
Sintering of Potassium Doped Hydroxy-Fluorapatite Bioceramics

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Powder Characterization
2.3. Sintering and Mechanical Assays
3. Results
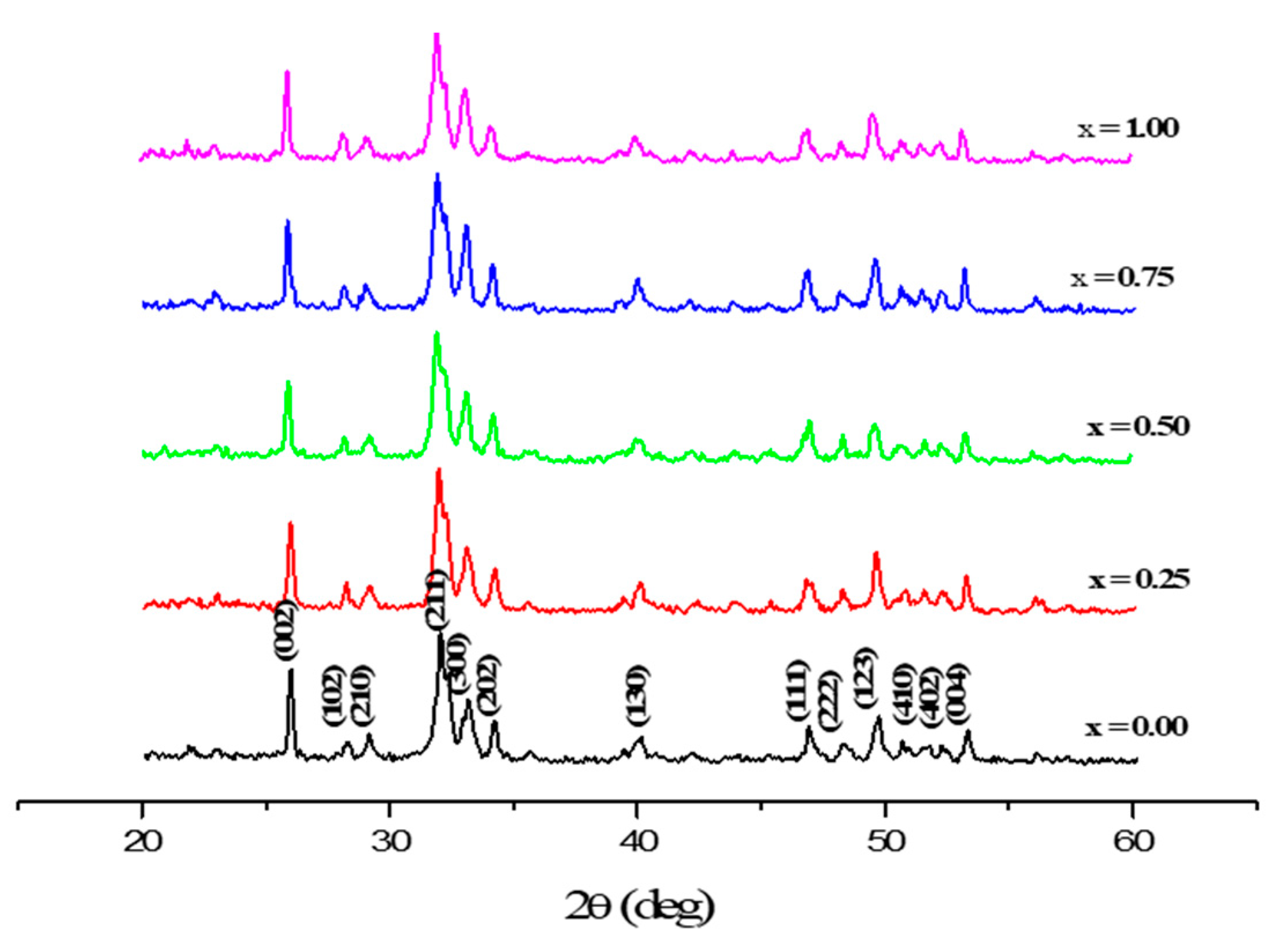
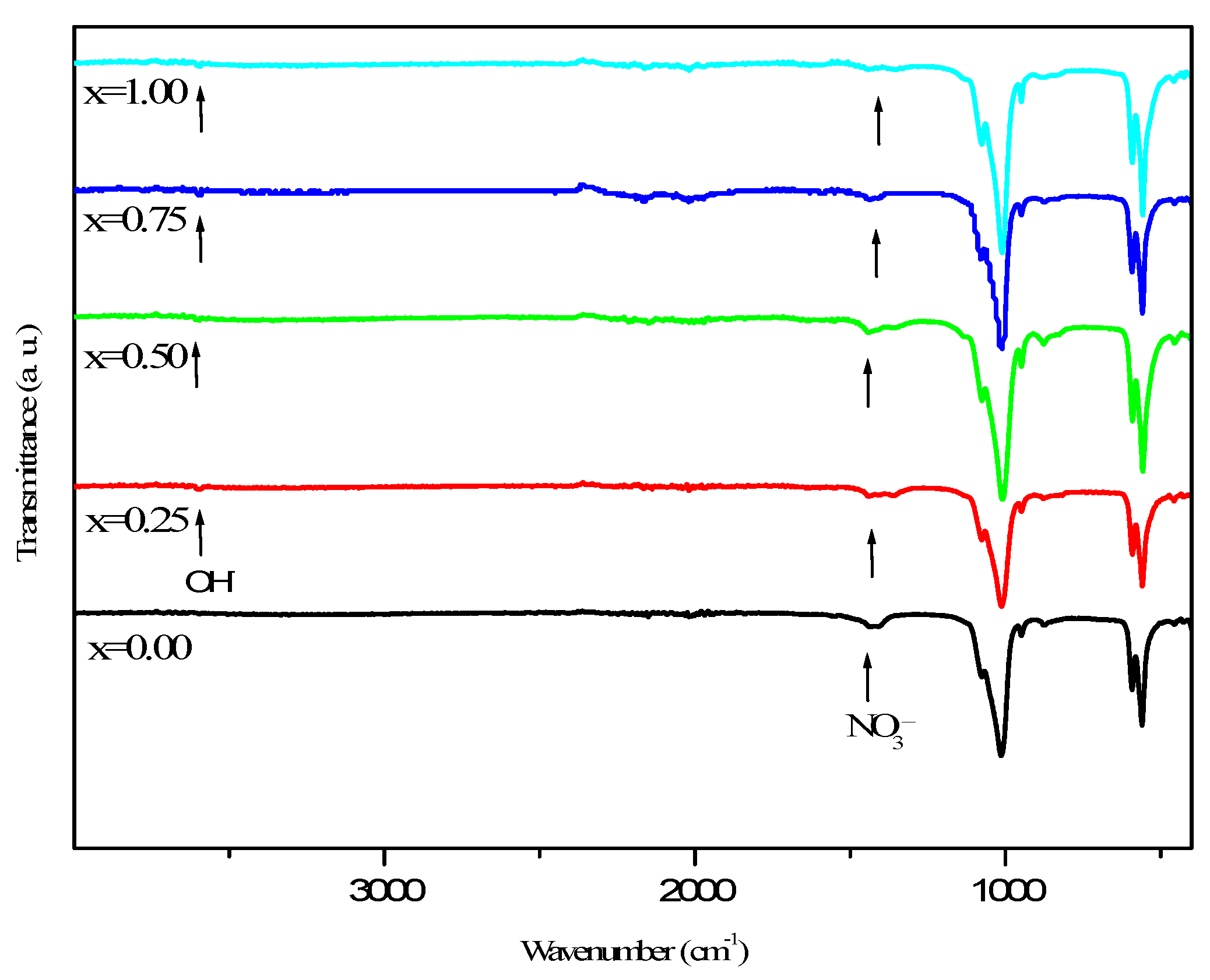
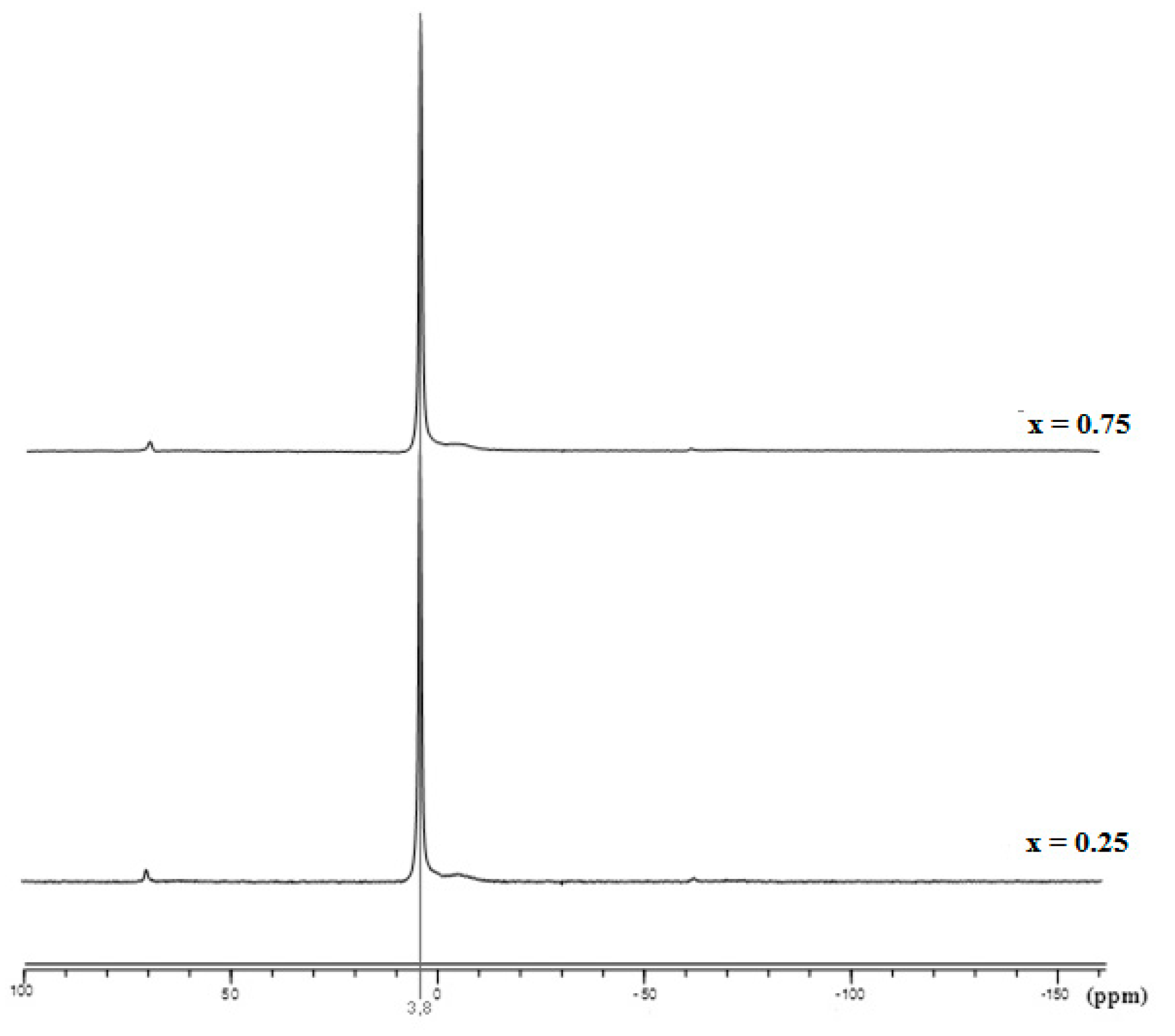
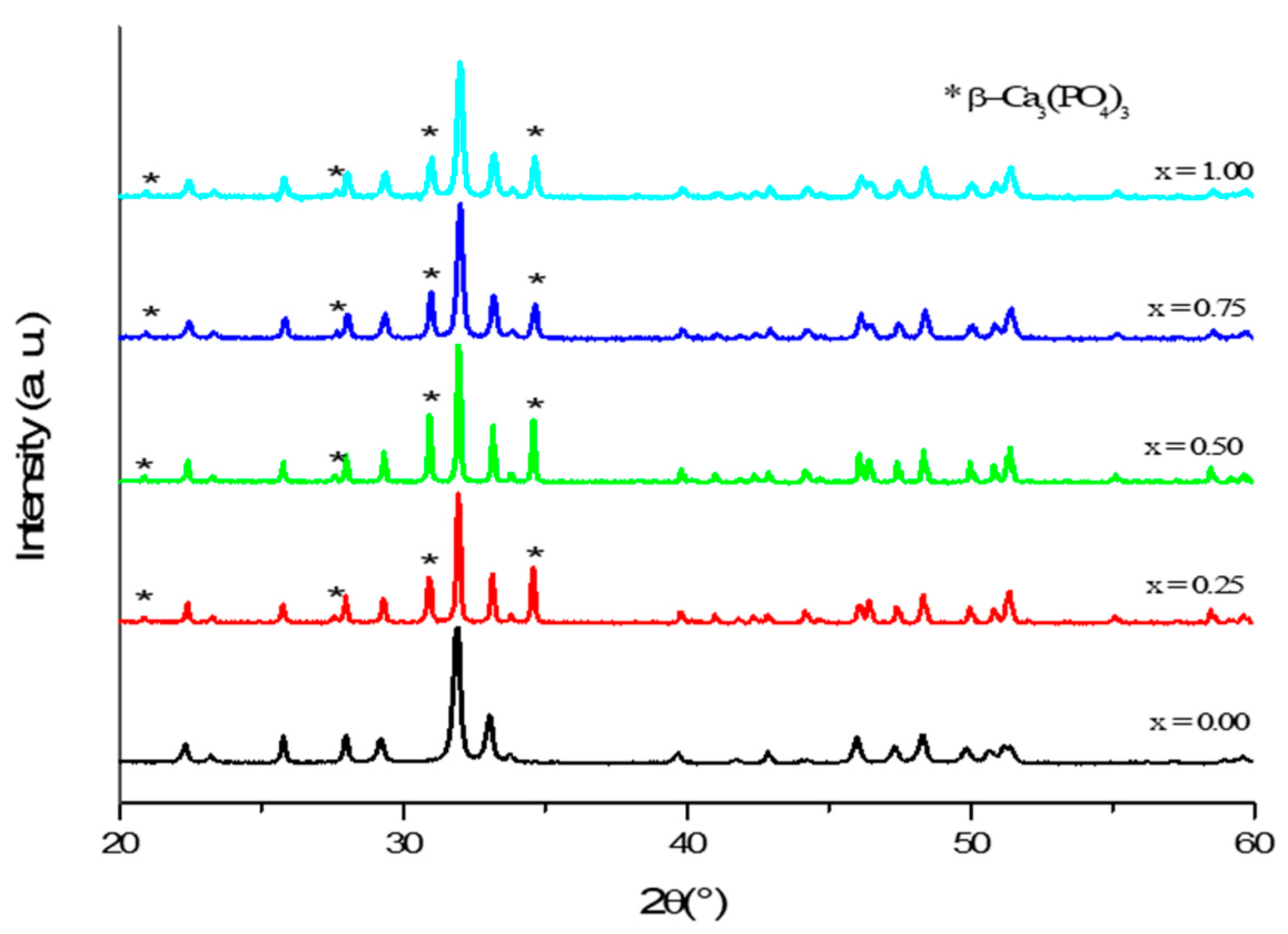
3.1. Phase Analysis
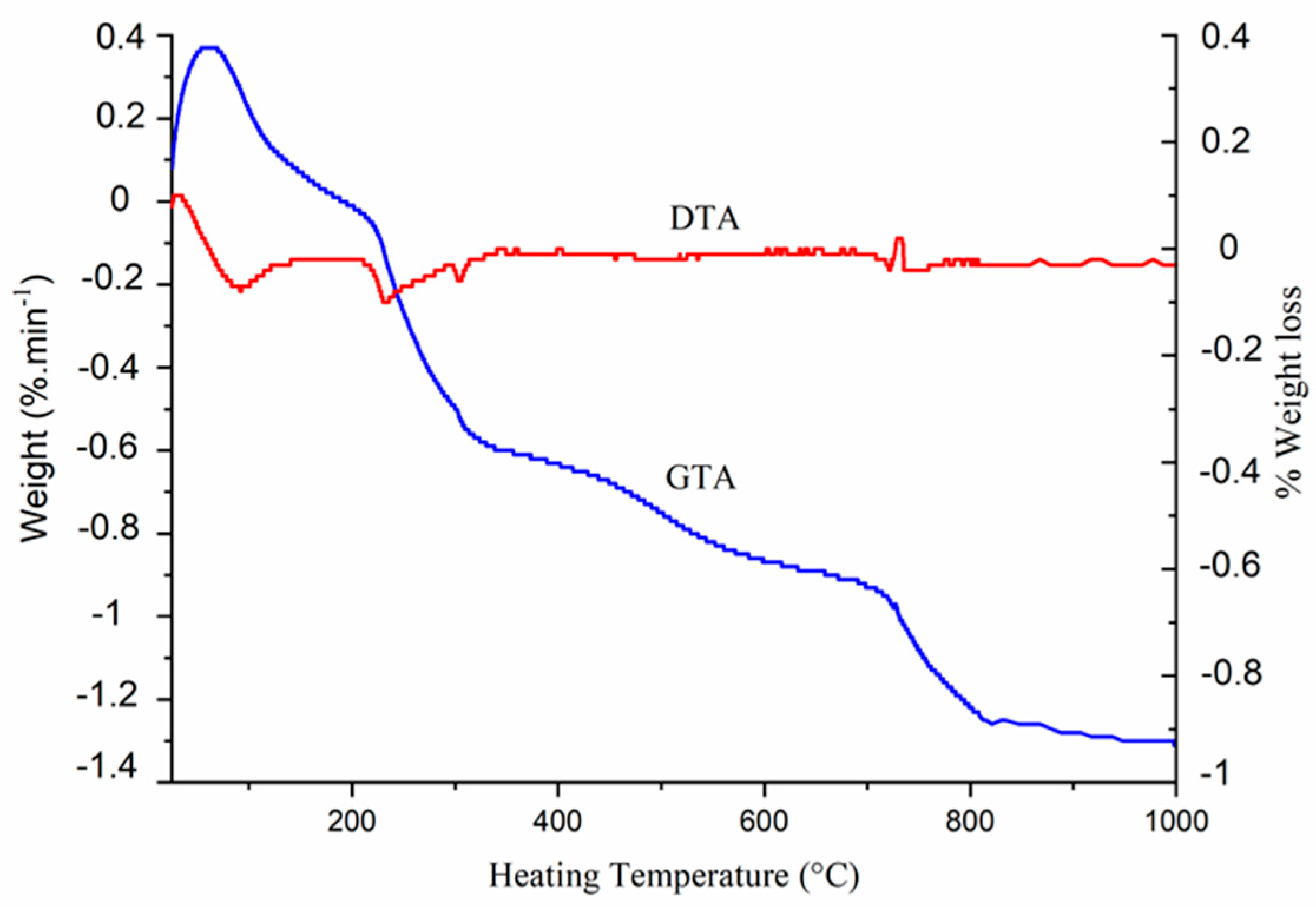
3.2. Thermal Behavior
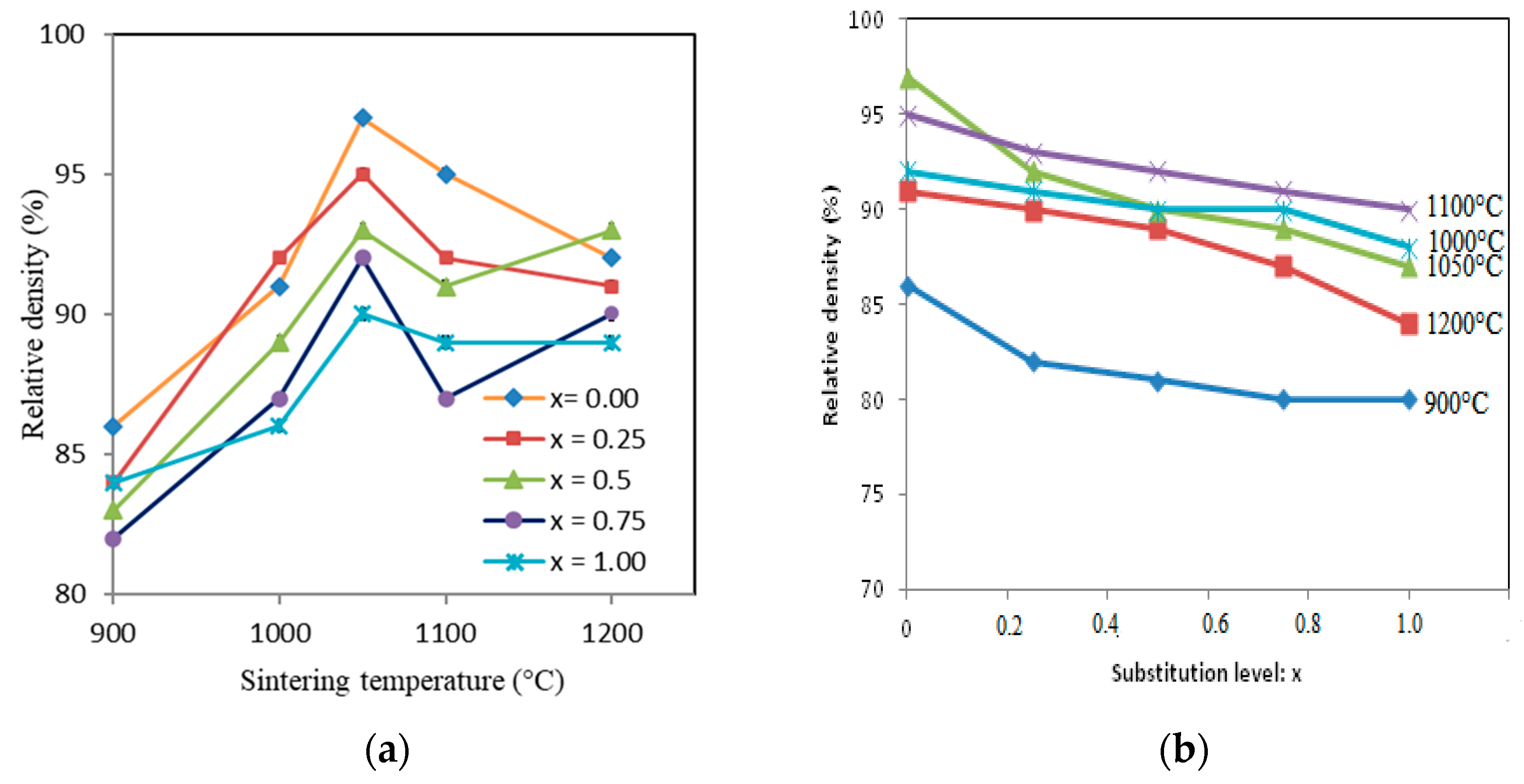
3.3. Sintering
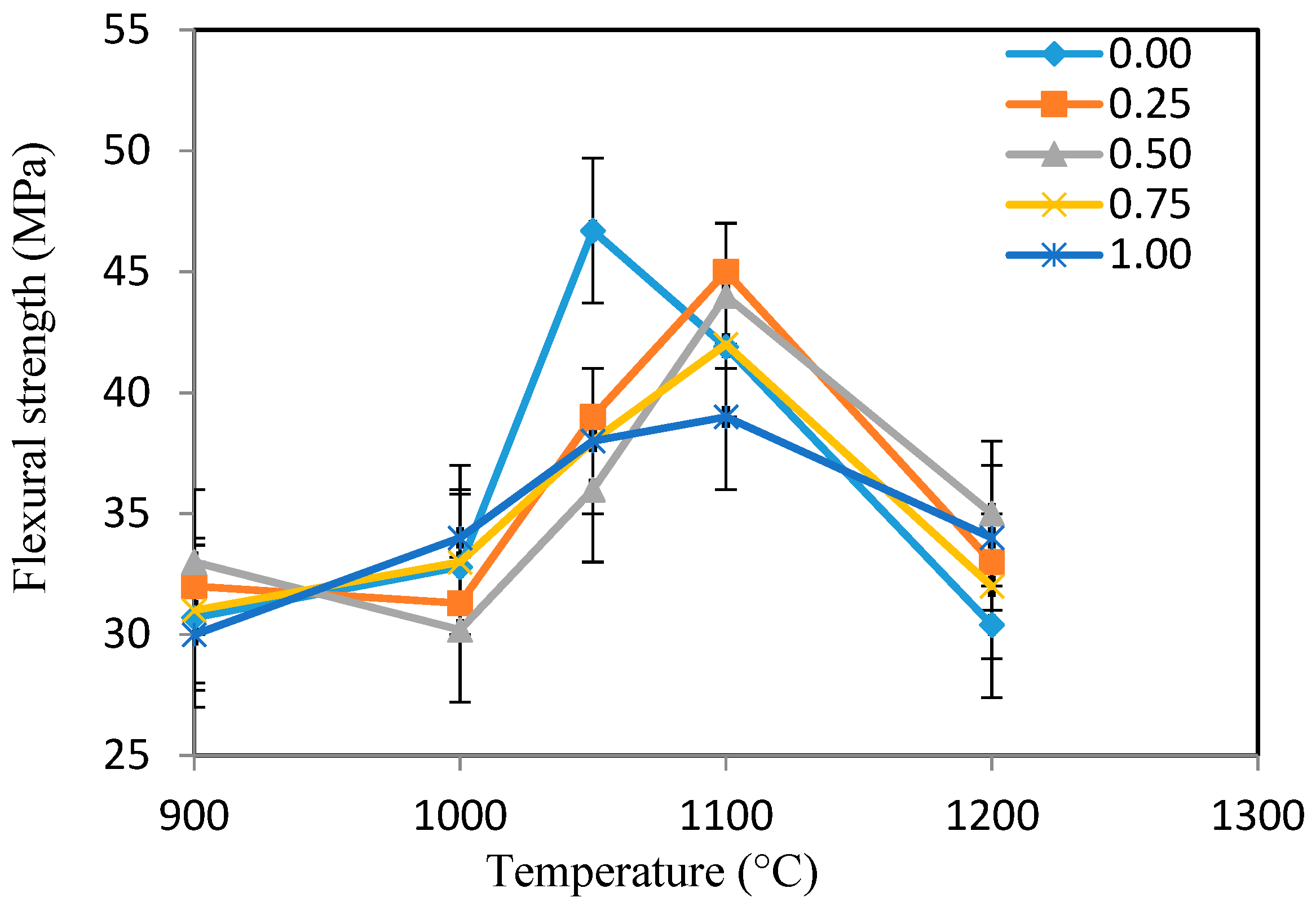
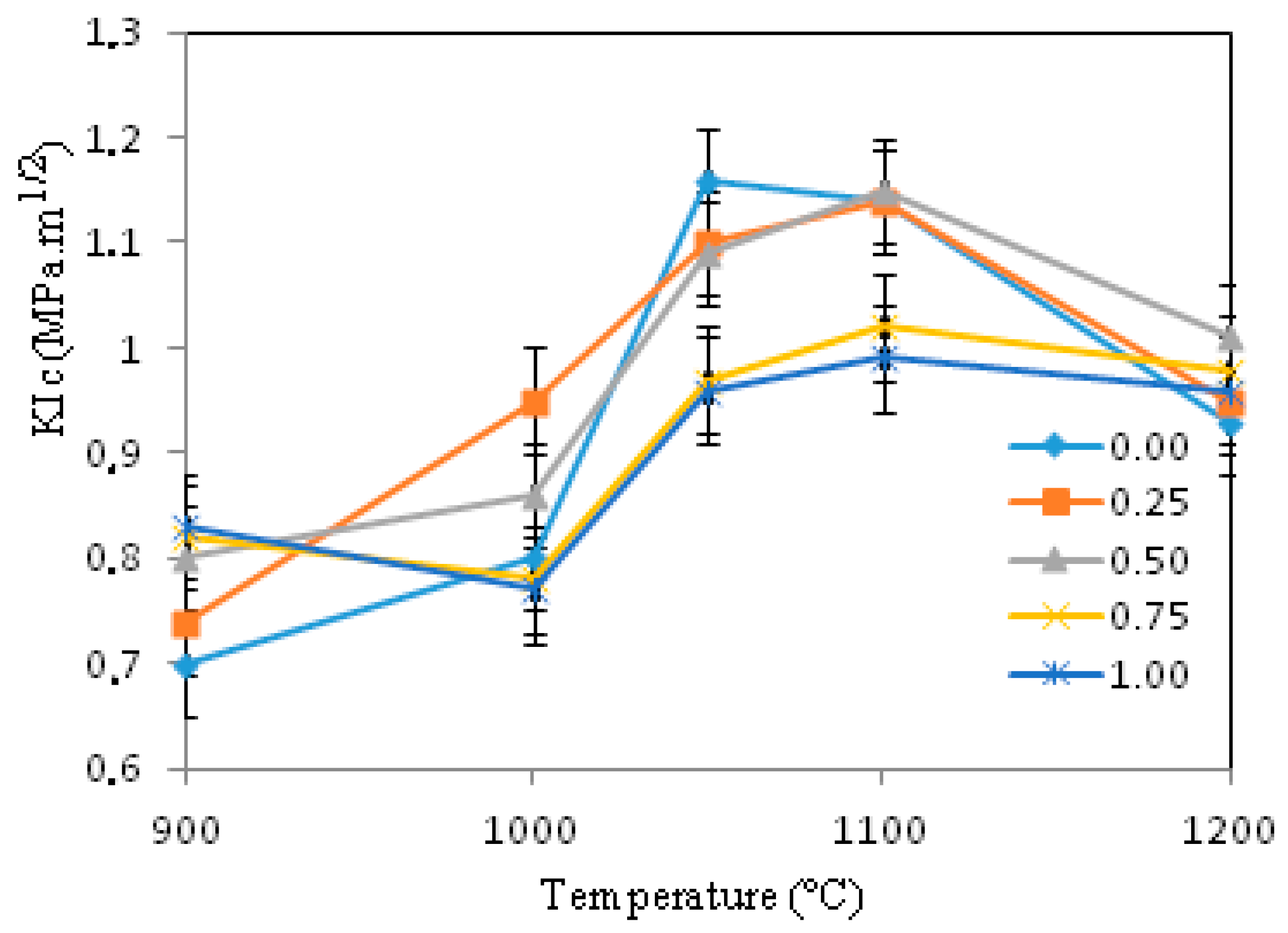
3.4. Mechanical Characterizations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boskey, A.L. Mineralization of bones and teeth. Elements 2007, 3, 385–391. [Google Scholar] [CrossRef]
- Young, M.F. Bone matrix proteins: Their function, regulation, and relationship to osteoporosis. Osteoporos. Int. 2003, 14 (Suppl. 3), S35–S42. [Google Scholar] [CrossRef]
- Currey, J.D. The structure and mechanics of bone. J. Mater. Sci. 2012, 47, 41–54. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C.; Drouet, C.; Glimcher, M.J. Bone mineral: Update on chemical composition and structure. Osteoporos. Int. 2009, 20, 1013–1021. [Google Scholar] [CrossRef]
- Bolger, M.W.; Romanowicz, G.E.; Kohn, D.H. Advancements in composition and structural characterization of bone to inform mechanical outcomes and modeling. Curr. Opin. Biomed. Eng. 2019, 11, 76–84. [Google Scholar] [CrossRef]
- Jarcho, M. Calcium phosphate ceramics as hard tissue prosthetics. Clin. Orthop. Relat. Res. 1981, 157, 259–278. [Google Scholar] [CrossRef]
- Daculsi, G.; Passuti, N.; Martin, S. Macroporous calcium phosphate ceramic for long bone surgery in humans and dogs. Clinical and histological study. J. Biomed. Mater. Res. 1990, 24, 379–396. [Google Scholar] [CrossRef]
- Khairoun, I.; Boltong, M.G.; Driessens, F.C.M.; Planell, J.A. Some factors controlling the injectability of calcium phosphate bone cements. J. Mater. Sci. Mater. Med. 1998, 9, 425–428. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C.; Drouet, C.; Sfihi, H.; Barroug, A. Physico-chemical properties of nanocrystalline apatites: Implications for biominerals and biomaterials. Mater. Sci. Eng. C 2007, 27, 198–205. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates in nature, biology and medicine. Materials 2009, 2, 399–498. [Google Scholar] [CrossRef]
- Cantaert, B.; Beniash, E.; Meldrum, F.C. The role of polyaspartic acid in the precipitation of calcium phosphate in confinement. J. Mater. Chem. B 2013, 1, 6586–6595. [Google Scholar] [CrossRef]
- He, F.; Yang, G.; Wang, X.; Zhao, S. Effect of electrochemically deposited nanohydroxyapatite on bone bonding of sandblasted/dual acid-etched titanium implant. Int. J. Oral Maxillofac. Implant. 2009, 24, 790–799. [Google Scholar]
- Damien, E.; Revell, P.A. Coralline hydroxyapatite bone graft substitute: A review of experimental studies and biomedical applications. Appl. Biomater. Biomech. 2004, 2, 65–73. [Google Scholar]
- Bucholz, R.W.; Carlton, A.; Holmes, R.E. Hydroxyapatite and tricalcium phosphate bone graft substitutes. Orthop. Clin. N. Am. 1987, 18, 323–334. [Google Scholar] [CrossRef]
- Chang, M.C.; Tanaka, J. FT-IR study for hydroxyapatite/collagen nanocomposite cross-linked by glutaraldehyde. Biomaterials 2002, 23, 4811–4818. [Google Scholar] [CrossRef]
- Nassif, N.; Martineau, F.; Syzgantseva, O.; Gobeaux, F.; Willinger, M.; Coradin, T.; Cassaignon, S.; Azaïs, T.; Giraud-Guille, M.M. In vivo inspired conditions to synthesize biomimetic hydroxyapatite. Chem. Mater. 2010, 22, 3653–3663. [Google Scholar] [CrossRef]
- Aoba, T. The effect of fluoride on apatite structure and growth. Crit. Rev. Oral. Biol. Med. 1997, 8, 136–153. [Google Scholar] [CrossRef]
- Featherstone, J.D. Fluoride, remineralization and root caries. Am. J. Dent. 1994, 7, 271–274. [Google Scholar]
- Borkowski, L.; Przekora, A.; Belcarz, A.; Palka, K.; Jozefaciuk, G.; Lubek, T.; Jojczuk, M.; Nogalski, A.; Ginalska, G. Fluorapatite ceramics for bone tissue regeneration: Synthesis, characterization and assessment of biomedical potential. Mater. Sci. Eng. C 2020, 116, 111211. [Google Scholar] [CrossRef]
- Ten Cate, J.M.; Featherstone, J.D. Mechanistic aspects of the interactions between fluoride and dental enamel. Rev. Oral. Biol. Med. 1991, 2, 283–296. [Google Scholar] [CrossRef]
- Jones, S.P.; Cheuk, G.C.; Georgiou, G.; Moles, D.R. Comparison of fluoridated apatites with pure hydroxyapatite as potential biomimetic alternatives to enamel for laboratory-based bond strength studies. Aust. Orthodontic J. 2009, 25, 12–18. [Google Scholar]
- Nikčević, I.; Jokanović, V.; Mitrić, M.; Nedić, Z.; Makovec, D.; Uskoković, D. Mechanochemical synthesis of nanostructured fluorapatite/fluorhydroxyapatite and carbonated fluorapatite/fluorhydroxyapatite. J. Solid State Chem. 2004, 177, 2565–2574. [Google Scholar] [CrossRef]
- Azami, M.; Jalilifiroozinezhad, S.; Mozafari, M.; Rabiee, M. Synthesis and solubility of calcium fluoride/hydroxy-fluorapatite nanocrystals for dental applications. Ceram. Int. 2011, 37, 2007–2014. [Google Scholar] [CrossRef]
- Ajcharanukul, O.; Kraivaphan, P.; Wanachantararak, S.; Vongsavan, N.; Matthews, B. Effects of potassium ions on dentine sensitivity in man. Arch. Oral. Biol. 2007, 52, 632–639. [Google Scholar] [CrossRef]
- Zhu, K.; Devine, A.; Prince, R.L. The effects of high potassium consumption on bone mineral density in a prospective cohort study of elderly postmenopausal women. Osteoporos. Int. 2009, 20, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Bushinsky, D.A.; Riordon, D.R.; Chan, J.S. Decreased potassium stimulates bone resorption. Am. J. Physiol. 1997, 272, F781–F788. [Google Scholar] [CrossRef]
- Wiesmann, H.P.; Plate, U.; Zierold, K.; Hohling, H.J. Potassium is involved in apatite biomineralization. J. Dent. Res. 1998, 77, 1654–1657. [Google Scholar] [CrossRef]
- Kannan, S.; Ventura, J.M.G.; Ferreira, J.M.F. Synthesis and thermal stability of potassium substituted hydroxyapatites and hydroxyapatite/β-tricalciumphosphate mixtures. Ceram. Int. 2007, 33, 1489–1494. [Google Scholar] [CrossRef]
- Yokota, T.; Honda, M.; Aizawa, M. Fabrication of potassium-substituted hydroxyapatite ceramics via ultrasonic spray-pyrolysis route. Phosphorus Res. Bul. 2017, 33, 35–40. [Google Scholar] [CrossRef][Green Version]
- Leroy, N.; Bres, E. Structure and substitutions in fluorapatite. Eur. Cell Mater. 2001, 2, 36–48. [Google Scholar] [CrossRef]
- El Feki, H.; Amami, M.; Ben Salah, A.; Jemal, M. Synthesis of potassium chloroapatites, IR, X-ray and Raman studies. Phys. Stat. Sol. 2004, 1, 1985–1988. [Google Scholar] [CrossRef]
- El Feki, H.; Naddari, T.; Savariault, J.M.; Ben Salah, A. Localisation of potassium in substituted lead hydroxyapatite: Pb9.30K0.60(PO4)6(OH)1.20 by X-ray diffraction. Solid State Sci. 2000, 2, 725–733. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1994; Volume 18, 404p. [Google Scholar]
- Evans, A.G.; Charles, E.A. Fracture toughness determination by indentation. J. Am. Ceram. Soc. 1976, 59, 371–372. [Google Scholar] [CrossRef]
- Hidouri, M.; Dorozhkin, S.V.; Albeladi, N. Thermal behavior, sintering and mechanical characterization of multiple ion-substituted hydroxyapatite bioceramics. J. Inorg. Organomet. Polym. Mater. 2018, 29, 87–100. [Google Scholar] [CrossRef]
- Okazaki, M. Heterogeneous synthesis of fluoridated hydroxyapatites. Biomaterials 1992, 13, 749–754. [Google Scholar] [CrossRef]
- Nsar, S.; Hassine, A.; Bouzouita, K. Sintering and mechanical properties of magnesium and fluorine co-substituted hydroxyapatites. J. Biomater. Nanobiotechnol. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Hata, M.; Okada, K.; Iwai, S. Cadmium hydroxyapatite. Acta Cryst. B 1978, 34, 3062–3064. [Google Scholar] [CrossRef]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Hidouri, M.; Dorozhkin, S.V. Structure and thermal stability of sodium and carbonate-co-substituted strontium hydroxyfluorapatites. N. J. Chem. 2018, 42, 8469–8477. [Google Scholar] [CrossRef]
- Xu, J.L.; Khor, K.A. Chemical analysis of silica doped hydroxyapatite biomaterials consolidated by a spark plasma sintering method. Inorg. Biochem. 2007, 101, 187–195. [Google Scholar] [CrossRef]
- Kingery, W.D.; Bowen, H.K.; Uhlmann, D.R. Introduction to Ceramics; Wiley: New York, NY, USA, 1976; 1056p. [Google Scholar]
- Martin, R.I.; Brown, P.W. Mechanical properties of hydroxyapatite formed at physiological temperature. J. Mater. Sci. Mater. Med. 1995, 6, 138–143. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x | a (pm) | c (pm) | V (106pm3) | D300 | D002 | D022 |
|---|---|---|---|---|---|---|
| 0.00 | 934.3(1) | 685.4(1) | 517.0(1) | 462(2) | 437(2) | 423(4) |
| 0.25 | 932.4(2) | 685.2(2) | 515.4(2) | 448(2) | 432(3) | 419(3) |
| 0.50 | 931.8(1) | 685.7(1) | 514.69(3) | 434(3) | 442(2) | 408(2) |
| 0.75 | 931.3(1) | 686.1(1) | 515.32(1) | 413(3) | 436(3) | 402(3) |
| 1.00 | 930.7(2) | 686.3(2) | 514.81(2) | 407(4) | 448(4) | 393(3) |
| x | Ca | K | P | F |
Ca/P Molar Ratio |
(K + Ca)/P Molar Ratio |
|---|---|---|---|---|---|---|
| 0.00 | 9.89(2) | - | 5.94(3) | 1.97(2) | 1.67(3) | 1.66(3) |
| 0.25 | 9.70(3) | 0.22(2) | 5.95(3) | 1.46(3) | 1.64(3) | 1.66(3) |
| 0.5 | 9.42(3) | 0.45(3) | 5.94(3) | 0.95(2) | 1.59(3) | 1.66(3) |
| 0.75 | 9.20(2) | 0.71(3) | 5.95(2) | 0.47(3) | 1.54(2) | 1.66(3) |
| 1.00 | 8.95(2) | 0.95(3) | 5.94(3) | traces | 1.50(3) | 1.66(3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slimen, J.B.; Hidouri, M.; Ghouma, M.; Salem, E.B.; Dorozhkin, S.V. Sintering of Potassium Doped Hydroxy-Fluorapatite Bioceramics. Coatings 2021, 11, 858. https://doi.org/10.3390/coatings11070858
Slimen JB, Hidouri M, Ghouma M, Salem EB, Dorozhkin SV. Sintering of Potassium Doped Hydroxy-Fluorapatite Bioceramics. Coatings. 2021; 11(7):858. https://doi.org/10.3390/coatings11070858
Chicago/Turabian StyleSlimen, Jihen Ben, Mustapha Hidouri, Marwa Ghouma, Ezzedine Ben Salem, and Sergey V. Dorozhkin. 2021. "Sintering of Potassium Doped Hydroxy-Fluorapatite Bioceramics" Coatings 11, no. 7: 858. https://doi.org/10.3390/coatings11070858
APA StyleSlimen, J. B., Hidouri, M., Ghouma, M., Salem, E. B., & Dorozhkin, S. V. (2021). Sintering of Potassium Doped Hydroxy-Fluorapatite Bioceramics. Coatings, 11(7), 858. https://doi.org/10.3390/coatings11070858