technol. A 1984, 2, 500–503. [Google Scholar] [CrossRef]
- Kelly, P.J.; Arnell, R.D. Magnetron sputtering: A review of recent developments and applications. Vacuum 2000, 56, 159–172. [Google Scholar] [CrossRef]
- Mahieu, S.; Depla, D. Reactive sputter deposition of TiN layers: Modelling the growth by characterization of particle fluxes towards the substrate. J. Phys. D Appl. Phys. 2009, 42, 053002. [Google Scholar] [CrossRef]
- Anders, A. A structure zone diagram including plasma-based deposition and ion etching. Thin Solid Films 2010, 518, 4087–4090. [Google Scholar] [CrossRef]
- Petrov, I.; Barna, P.B.; Hultman, L.; Greene, J.E. Microstructural evolution during film growth. J. Vac. Sci. Technol. A 2003, 21, S117–S128. [Google Scholar] [CrossRef]
- Mahieu, S.; Ghekiere, P.; Depla, D.; De Gryse, R. Biaxial alignment in sputter deposited thin films. Thin Solid Films 2006, 515, 1229–1249. [Google Scholar] [CrossRef]
- Haile, J.M. Molecular Dynamics Simulation: Elementary Methods; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1992. [Google Scholar]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Barth, J.V.; Brune, H. Atomare Prozesse an Oberflächen. Phys. Unserer Zeit 1998, 29, 251–260. [Google Scholar] [CrossRef]
- Baskes, M.I. Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B 1992, 46, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Ercolessi, F.; Adams, J.B. Interatomic Potentials from First-Principles Calculations: The Force-Matching Method. Europhys. Lett. 1994, 26, 583. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Lee, B.-J. Modified embedded-atom method interatomic potentials for the Ti–C and Ti–N binary systems. Acta Mater. 2008, 56, 3481–3489. [Google Scholar] [CrossRef]
- Kim, H.-K.; Jung, W.-S.; Lee, B.-J. Modified embedded-atom method interatomic potentials for the Fe–Ti–C and Fe–Ti–N ternary systems. Acta Mater. 2009, 57, 3140–3147. [Google Scholar] [CrossRef]
- Rogovoi, Y.I. Metal-metal and metal-nitrogen bond potentials in cubic mononitrides. Powder Metall. Met. Ceram. 1997, 36, 518–525. [Google Scholar] [CrossRef]
- Yu, H.; Sun, F. A modified embedded atom method interatomic potential for the Ti–N system. Phys. B Condens. Matter 2009, 404, 1692–1694. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar]
- Gades, H.; Urbassek, H.M. Pair versus many-body potentials in atomic emission processes from a Cu surface. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1992, 69, 232–241. [Google Scholar] [CrossRef]
- Montalenti, F.; Voter, A.F. Applying Accelerated Molecular Dynamics to Crystal Growth. Phys. Status Solidi (b) 2001, 226, 21–27. [Google Scholar] [CrossRef]
- Divi, S.; Chatterjee, A. Study of Silicon Thin Film Growth at High Deposition Rates Using Parallel Replica Molecular Dynamics Simulations. Energy Procedia 2014, 54, 270–280. [Google Scholar] [CrossRef]
- Sorensen, M.R.; Voter, A.F. Temperature-accelerated dynamics for simulation of infrequent events. J. Chem. Phys. 2000, 112, 9599–9606. [Google Scholar] [CrossRef]
- Zamora, R.J.; Uberuaga, B.P.; Perez, D.; Voter, A.F. The Modern Temperature-Accelerated Dynamics Approach. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 87–110. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, W.; Yip, S. (Eds.) Theory and Methods for Materials Modeling: An Introduction. In Handbook of Materials Modeling: Methods: Theory and Modeling; Springer International Publishing: Cham, Switzerland, 2020; pp. 3–12. [Google Scholar]
- Voter, A.F. Hyperdynamics: Accelerated Molecular Dynamics of Infrequent Events. Phys. Rev. Lett. 1997, 78, 3908–3911. [Google Scholar] [CrossRef]
- Ebina, H.; Fukuhara, S.; Shibuta, Y. Accelerated molecular dynamics simulation of vacancy diffusion in substitutional alloy with collective variable-driven hyperdynamics. Comput. Mater. Sci. 2021, 196, 110577. [Google Scholar] [CrossRef]
- Voter, A.F. Parallel replica method for dynamics of infrequent events. Phys. Rev. B 1998, 57, R13985–R13988. [Google Scholar] [CrossRef]
- Perriot, R.; Uberuaga, B.P.; Zamora, R.J.; Perez, D.; Voter, A.F. Evidence for percolation diffusion of cations and reordering in disordered pyrochlore from accelerated molecular dynamics. Nat. Commun. 2017, 8, 618. [Google Scholar] [CrossRef]
- Duda, E.V.; Kornich, G.V. Method for construction of a biased potential for hyperdynamic simulation of atomic systems. Phys. Solid State 2017, 59, 1900–1905. [Google Scholar] [CrossRef]
- Duda, E.V.; Kornich, G.V. Construction of a Changed Potential of Interatomic Interaction in the Case of Temperature-Accelerated Dynamics Simulation. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2018, 12, 825–833. [Google Scholar] [CrossRef]
- Duda, E.V.; Kornich, G.V. On the Combination of Methods of Temperature-Accelerated Dynamics and Hyperdynamics. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2019, 13, 667–669. [Google Scholar] [CrossRef]
- Derby, B.; Cooper, J.; Lach, T.; Martinez, E.; Kim, H.; Baldwin, J.K.; Kaoumi, D.; Edwards, D.J.; Schreiber, D.K.; Uberuaga, B.P.; et al. A pathway to synthesizing single-crystal Fe and FeCr films. Surf. Coat. Technol. 2020, 403, 126346. [Google Scholar] [CrossRef]
- Uberuaga, B.P.; Perez, D.; Voter, A.F. Accelerated Molecular Dynamics Methods for Long-Time Simulations in Materials. In Computational Materials, Chemistry, and Biochemistry: From Bold Initiatives to the Last Mile: In Honor of William A. Goddard’s Contributions to Science and Engineering; Shankar, S., Muller, R., Chen, G.H., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 137–156. [Google Scholar]
- Neyts, E.C.; Brault, P. Molecular Dynamics Simulations for Plasma-Surface Interactions. Plasma Process. Polym. 2017, 14, 1600145. [Google Scholar] [CrossRef]
- Fichthorn, K.A.; Miron, R.A.; Wang, Y.; Tiwary, Y. Accelerated molecular dynamics simulation of thin-film growth with the bond-boost method. J. Phys. Condens. Matter 2009, 21, 084212. [Google Scholar] [CrossRef] [PubMed]
- Duda, E.; Kornich, G. Simulation of Vacancy Diffusion in a Crystal by the Method of Temperature-Accelerated Dynamics. Met. Adv. Technol. 2020, 42, 341–350. [Google Scholar] [CrossRef]
- Duda, E.V.; Kornich, G.V. Hyperdynamics Simulation of the Diffusion of a Vacancy in a Crystal. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2020, 14, 1205–1207. [Google Scholar] [CrossRef]
- Gilmer, G.H.; Huang, H.; Roland, C. Thin film deposition: Fundamentals and modeling. Comput. Mater. Sci. 1998, 12, 354–380. [Google Scholar] [CrossRef]
- Gilmer, G.H.; Huang, H.; de la Rubia, T.D.; Torre, J.D.; Baumann, F. Lattice Monte Carlo models of thin film deposition. Thin Solid Films 2000, 365, 189–200. [Google Scholar] [CrossRef]
- Baumann, F.H.; Chopp, D.L.; de la Rubia, T.D.; Gilmer, G.H.; Greene, J.E.; Huang, H.; Kodambaka, S.; O’Sullivan, P.; Petrov, I. Multiscale Modeling of Thin-Film Deposition: Applications to Si Device Processing. MRS Bull. 2001, 26, 182–189. [Google Scholar] [CrossRef]
- Nita, F.; Mastail, C.; Abadias, G. Three-dimensional kinetic Monte Carlo simulations of cubic transition metal nitride thin film growth. Phys. Rev. B 2016, 93, 064107. [Google Scholar] [CrossRef]
- Rubinstein, R.Y.; Kroese, D.P. Simulation and the Monte Carlo method. In Wiley Series in Probability and Statistics, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Cheimarios, N.; To, D.; Kokkoris, G.; Memos, G.; Boudouvis, A.G. Monte Carlo and Kinetic Monte Carlo Models for Deposition Processes: A Review of Recent Works. Front. Phys. 2021, 9, 631918. [Google Scholar] [CrossRef]
- Sangiovanni, D.G.; Edström, D.; Hultman, L.; Chirita, V.; Petrov, I.; Greene, J.E. Dynamics of Ti, N, and TiNₓ (x = 1–3) admolecule transport on TiN(001) surfaces. Phys. Rev. B 2012, 86, 155443. [Google Scholar] [CrossRef]
- Mareus, R.; Mastail, C.; Nita, F.; Michel, A.; Abadias, G. Effect of temperature on the growth of TiN thin films by oblique angle sputter-deposition: A three-dimensional atomistic computational study. Comput. Mater. Sci. 2021, 197, 110662. [Google Scholar] [CrossRef]
- Kim, S.; An, H.; Oh, S.; Jung, J.; Kim, B.; Nam, S.K.; Han, S. Atomistic kinetic Monte Carlo simulation on atomic layer deposition of TiN thin film. Comput. Mater. Sci. 2022, 213, 111620. [Google Scholar] [CrossRef]
- Fu, T.; Peng, X.; Huang, C.; Yin, D.; Li, Q.; Wang, Z. Molecular dynamics simulation of VN thin films under indentation. Appl. Surf. Sci. 2015, 357, 643–650. [Google Scholar] [CrossRef]
- Helmersson, U.; Todorova, S.; Barnett, S.A.; Sundgren, J.; Markert, L.C.; Greene, J.E. Growth of single-crystal TiN/VN strained-layer superlattices with extremely high mechanical hardness. J. Appl. Phys. 1987, 62, 481–484. [Google Scholar] [CrossRef]
- Fu, T.; Peng, X.; Zhao, Y.; Li, T.; Li, Q.; Wang, Z. Molecular dynamics simulation of deformation twin in rocksalt vanadium nitride. J. Alloys Compd. 2016, 675, 128–133. [Google Scholar] [CrossRef]
- Fu, T.; Peng, X.; Wan, C.; Lin, Z.; Chen, X.; Hu, N.; Wang, Z. Molecular dynamics simulation of plasticity in VN(001) crystals under nanoindentation with a spherical indenter. Appl. Surf. Sci. 2017, 392, 942–949. [Google Scholar] [CrossRef]
- Edström, D.; Sangiovanni, D.G.; Hultman, L.; Petrov, I.; Greene, J.E.; Chirita, V. Large-scale molecular dynamics simulations of TiN/TiN(001) epitaxial film growth. J. Vac. Sci. Technol. A 2016, 34, 041509. [Google Scholar] [CrossRef]
- Sait, R.A.; Cross, R.B.M. Synthesis and characterization of sputtered titanium nitride as a nucleation layer for novel neural electrode coatings. Appl. Surf. Sci. 2017, 424, 290–298. [Google Scholar] [CrossRef]
- Hernández, N.C.; Sanz, J.F. Interaction potentials from periodic density-functional theory calculations: Molecular-dynamics simulations of Au clusters deposited on the TiN (001) surface. J. Chem. Phys. 2005, 123, 244706. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Miura, H. Molecular dynamics analysis of adhesion strength of interfaces between thin films. J. Mater. Res. 2001, 16, 1789–1794. [Google Scholar] [CrossRef]
- Xu, Z.H.; Yuan, L.; Shan, D.B.; Guo, B. A molecular dynamics simulation of TiN film growth on TiN(001). Comput. Mater. Sci. 2011, 50, 1432–1436. [Google Scholar] [CrossRef]
- Li, J.; Lin, J.; Ma, Q.; Luan, H.; Zhu, L.; Bai, R.; Dong, G.; Wang, D.; Guan, Y.; Zhang, X. Molecular Dynamics Simulation of the Incident Energy Effect on the Properties of TiN Films. Coatings 2023, 13, 794. [Google Scholar] [CrossRef]
- Amini, H.; Gholizadeh, P.; Poursaeidi, E.; Davoodi, J. A molecular dynamics simulation of Ti–TiN multilayer deposition on FeCrNi(001) alloy substrate. Vacuum 2021, 193, 110519. [Google Scholar] [CrossRef]
- Methary, J.; Sathiyanarayanan, R.; Li, R.; Stout, P.J. Stoichiometry tuning of TaN films through ion treatment: Molecular dynamics study. J. Vac. Sci. Technol. A 2021, 39, 062601. [Google Scholar] [CrossRef]
- Nikravesh, M.; Akbari, G.H.; Poladi, A. A comprehensive study on the surface tribology of Ta thin film using molecular dynamics simulation: The effect of TaN interlayer, power and temperature. Tribol. Int. 2017, 105, 185–192. [Google Scholar] [CrossRef]
- Firouzabadi, S.S.; Dehghani, K.; Naderi, M.; Mahboubi, F. Numerical investigation of sputtering power effect on nano-tribological properties of tantalum-nitride film using molecular dynamics simulation. Appl. Surf. Sci. 2016, 367, 197–204. [Google Scholar] [CrossRef]
- Pogrebnjak, A.; Ivashchenko, V.; Bondar, O.; Beresnev, V.; Sobol, O.; Załęski, K.; Jurga, S.; Coy, E.; Konarski, P.; Postolnyi, B. Multilayered vacuum-arc nanocomposite TiN/ZrN coatings before and after annealing: Structure, properties, first-principles calculations. Mater. Charact. 2017, 134, 55–63. [Google Scholar] [CrossRef]
- Pogrebnjak, A.D.; Eyidi, D.; Abadias, G.; Bondar, O.V.; Beresnev, V.M.; Sobol, O.V. Structure and properties of arc evaporated nanoscale TiN/MoN multilayered systems. Int. J. Refract. Met. Hard Mater. 2015, 48, 222–228. [Google Scholar] [CrossRef]
- Jiang, E.; Sun, C.; Li, J.; Liu, Y. The structures and magnetic properties of FeN films prepared by the facing targets sputtering method. J. Appl. Phys. 1989, 65, 1659–1663. [Google Scholar] [CrossRef]
- Heryadi, D.; Schwingenschlögl, U. Formation dynamics of FeN thin films on Cu(100). Chem. Phys. Lett. 2012, 523, 78–82. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, G.; Wang, J.-P. Study of γ\textasciiacutex-F4N Annealing Process Through Molecular Dynamics Modeling. In TMS 2022 151st Annual Meeting & Exhibition Supplemental Proceedings; Springer: Cham, Switzerland, 2022. [Google Scholar]
- Amaya-Roncancio, S.; Arias-Mateus, D.F.; Segura-Giraldo, B.; la Roche, J.D.; Restrepo-Parra, E. Molecular dynamics simulation of the nanoindentation process in Cr/CrN and (Cr/CrN)2 thin films. Contemp. Eng. Sci. 2018, 11, 4617–4635. [Google Scholar] [CrossRef]
- Kang, Q.; Wang, G.; Liu, Q.; Sui, X.; Liu, Y.; Chen, Y.; Luo, S.; Li, Z. Investigation for oxidation mechanism of CrN: A combination of DFT and ab initio molecular dynamics study. J. Alloys Compd. 2021, 885, 160940. [Google Scholar] [CrossRef]
- Guo, F.; Wang, J.; Du, Y.; Holec, D.; Ou, P.; Zhou, H.; Chen, L.; Kong, Y. Structural evolution of oxygen on the surface of TiAlN: Ab initio molecular dynamics simulations. Appl. Surf. Sci. 2019, 470, 520–525. [Google Scholar] [CrossRef]
- van de Walle, A.; Tiwary, P.; de Jong, M.; Olmsted, D.L.; Asta, M.; Dick, A.; Shin, D.; Wang, Y.; Chen, L.-Q.; Liu, Z.-K. Efficient stochastic generation of special quasirandom structures. Calphad 2013, 42, 13–18. [Google Scholar] [CrossRef]
- Pogrebnjak, A.D.; Bondar, O.V.; Abadias, G.; Ivashchenko, V.; Sobol, O.V.; Jurga, S.; Coy, E. Structural and mechanical properties of NbN and Nb-Si-N films: Experiment and molecular dynamics simulations. Ceram. Int. 2016, 42, 11743–11756. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goncharov, A.; Yunda, A.; Kolinko, I.; Kornich, G.; Shyrokorad, D. Modeling the Deposition of Thin Films of Transition Metal Nitrides. Coatings 2023, 13, 2035. https://doi.org/10.3390/coatings13122035
Goncharov A, Yunda A, Kolinko I, Kornich G, Shyrokorad D. Modeling the Deposition of Thin Films of Transition Metal Nitrides. Coatings. 2023; 13(12):2035. https://doi.org/10.3390/coatings13122035
Chicago/Turabian StyleGoncharov, Alexander, Andrii Yunda, Ivan Kolinko, Grygoriy Kornich, and Dmytro Shyrokorad. 2023. "Modeling the Deposition of Thin Films of Transition Metal Nitrides" Coatings 13, no. 12: 2035. https://doi.org/10.3390/coatings13122035
APA StyleGoncharov, A., Yunda, A., Kolinko, I., Kornich, G., & Shyrokorad, D. (2023). Modeling the Deposition of Thin Films of Transition Metal Nitrides. Coatings, 13(12), 2035. https://doi.org/10.3390/coatings13122035