Putative Antimicrobial Peptides of the Posterior Salivary Glands from the Cephalopod Octopus vulgaris Revealed by Exploring a Composite Protein Database

 ,
,  ,
, 
 , ,
, ,  ,
, 

Abstract
1. Introduction
2. Results and Discussion
2.1. LC-MS/MS Analyses and Protein Identification
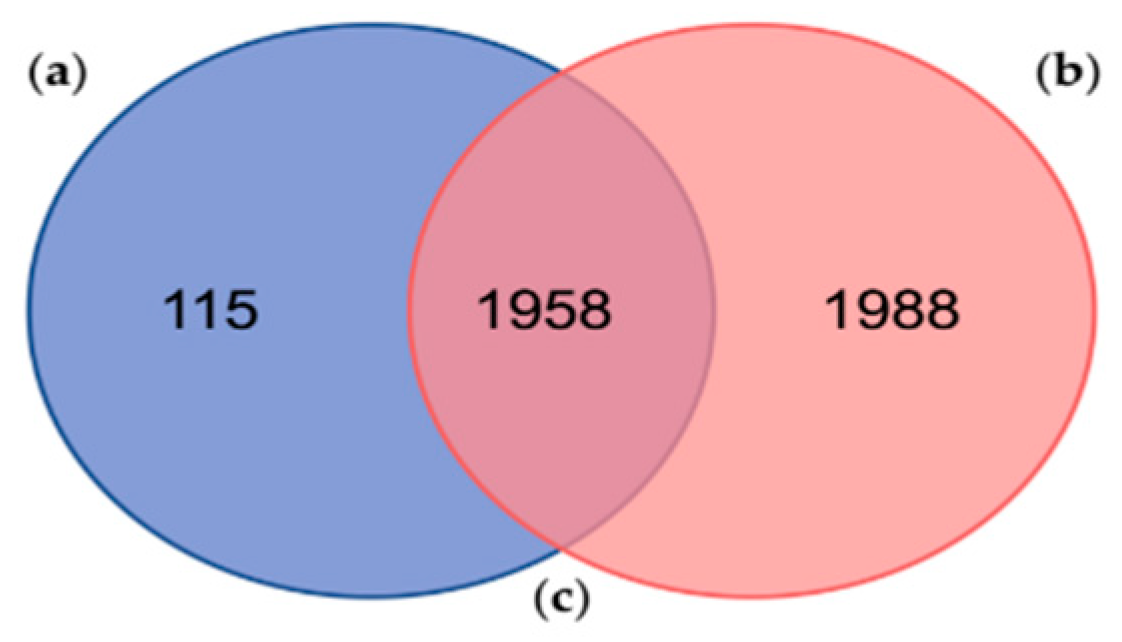
2.2. Comparison with a Similar Proteogenomic Study of the PSGs from O. vulgaris
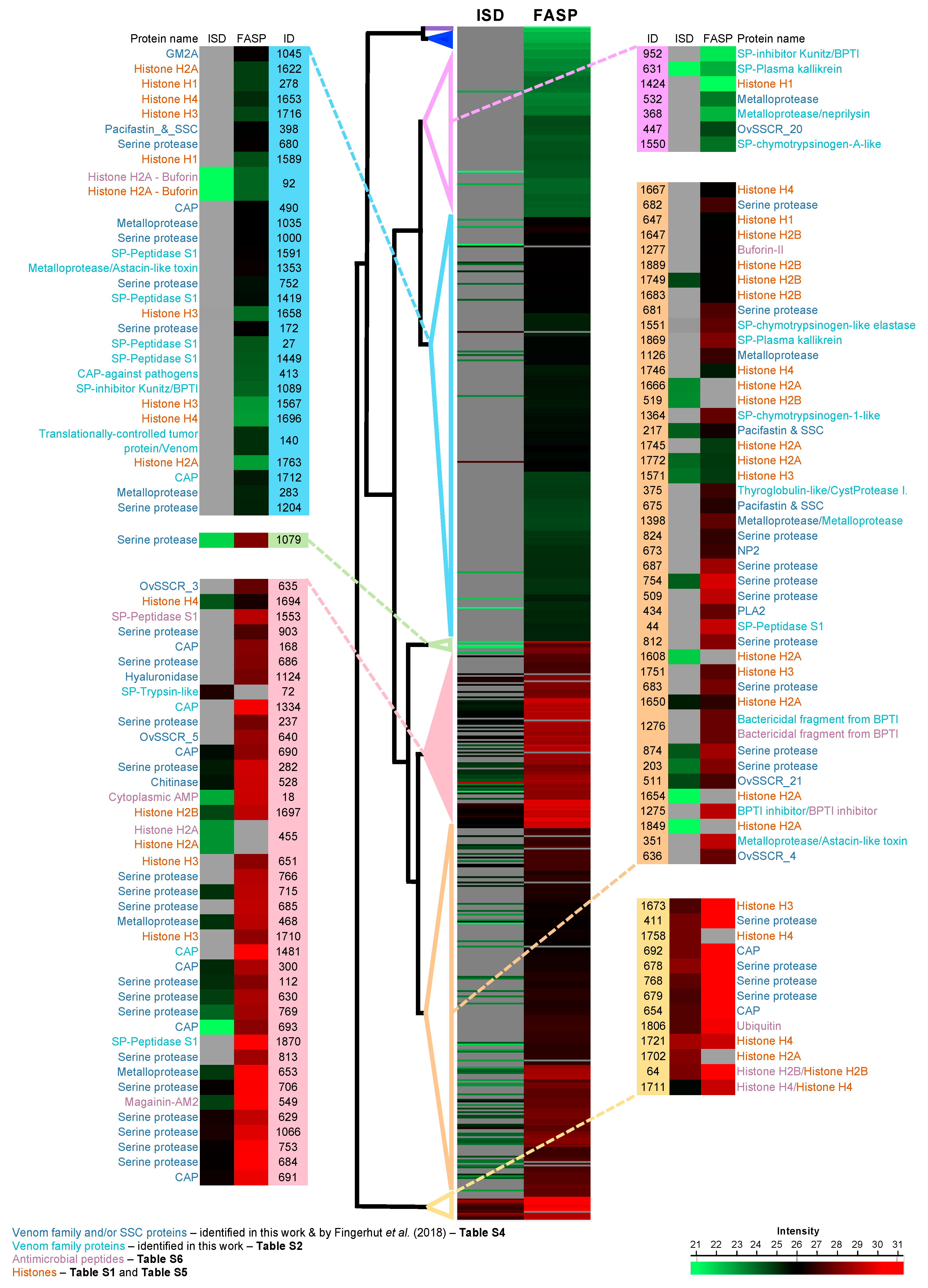
2.3. Leading Proteins Found with Homology to Venom Protein Families
2.4. Histones and Antimicrobial Peptides Found in the Proteome of the Psgs from Octopus vulgaris
Are Amps and Histones Part of the Host Defense Barrier in the Salivary Apparatus of the Octopus vulgaris?
3. Materials and Methods
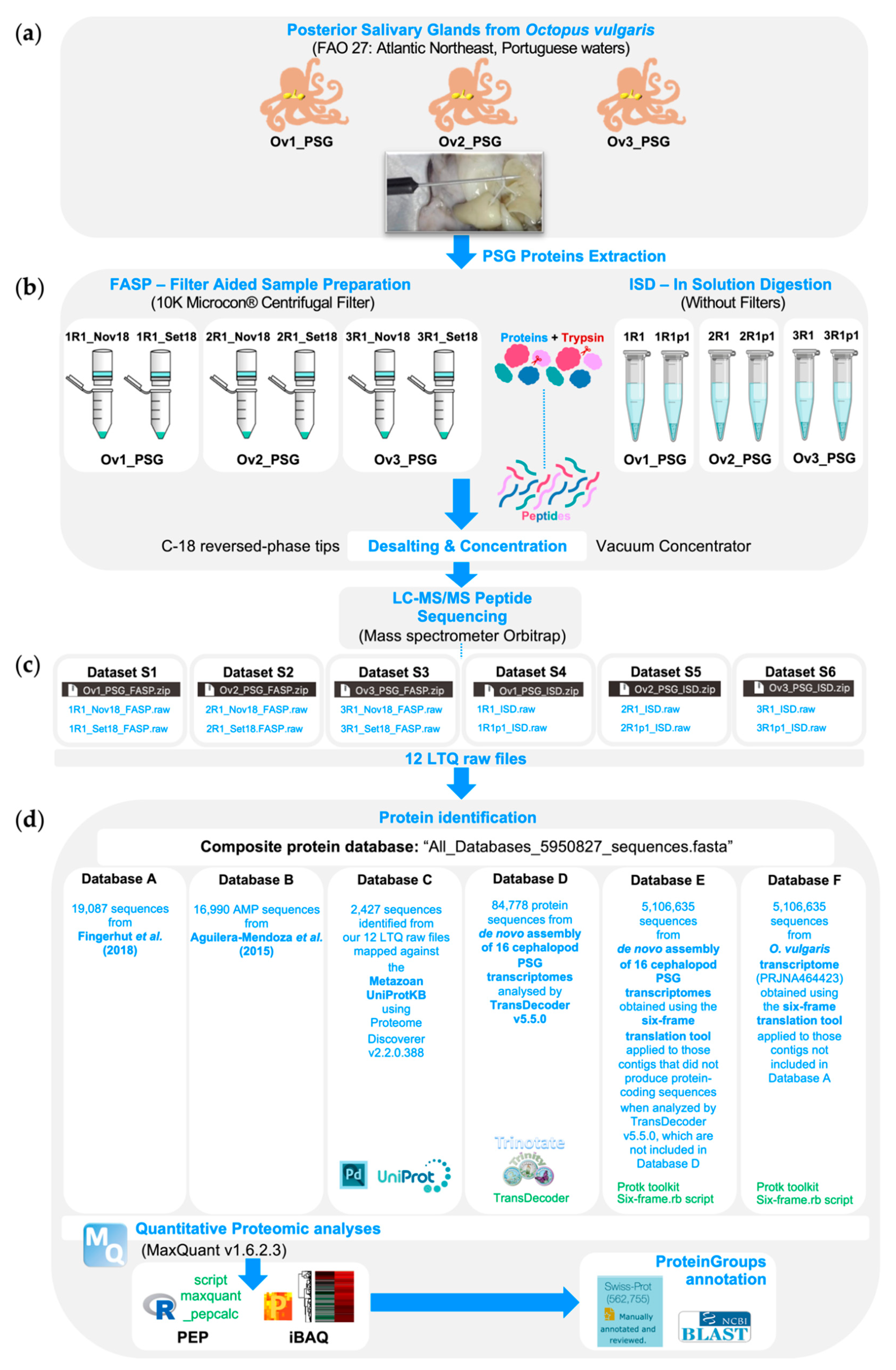
3.1. O. vulgaris Sampling and Protein Extraction
3.2. Sample Preparation for LC-MS/MS Analyses
3.2.1. Filter Aided Sample Preparation
3.2.2. In Solution Digestion
3.3. LC-MS/MS Analyses
3.4. Protein Identification
3.4.1. Quantitative Proteomic Analyses
3.4.2. Maxquant Proteingroups Annotation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Almeida, D.; Maldonado, E.; Vasconcelos, V.; Antunes, A. Adaptation of the Mitochondrial Genome in Cephalopods: Enhancing Proton Translocation Channels and the Subunit Interactions. PLoS ONE 2015, 10, e0135405. [Google Scholar] [CrossRef] [PubMed]
- Albertin, C.B.; Bonnaud, L.; Brown, C.T.; Crookes-Goodson, W.J.; da Fonseca, R.R.; Di Cristo, C.; Dilkes, B.P.; Edsinger-Gonzales, E.; Freeman, R.M.; Hanlon, R.T.; et al. Cephalopod genomics: A plan of strategies and organization. Stand. Genom. Sci. 2012, 7, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Hoving, H.-J.T.; Perez, J.A.A.; Bolstad, K.S.R.; Braid, H.E.; Evans, A.B.; Fuchs, D.; Judkins, H.; Kelly, J.T.; Marian, J.E.A.R.; Nakajima, R.; et al. The Study of Deep-Sea Cephalopods. In Advances in Marine Biology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 67, pp. 235–359. ISBN 0065-2881. [Google Scholar]
- Nateewathana, A.; Munprasit, A.; Dithachey, P. Systematics and distribution of oceanic cephalopods in the South China Sea, Area III: Western Philippines. In Proceedings of the Third Technical Seminar on Marine Fishery Resources Survey in the South China Sea, Area III, Western Philippines, Philippines, 13–15 July 1999; Secretariat, Southeast Asian Fisheries Development Center: Bangkok, Thailand, 2000; pp. 76–100. [Google Scholar]
- Seibel, B.A.; Thuesen, E.V.; Childress, J.J. Flight of the vampire: Ontogenetic gait-transition in vampyroteuthis infernalis (Cephalopoda: Vampyromorpha). J. Exp. Biol. 1998, 201, 2413–2424. [Google Scholar] [PubMed]
- Allcock, A.L.; Cooke, I.R.; Strugnell, J.M. What can the mitochondrial genome reveal about higher-level phylogeny of the molluscan class Cephalopoda? Zool. J. Linn. Soc. 2011, 161, 573–586. [Google Scholar] [CrossRef]
- Boyle, P.; Rodhouse, P. Cephalopods: Ecology and Fisheries; Boyle, P., Rodhouse, P., Eds.; Wiley-Blackwell: Oxford, UK, 2008; ISBN 978-1-405-14543-5. [Google Scholar]
- Nesis, K.N. Distribution of recent Cephalopoda and implications for Plio-Pleistocene events. Berliner Paläobiologische Abhandlungen 2003, 3, 199–224. [Google Scholar]
- Winkelmann, I.; Campos, P.F.; Strugnell, J.; Cherel, Y.; Smith, P.J.; Kubodera, T.; Allcock, L.; Kampmann, M.; Schroeder, H.; Guerra, A.; et al. Mitochondrial genome diversity and population structure of the giant squid Architeuthis: Genetics sheds new light on one of the most enigmatic marine species. Proc. R. Soc. B Biol. Sci. 2013, 280, 20130273. [Google Scholar] [CrossRef]
- Hanlon, R.T.; Messenger, J.B. Cephalopod Behaviour, 2nd ed.; Cambridge University Press: Cambridge, UK, 2018; ISBN 9780511843600. [Google Scholar]
- Wells, M.J. Octopus: Physiology and Behaviour of an Advanced Invertebrate, 1st ed.; Chapman and Hall: London, UK, 1978; ISBN 9780470991978. [Google Scholar]
- Villanueva, R.; Perricone, V.; Fiorito, G. Cephalopods as Predators: A Short Journey among Behavioral Flexibilities, Adaptions, and Feeding Habits. Front. Physiol. 2017, 8, 598. [Google Scholar] [CrossRef]
- Cooke, I.R.; Whitelaw, B.; Norman, M.; Caruana, N.; Strugnell, J.M. Toxicity in Cephalopods. In Evolution of Venomous Animals and Their Toxins; Springer: Dordrecht, The Netherlands, 2015; pp. 1–15. [Google Scholar]
- Ponte, G.; Modica, M.V. Salivary Glands in Predatory Mollusks: Evolutionary Considerations. Front. Physiol. 2017, 8, 580. [Google Scholar] [CrossRef]
- Ghiretti, F. Cephalotoxin: The Crab-paralysing Agent of the Posterior Salivary Glands of Cephalopods. Nature 1959, 183, 1192–1193. [Google Scholar] [CrossRef]
- Kanda, A.; Iwakoshi-Ukena, E.; Takuwa-Kuroda, K.; Minakata, H. Isolation and characterization of novel tachykinins from the posterior salivary gland of the common octopus Octopus vulgaris. Peptides 2003, 24, 35–43. [Google Scholar] [CrossRef]
- Ueda, A.; Nagai, H.; Ishida, M.; Nagashima, Y.; Shiomi, K. Purification and molecular cloning of SE-cephalotoxin, a novel proteinaceous toxin from the posterior salivary gland of cuttlefish Sepia esculenta. Toxicon 2008, 52, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Ruder, T.; Sunagar, K.; Undheim, E.A.B.; Ali, S.A.; Wai, T.-C.; Low, D.H.W.; Jackson, T.N.W.; King, G.F.; Antunes, A.; Fry, B.G. Molecular Phylogeny and Evolution of the Proteins Encoded by Coleoid (Cuttlefish, Octopus, and Squid) Posterior Venom Glands. J. Mol. Evol. 2013, 76, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, B.L.; Strugnell, J.M.; Faou, P.; da Fonseca, R.R.; Hall, N.E.; Norman, M.; Finn, J.; Cooke, I.R. Combined Transcriptomic and Proteomic Analysis of the Posterior Salivary Gland from the Southern Blue-Ringed Octopus and the Southern Sand Octopus. J. Proteome Res. 2016, 15, 3284–3297. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Norman, J.A. Tentacles of Venom: Toxic Protein Convergence in the Kingdom Animalia. J. Mol. Evol. 2009, 68, 311–321. [Google Scholar] [CrossRef]
- Cornet, V.; Henry, J.; Corre, E.; Le Corguille, G.; Zanuttini, B.; Zatylny-Gaudin, C. Dual role of the cuttlefish salivary proteome in defense and predation. J. Proteom. 2014, 108, 209–222. [Google Scholar] [CrossRef]
- Fingerhut, L.C.H.W.; Strugnell, J.M.; Faou, P.; Labiaga, Á.R.; Zhang, J.; Cooke, I.R. Shotgun Proteomics Analysis of Saliva and Salivary Gland Tissue from the Common Octopus Octopus vulgaris. J. Proteome Res. 2018, 17, 3866–3876. [Google Scholar] [CrossRef]
- Domínguez-Pérez, D.; Durban, J.; Agüero-Chapin, G.; López, J.T.; Molina-Ruiz, R.; Almeida, D.; Calvete, J.J.; Vasconcelos, V.; Antunes, A. The Harderian gland transcriptomes of Caraiba andreae, Cubophis cantherigerus and Tretanorhinus variabilis, three colubroid snakes from Cuba. Genomics 2019, 111, 1720–1727. [Google Scholar] [CrossRef]
- Sunagar, K.; Fry, B.G.; Jackson, T.N.W.; Casewell, N.R.; Undheim, E.A.B.; Vidal, N.; Ali, S.A.; King, G.F.; Vasudevan, K.; Vasconcelos, V.; et al. Correction: Molecular Evolution of Vertebrate Neurotrophins: Co-Option of the Highly Conserved Nerve Growth Factor Gene into the Advanced Snake Venom Arsenalf. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Galpert, D.; Molina-Ruiz, R.; Ancede-Gallardo, E.; Pérez-Machado, G.; De la Riva, G.A.; Antunes, A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules 2019, 10, 26. [Google Scholar] [CrossRef]
- Ledoux, J.-B.; Antunes, A. Beyond the beaten path: Improving natural products bioprospecting using an eco-evolutionary framework—The case of the octocorals. Crit. Rev. Biotechnol. 2018, 38, 184–198. [Google Scholar] [CrossRef]
- Ruder, T.; Ali, S.A.; Ormerod, K.; Brust, A.; Roymanchadi, M.-L.; Ventura, S.; Undheim, E.A.B.; Jackson, T.N.W.; Mercier, A.J.; King, G.F.; et al. Functional characterization on invertebrate and vertebrate tissues of tachykinin peptides from octopus venoms. Peptides 2013, 47, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, A.; Erspamer, V. The isolation and amino acid sequence of eledoisin, the active endecapeptide of the posterior salivary glands of Eledone. Arch. Biochem. Biophys. 1963, 101, 56–65. [Google Scholar] [CrossRef]
- Norman, M.D.; Reid, A.L. Guide to Squid, Cuttlefish and Octopuses of Australasia; CSIRO Publishing: Clayton South, Australia, 2000; ISBN 9780643065772. [Google Scholar]
- Laustsen, A.H. Toxin synergism in snake venoms. Toxin Rev. 2016, 35, 165–170. [Google Scholar] [CrossRef]
- Undheim, E.; Sunagar, K.; Herzig, V.; Kely, L.; Low, D.; Jackson, T.; Jones, A.; Kurniawan, N.; King, G.; Ali, S.; et al. A Proteomics and Transcriptomics Investigation of the Venom from the Barychelid Spider Trittame loki (Brush-Foot Trapdoor). Toxins 2013, 5, 2488–2503. [Google Scholar] [CrossRef]
- Domínguez-Pérez, D.; Campos, A.; Rodríguez, A.A.; Turkina, M.V.; Ribeiro, T.; Osorio, H.; Vasconcelos, V.; Antunes, A. Proteomic Analyses of the Unexplored Sea Anemone Bunodactis verrucosa. Mar. Drugs 2018, 16, 42. [Google Scholar] [CrossRef]
- Sutherland, S.K.; Lane, W.R. Toxins and mode of envenomation of the common ringed or blue-banded octopus. Med. J. Aust. 1969, 893–898. [Google Scholar] [CrossRef]
- Houyvet, B.; Zanuttini, B.; Corre, E.; Le Corguillé, G.; Henry, J.; Zatylny-Gaudin, C. Design of antimicrobial peptides from a cuttlefish database. Amin. Acids 2018, 50, 1573–1582. [Google Scholar] [CrossRef]
- Matos, A.; Domínguez-Pérez, D.; Almeida, D.; Agüero-Chapin, G.; Campos, A.; Osório, H.; Vasconcelos, V.; Antunes, A. Shotgun Proteomics of Ascidians Tunic Gives New Insights on Host–Microbe Interactions by Revealing Diverse Antimicrobial Peptides. Mar. Drugs 2020, 18, 362. [Google Scholar] [CrossRef]
- Maselli, V.; Galdiero, E.; Salzano, A.M.; Scaloni, A.; Maione, A.; Falanga, A.; Naviglio, D.; Guida, M.; Di Cosmo, A.; Galdiero, S. OctoPartenopin: Identification and Preliminary Characterization of a Novel Antimicrobial Peptide from the Suckers of Octopus vulgaris. Mar. Drugs 2020, 18, 380. [Google Scholar] [CrossRef]
- Besednova, N.N.; Kovalev, N.N.; Zaporozhets, T.S.; Kuznetsova, T.A.; Gazha, A.K. Cephalopods as a Source of New Antimicrobial Substances. Antibiotiki i Khimioterapiia = Antibiot. Chemoterapy [sic] 2016, 61, 32–42. [Google Scholar]
- Besednova, N.N.; Zaporozhets, T.S.; Kovalev, N.N.; Makarenkova, I.D.; Yakovlev, Y.M. Cephalopods: The potential for their use in medicine. Russ. J. Mar. Biol. 2017, 43, 101–110. [Google Scholar] [CrossRef]
- Monolisha, S.; Mani, A.E.; Patterson, J.; Edward, J.K.P. Molecular characterization and antimicrobial activity of Octopus aegina and Octopus dolfusii in gulf of Mannar coast. Int. J. Pharm. Sci. Res. 2013, 4, 3582–3587. [Google Scholar]
- Hoeksema, M.; van Eijk, M.; Haagsman, H.P.; Hartshorn, K.L. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol. 2016, 11, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Jodoin, J.; Hincke, M.T. Histone H5 is a potent Antimicrobial Agent and a template for novel Antimicrobial Peptides. Sci. Rep. 2018, 8, 2411. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Iwamuro, S. Potential Roles of Histones in Host Defense as Antimicrobial Agents. Infect. Disord. Drug Targets 2008, 8, 195–205. [Google Scholar] [CrossRef]
- Kim, H.S.; Yoon, H.; Minn, I.; Park, C.B.; Lee, W.T.; Zasloff, M.; Kim, S.C. Pepsin-Mediated Processing of the Cytoplasmic Histone H2A to Strong Antimicrobial Peptide Buforin I. J. Immunol. 2000, 165, 3268–3274. [Google Scholar] [CrossRef]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Tellez-Ibarra, R.; Llorente-Quesada, M.T.; Salgado, J.; Barigye, S.J.; Liu, J. Overlap and diversity in antimicrobial peptide databases: Compiling a non-redundant set of sequences. Bioinformatics 2015, 31, 2553–2559. [Google Scholar] [CrossRef][Green Version]
- Masso-Silva, J.; Diamond, G. Antimicrobial Peptides from Fish. Pharmaceuticals 2014, 7, 265–310. [Google Scholar] [CrossRef]
- Almeida, D.; Domínguez-Pérez, D.; Matos, A.; Agüero-Chapin, G.; Castaño, Y.; Vasconcelos, V.; Campos, A.; Antunes, A. Data employed in the construction of a composite protein database for proteogenomic analyses of cephalopods salivary apparatus. Data. submitted.
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.A.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The Toxicogenomic Multiverse: Convergent Recruitment of Proteins Into Animal Venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef]
- Grisley, M.S. Separation and partial characterization of SALIVARY enzymes expressed during prey handling in the octopus eledone cirrhosa. Comp. Biochem. Physiol. Part B Comp. Biochem. 1993, 105, 183–192. [Google Scholar] [CrossRef]
- Gibbs, G.M.; Roelants, K.; O’Bryan, M.K. The CAP Superfamily: Cysteine-Rich Secretory Proteins, Antigen 5, and Pathogenesis-Related 1 Proteins—Roles in Reproduction, Cancer, and Immune Defense. Endocr. Rev. 2008, 29, 865–897. [Google Scholar] [CrossRef]
- Takeda, S.; Takeya, H.; Iwanaga, S. Snake venom metalloproteinases: Structure, function and relevance to the mammalian ADAM/ADAMTS family proteins. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.M.; Rucavado, A.; Escalante, T.; Díaz, C. Hemorrhage induced by snake venom metalloproteinases: Biochemical and biophysical mechanisms involved in microvessel damage. Toxicon 2005, 45, 997–1011. [Google Scholar] [CrossRef]
- Fry, B.G.; Undheim, E.A.B.; Ali, S.A.; Jackson, T.N.W.; Debono, J.; Scheib, H.; Ruder, T.; Morgenstern, D.; Cadwallader, L.; Whitehead, D.; et al. Squeezers and Leaf-cutters: Differential Diversification and Degeneration of the Venom System in Toxicoferan Reptiles. Mol. Cell. Proteom. 2013, 12, 1881–1899. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Yang, Z.; Maldonado, E.; Li, C.; Zhang, G.; Gilbert, M.T.P.; Jarvis, E.D.; O’Brien, S.J.; Johnson, W.E.; Antunes, A. Olfactory Receptor Subgenomes Linked with Broad Ecological Adaptations in Sauropsida. Mol. Biol. Evol. 2015, 32, 2832–2843. [Google Scholar] [CrossRef] [PubMed]
- Sarras, M.P.; Yan, L.; Leontovich, A.; Zhang, J.S. Structure, expression, and developmental function of early divergent forms of metalloproteinases in Hydra. Cell Res. 2002, 12, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Grisley, M.S.; Boyle, P.R. Chitinase, a new enzyme in octopus saliva. Comp. Biochem. Physiol. Part B Comp. Biochem. 1990, 95, 311–316. [Google Scholar] [CrossRef]
- Hamid, R.; Khan, M.A.; Ahmad, M.; Ahmad, M.M.; Abdin, M.Z.; Musarrat, J.; Javed, S. Chitinases: An update. J. Pharm. Bioallied Sci. 2013, 5, 21. [Google Scholar]
- Kini, R.M. Excitement ahead: Structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon 2003, 42, 827–840. [Google Scholar] [CrossRef]
- Undheim, E.A.B.; Georgieva, D.N.; Thoen, H.H.; Norman, J.A.; Mork, J.; Betzel, C.; Fry, B.G. Venom on ice: First insights into Antarctic octopus venoms. Toxicon 2010, 56, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Pavasovic, A.; Norton, R.S. Sea Anemones: Quiet Achievers in the Field of Peptide Toxins. Toxins 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Shiomi, K. Peptide Toxins in Sea Anemones: Structural and Functional Aspects. Mar. Biotechnol. 2006, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schweitz, H.; Bruhn, T.; Guillemare, E.; Moinier, D.; Lancelin, J.-M.; Béress, L.; Lazdunski, M. Kalicludines and Kaliseptine: Two different classes of sea anemone toxins for voltage-sensitive K+ channels. J. Biol. Chem. 1995, 270, 25121–25126. [Google Scholar] [CrossRef]
- Domínguez-Pérez, D.; Diaz-Garcia, C.; García-Delgado, N.; Sierra-Gómez, Y.; Castañeda, O.; Antunes, A. Insights into the Toxicological Properties of a Low Molecular Weight Fraction from Zoanthus sociatus (Cnidaria). Mar. Drugs 2013, 11, 2873–2881. [Google Scholar] [CrossRef]
- Masci, P.P.; Whitaker, A.N.; Sparrow, L.G.; de Jersey, J.; Winzor, D.J.; Watters, D.J.; Lavin, M.F.; Gaffney, P.J. Textilinins from Pseudonaja textilis textilis. Characterization of two plasmin inhibitors that reduce bleeding in an animal model. Blood Coagul. Fibrinolysis 2000, 11, 385–393. [Google Scholar] [CrossRef]
- Flight, S.M.; Johnson, L.A.; Du, Q.S.; Warner, R.L.; Trabi, M.; Gaffney, P.J.; Lavin, M.F.; de Jersey, J.; Masci, P.P. Textilinin-1, an alternative anti-bleeding agent to aprotinin: Importance of plasmin inhibition in controlling blood loss. Br. J. Haematol. 2009, 145, 207–211. [Google Scholar] [CrossRef]
- Shafqat, J.; Zaidi, Z.H.; Jörnvall, H. Purification and characterization of a chymotrypsin Kunitz inhibitor type of polypeptide fro the venom of cobra ( Naja naja naja ). FEBS Lett. 1990, 275, 6–8. [Google Scholar] [CrossRef]
- Siddiqi, A.R.; Zaidi, Z.H.; Jörnvall, H. Purification and characterization of a Kunitz-type trypsin inhibitor from Leaf-nosed viper venom. FEBS Lett. 1991, 294, 141–143. [Google Scholar] [CrossRef]
- Lu, J.; Yang, H.; Yu, H.; Gao, W.; Lai, R.; Liu, J.; Liang, X. A novel serine protease inhibitor from Bungarus fasciatus venom. Peptides 2008, 29, 369–374. [Google Scholar] [CrossRef]
- Choo, Y.M.; Lee, K.S.; Yoon, H.J.; Qiu, Y.; Wan, H.; Sohn, M.R.; Sohn, H.D.; Jin, B.R. Antifibrinolytic Role of a Bee Venom Serine Protease Inhibitor That Acts as a Plasmin Inhibitor. PLoS ONE 2012, 7, e32269. [Google Scholar] [CrossRef]
- Lenarcic, B.; Turk, V. Thyroglobulin Type-1 Domains in Equistatin Inhibit Both Papain-like Cysteine Proteinases and Cathepsin D. J. Biol. Chem. 1999, 274, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Lenarčič, B.; Ritonja, A.; Štrukelj, B.; Turk, B.; Turk, V. Equistatin, a New Inhibitor of Cysteine Proteinases from Actinia equina, Is Structurally Related to Thyroglobulin Type-1 Domain. J. Biol. Chem. 1997, 272, 13899–13903. [Google Scholar] [CrossRef]
- Simonet, G.; Claeys, I.; Broeck, J. Vanden Structural and functional properties of a novel serine protease inhibiting peptide family in arthropods. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2002, 132, 247–255. [Google Scholar] [CrossRef]
- Mer, G.; Kellenberger, C.; Koehl, P.; Stote, R.; Sorokine, O.; Van Dorsselaer, A.; Luu, B.; Hietter, H.; Lefevre, J.-F. Solution Structure of PMP-D2, a 35-Residue Peptide Isolated from the Insect Locusta migratoria. Biochemistry 1994, 33, 15397–15407. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.H.; Gorton, V.J.; Harding, L.; Patel, D.; Pacey, S.; Kellenberger, C.; Hietter, H.; Bermudez, I. Inhibition of Neuronal High Voltage-activated Calcium Channels by Insect Peptides: A Comparison with the Actions of $ω$-Conotoxin GVIA. Neuropharmacology 1997, 36, 195–208. [Google Scholar] [CrossRef]
- Ghosh, S. Sialic acid and biology of life: An introduction. In Sialic Acids and Sialoglycoconjugates in the Biology of Life, Health and Disease; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–61. [Google Scholar]
- Margres, M.J.; Aronow, K.; Loyacano, J.; Rokyta, D.R. The venom-gland transcriptome of the eastern coral snake (Micrurus fulvius) reveals high venom complexity in the intragenomic evolution of venoms. BMC Genom.oS On 2013, 14, 531. [Google Scholar] [CrossRef]
- Hayes, J.J.; Clark, D.J.; Wolffe, A.P. Histone contributions to the structure of DNA in the nucleosome. Proc. Natl. Acad. Sci. USA 1991, 88, 6829–6833. [Google Scholar] [CrossRef]
- Maeshima, K.; Eltsov, M. Packaging the Genome: The Structure of Mitotic Chromosomes. J. Biochem. 2008, 143, 145–153. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Twenty-Five Years of the Nucleosome, Fundamental Particle of the Eukaryote Chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef]
- Alex, A.; Silva, V.; Vasconcelos, V.; Antunes, A. Evidence of Unique and Generalist Microbes in Distantly Related Sympatric Intertidal Marine Sponges (Porifera: Demospongiae). PLoS ONE 2013, 8, e80653. [Google Scholar] [CrossRef]
- Alex, A.; Antunes, A. Pyrosequencing Characterization of the Microbiota from Atlantic Intertidal Marine Sponges Reveals High Microbial Diversity and the Lack of Co-Occurrence Patterns. PLoS ONE 2015, 10, e0127455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.; Vasconcelos, V.; Tamagnini, P.; Santos, A.; Antunes, A. Unusual Symbiotic Cyanobacteria Association in the Genetically Diverse Intertidal Marine Sponge Hymeniacidon perlevis (Demospongiae, Halichondrida). PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Takamura, Y.; Kawakami, T.; Aimoto, S.; Saito, H.; Mukai, T. Effect of amino acid distribution of amphipathic helical peptide derived from human apolipoprotein A-I on membrane curvature sensing. FEBS Lett. 2013, 587, 510–515. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, M.S.; Kim, S.C. A novel antimicrobial peptide from Bufo bufo gargarizans. Biochem. Biophys. Res. Commun. 1996, 218, 408–413. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of Action of the Antimicrobial Peptide Buforin II: Buforin II Kills Microorganisms by Penetrating the Cell Membrane and Inhibiting Cellular Functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef]
- Seo, J.-K.; Lee, M.J.; Go, H.-J.; Kim, G.D.; Jeong, H.D.; Nam, B.-H.; Park, N.G. Purification and antimicrobial function of ubiquitin isolated from the gill of Pacific oyster, Crassostrea gigas. Mol. Immunol. 2013, 53, 88–98. [Google Scholar] [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.N. Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog. Neurobiol. 2004, 73, 311–357. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, D.H.; Goldstein, G.; Niall, H.D. The complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells. Biochemistry 1975, 14, 2214–2218. [Google Scholar] [CrossRef]
- Busch, H. [23] Ubiquitination of Proteins. In Methods Enzymol; Elsevier: Amsterdam, The Netherlands, 1984; Volume 106, pp. 238–262. ISBN 9780121820060. [Google Scholar]
- Siegelman, M.; Bond, M.; Gallatin, W.; St John, T.; Smith, H.; Fried, V.; Weissman, I. Cell surface molecule associated with lymphocyte homing is a ubiquitinated branched-chain glycoprotein. Science 1986, 231, 823–829. [Google Scholar] [CrossRef]
- Audhya, T.; Goldstein, G. Thymopoietin and ubiquitin. In Methods Enzymol; Elsevier: Amsterdam, The Netherlands, 1985; Volume 116, pp. 279–291. ISSN 0076-6879 (Print) 0076-6879 (Linking). [Google Scholar]
- Alonso, S.; Pethe, K.; Russell, D.G.; Purdy, G.E. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. USA 2007, 104, 6031–6036. [Google Scholar] [CrossRef] [PubMed]
- Bachère, E.; Rosa, R.D.; Schmitt, P.; Poirier, A.C.; Merou, N.; Charrière, G.M.; Destoumieux-Garzón, D. The new insights into the oyster antimicrobial defense: Cellular, molecular and genetic view. Fish Shellfish Immunol. 2015, 46, 50–64. [Google Scholar] [CrossRef]
- Wang, Y.; Griffiths, W.J.; Jörnvall, H.; Agerberth, B.; Johansson, J. Antibacterial peptides in stimulated human granulocytes. Eur. J. Biochem. 2002, 269, 512–518. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Molina-Ruiz, R.; Maldonado, E.; de la Riva, G.; Sánchez-Rodríguez, A.; Vasconcelos, V.; Antunes, A. Exploring the Adenylation Domain Repertoire of Nonribosomal Peptide Synthetases Using an Ensemble of Sequence-Search Methods. PLoS ONE 2013, 8, e65926. [Google Scholar] [CrossRef]
- Wang, B.; Harwig, S.S.; Lehrer, R.I. [Rat bladder ubiquitin-like molecule: Isolation, purification and N-terminal sequencing]. J. West China Univ. Med. Sci. 1993, 24, 127–130. [Google Scholar]
- Kieffer, A.; Goumon, Y.; Ruh, O.; Chasserot-Golaz, S.; Nullans, G.; Gasnier, C.; Aunis, D.; Metz-Boutigue, M. The N- and C-terminal fragments of ubiquitin are important for the antimicrobial activities. FASEB J. 2003, 17, 776–778. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, S.Y.; Park, S.-C.; Shin, S.Y.; Choi, S.J.; Park, Y.; Hahm, K.-S. Purification and antimicrobial activity studies of the N-terminal fragment of ubiquitin from human amniotic fluid. Biochim. Biophys. Acta Proteins Proteom. 2007, 1774, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Bleackley, M.R.; Hayes, B.M.; Parisi, K.; Saiyed, T.; Traven, A.; Potter, I.D.; van der Weerden, N.L.; Anderson, M.A. Bovine pancreatic trypsin inhibitor is a new antifungal peptide that inhibits cellular magnesium uptake. Mol. Microbiol. 2014, 92, 1188–1197. [Google Scholar] [CrossRef]
- Conlon, J.M.; Kim, J.B. A Protease Inhibitor of the Kunitz Family from Skin Secretions of the Tomato Frog, Dyscophus guineti (Microhylidae). Biochem. Biophys. Res. Commun. 2000, 279, 961–964. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Al-Ghaferi, N.; Ahmed, E.; Meetani, M.A.; Leprince, J.; Nielsen, P.F. Orthologs of magainin, PGLa, procaerulein-derived, and proxenopsin-derived peptides from skin secretions of the octoploid frog Xenopus amieti (Pipidae). Peptides 2010, 31, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.; Maldonado, E.; Khan, I.; Silva, L.; Gilbert, M.T.P.; Zhang, G.; Jarvis, E.D.; O’Brien, S.J.; Johnson, W.E.; Antunes, A. Whole-Genome Identification, Phylogeny, and Evolution of the Cytochrome P450 Family 2 (CYP2) Subfamilies in Birds. Genome Biol. Evol. 2016, 8, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Meth. 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Custom Databases | # Protein Sequences 7 | % Protein Sequences Relatively To The Composite Database 9 | # Proteins Identified 10 | % Proteins Identified Relatively To The Total Of Proteins Identified 12 | # Leading Proteins 13 | % Leading Proteins Relatively To The Total Of Proteins Identified 14 | % (# Leading Proteins Relatively To The # Proteins Identified Per Db) 15 |
|---|---|---|---|---|---|---|---|
| A1 | 19,087 | 0.32 | 2073 | 20.58 | 1759 | 17.46 | 84.85 |
| B2 | 16,990 | 0.03 | 44 | 0.44 | 12 | 0.12 | 27.27 |
| C3 | 2427 | 0.04 | 1845 | 18.31 | 1180 | 11.71 | 63.96 |
| D4 | 84,778 | 1.42 | 5275 | 52.36 | 3075 | 30.52 | 58.29 |
| E5 | 5,106,635 | 85.81 | 700 | 6.95 | 249 | 2.47 | 35.57 |
| F6 | 720,910 | 1.21 | 138 | 1.37 | 70 | 0.69 | 50.72 |
| Total | 5,950,8278 | 100 | 10,07511 | 100 | 6345 | 62.98 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, D.; Domínguez-Pérez, D.; Matos, A.; Agüero-Chapin, G.; Osório, H.; Vasconcelos, V.; Campos, A.; Antunes, A. Putative Antimicrobial Peptides of the Posterior Salivary Glands from the Cephalopod Octopus vulgaris Revealed by Exploring a Composite Protein Database. Antibiotics 2020, 9, 757. https://doi.org/10.3390/antibiotics9110757
Almeida D, Domínguez-Pérez D, Matos A, Agüero-Chapin G, Osório H, Vasconcelos V, Campos A, Antunes A. Putative Antimicrobial Peptides of the Posterior Salivary Glands from the Cephalopod Octopus vulgaris Revealed by Exploring a Composite Protein Database. Antibiotics. 2020; 9(11):757. https://doi.org/10.3390/antibiotics9110757
Chicago/Turabian StyleAlmeida, Daniela, Dany Domínguez-Pérez, Ana Matos, Guillermin Agüero-Chapin, Hugo Osório, Vitor Vasconcelos, Alexandre Campos, and Agostinho Antunes. 2020. "Putative Antimicrobial Peptides of the Posterior Salivary Glands from the Cephalopod Octopus vulgaris Revealed by Exploring a Composite Protein Database" Antibiotics 9, no. 11: 757. https://doi.org/10.3390/antibiotics9110757
APA StyleAlmeida, D., Domínguez-Pérez, D., Matos, A., Agüero-Chapin, G., Osório, H., Vasconcelos, V., Campos, A., & Antunes, A. (2020). Putative Antimicrobial Peptides of the Posterior Salivary Glands from the Cephalopod Octopus vulgaris Revealed by Exploring a Composite Protein Database. Antibiotics, 9(11), 757. https://doi.org/10.3390/antibiotics9110757