Bacteriophages as Alternatives to Antibiotics in Clinical Care



Abstract
1. Introduction
2. Phage Biology
Specificity
3. Phage Pharmacology
3.1. Pharmacodynamics
3.2. Pharmacokinetics
4. Role of the Immune Response in Phage Therapy
5. Resistance to Phages
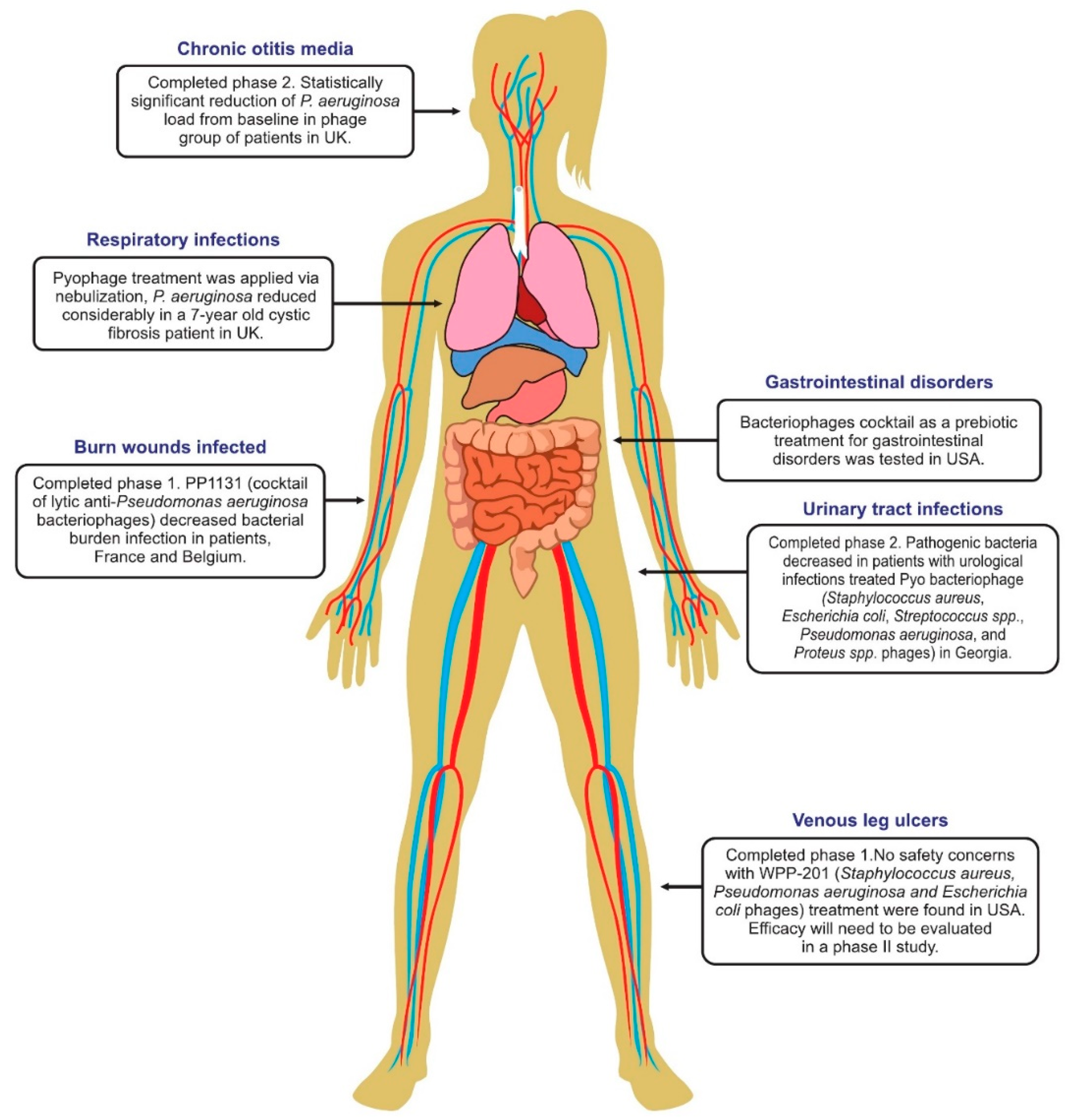
6. Phage Therapy Clinical Trials in Humans
6.1. Phage Treatment of Burns
6.2. Treatment of A Septicemia Patient with Acute Kidney Damage
6.3. Engineered Phages for Treatment of Mycobacteria in A Cystic Fibrosis Patient
6.4. Phage Therapy for Respiratory Infections
6.5. Phage Therapy for Urinary Tract Infections
6.6. Phage Therapy for Diarrhea
6.7. Treatment of Peri-Prosthetic Joint Infection
6.8. Treatment of Leg Ulcers
6.9. Therapy of Drug-Resistant Craniectomy Infection
6.10. Therapy of Ear Infections
7. Engineering and Other Genetic Technologies for Phage Therapy
8. The Medicinal Regulatory Status of Phages
9. Advantages and Disadvantages of Phage Therapy
9.1. Key Advantages
9.2. Key Disadvantages
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W. Bacteriophages: Evolution of the Majority. Theoretical Population Biology. Popul. Biol. 2002, 61, 471–480. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Międzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawełczyk, Z.; Rogóż, P.; Kłak, M.; Wojtasik, E.; et al. Clinical aspects of phage therapy. Adv. Virus Res. 2012, 83, 73–121. [Google Scholar] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Weber-Dabrowska, B.; Jónczyk-Matysiak, E.; ˙Zaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage Procurement for Therapeutic Purposes. Front. Microbiol. 2016, 7, 1177. [Google Scholar] [CrossRef] [PubMed]
- Aminov, R.; Caplin, J.; Chanishvili, N.; Coffey, A.; Cooper, I.; De Vos, D.; Doškar, J.; Friman, V.; Kurtböke, I.; Pantucek, R.; et al. Application of bacteriophages. Microbiol. Aust. 2018, 38, 63–66. [Google Scholar]
- NIH NIAID’s Antibacterial Resistance Program: Current Status and Future Directions. 2014. Available online: http://www.niaid.nih.gov/topics/antimicrobialresistane/documents/arstrategicplan2014.pdf (accessed on 23 September 2015).
- Moellering, R.C. NDM-1—A cause for worldwide concern. N. Eng. J. Med. 2010, 363, 2377–2379. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Lavigne, R. Introduction to Bacteriophages Biology and Diversity. In Bacteriophages in Control of Food and Waterborne Pathogens; Sabour, P.M., Griffiths, M.W., Eds.; American Society of Microbiology: Washington, DC, USA, 2010. [Google Scholar]
- Guttman, B.; Raya, R.; Kutter, E. Basic Phage Biology. In Bacteriophages: Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; CRC Press Florida: Boca Raton, FL, USA, 2011; pp. 29–66. [Google Scholar]
- Engelkirk, G.; Duben-Engelkirk, P. Burton’s Microbiology for the Health Sciences, 9th ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B.; Delattre, A.S.; Lavigne, R. Learning from bacteriophages—Advantages and limitations of phage and phage-encoded protein applications. Curr. Protein Pept. Sci. 2012, 13, 699–722. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, S.; Hauwa-Suleiman, B.; Ali Abbagana, B.; Alhaji-Mustafa, I.; Abbas-Musa, I. Novel Uses of Bacteriophages in the Treatment of Human Infections and Antibiotic Resistance. Am. J. Biosci. 2016, 4, 34–40. [Google Scholar] [CrossRef][Green Version]
- Available online: https://en.wikipedia.org/wiki/Listeria_phage_P100 (accessed on 1 August 2019).
- Friman, V.P.; Soanes-Brown, D.; Sierocinski, P.; Molin, S.; Johansen, H.K.; Merabishvili, M.; Pirnay, J.P.; De Vos, D.; Buckling, A. Pre-adapting parasitic phages to a pathogen leads to increased pathogen clearance and lowered resistance evolution with Pseudomonas aeruginosa cystic fibrosis bacterial isolates. J. Evol. Biol. 2016, 29, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Bhandari, D.; Dobson, R.; Billington, C. Potential for Bacteriophage Endolysins to Supplement or Replace Antibiotics in Food Production and Clinical Care. Antibiotics 2018, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Rafii, F.; Sutherland, J.B.; Cerniglia, C.E. Effects of treatment with antimicrobial agents on the human colonic microflora. Clin. Risk Manag. 2008, 4, 1343. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, K.; Gerstmans, H.; Saafan, H.; Dishisha, T.; Briers, Y. The Preclinical and Clinical Progress of Bacteriophages and Their Lytic Enzymes: The Parts are Easier than the Whole. Viruses 2019, 24, 11. [Google Scholar]
- Abedon, S.T.; Thomas-Abedon, C. Phage therapy Pharmacology. Cur. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef]
- Payne, R.J.H.; Phil, D.; Jansen, V.A. Phage therapy: The peculiar Kinetics of self-replicating pharmaceuticals. Clin. Pharm. Ther. 2000, 68, 225–230. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of phage therapy. Bacteriophage 2011, 2, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J.; Regoes, R.R. Pharmacodynamics of non-replicating viruses, bacteriocins and lysins. Proc. Biol. Sci. 2006, 273, 2703–2712. [Google Scholar] [CrossRef] [PubMed]
- McVay, C.S.; Velasquez, M.; Fralick, J.A. Phage therapy of Pseudomonas aeruginosa infection in a mouse burn wound model. Antimicrob. Agents Chemother. 2007, 51, 1934–1938. [Google Scholar] [CrossRef]
- Tanji, Y.; Shimada, T.; Fukudomi, H.; Miyanaga, K.; Nakai, Y.; Unno, H. Therapeutic use of phage cocktail for controlling Escherichia coli O157:H7 in gastrointestinal tract of mice. J. Biosci. Bioeng. 2005, 100, 280–287. [Google Scholar] [CrossRef]
- Semler, D.D.; Goudie, A.D.; Finlay, W.H.; Dennis, J.J. Aerosol phage therapy efficacy in Burkholderia cepacia complex respiratory infections. Antimicrob. Agents Chemother. 2014, 58, 4005–4013. [Google Scholar] [CrossRef]
- Brüssow, H. Phage therapy: The Escherichia coli experience. Microbiology 2005, 151, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.Y.; Jang, I.J.; Yoon, S.; Jang, K.; Yu, K.-S.; Cho, J.Y.; Seong, M.-W.; Jung, G.M.; Yoon, S.J.; Kang, S.H. Pharmacokinetics and tolerance of the phage endolysin-based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob. Agents Chemother. 2017, 24, 61. [Google Scholar] [CrossRef] [PubMed]
- Cassino, C.; Murphy, M.; Boyle, J.; Rotolo, J.; Wittekind, M. Results of the first in human study of lysin CF-301 evaluating the safety, tolerability and pharmacokinetic profile in healthy volunteers. In Proceedings of the 26th European Congress of Clinical Microbiology and Infectious Diseases, Amsterdam, The Netherlands, 9–12 April 2016. [Google Scholar]
- Sriram, B.; Chikkamadaiah, S.C.R.; Durgaiah, M.; Hariharan, S.; Jayaraman, R.; Kumar, S.; Maheshwari, U.; Nandish, P. Pharmacokinetics and efficacy of ectolysin P128 in a mouse model of systemic Methicillin resistant Staphylococcus aureus (MRSA) infection. In Proceedings of the ASM Microbe 2017, New Orleans, LA, USA, 1–5 June 2017. [Google Scholar]
- Dufour, N.; Delattre, R.; Ricard, J.D.; Debarbieux, L. The lysis of pathogenic Escherichia coli by bacteriophages releases less endotoxin than betalactams. Clin. Infect. Dis. 2017, 64, 1582–1588. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage endolysins: A novel anti-infective to control Gram-positive pathogens. Int. J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically engineered phages: A review of advances over the last decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Biswas, B.; Adhya, S.; Washart, P.; Paul, B.; Trostel, A.N.; Powell, B.; Carlton, R.; Merril, C.R. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect. Immun. 2002, 70, 204–210. [Google Scholar] [CrossRef]
- Fishman, M. Antibody formation in vitro. J. Exp. Med. 1961, 114, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, K.; Swita1a-Jelen, K.; Opolski, A.; Górski, A. Possible association between phages, Hoc protein, and the immune system. Arch. Virol. 2006, 151, 209–215. [Google Scholar] [CrossRef]
- Belleghem, J.D.; Clement, F.; Merabishvili, M.; Lavigne, R.; Vaneechoutte, M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 2017, 7, 8004. [Google Scholar] [CrossRef] [PubMed]
- Majewska, J.; Beta, W.; Lecion, D.; Hodyra-Stefaniak, K.; Kłopot, A.; Kaźmierczak, Z.; Miernikiewicz, P.; Piotrowicz, A.; Ciekot, J.; Owczarek, B.; et al. Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses 2015, 7, 4783–4799. [Google Scholar] [CrossRef] [PubMed]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapa, J.; Drab, M.; Jonczyk-Matysiak, E.; Lecion, D.; Kazmierczak, Z.; Beta, M.; Harhala, J.M.; Bubak, B.; et al. Mammalian host-versus-phage immune response determines phage fate in vivo. Sci. Rep. 2015, 5, 14802. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Davis, S.D.; Wedgwood, R.J. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J. Clin. Investig. 1971, 50, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Buckley, R.; Lugar, P. Diagnostic Immunization with Bacteriophage ΦX 174 in Patients with Common Variable Immunodeficiency/Hypogammaglobulinemia. Front. Immunol. 2014, 5, 410. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Floch, R.L.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Wallin, M.; Lin, Y.; Leung, S.S.Y.; Wang, H.; Morales, S.; Chan, H.K. Phage therapy for respiratory infections. Adv. Drug Deliv. Rev. 2018, 133, 76–86. [Google Scholar] [CrossRef]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Bacteriophage therapy: Clinical trials and regulatory hurdles. Front. Cell. Infect. Microbiol. 2018, 8, 376. [Google Scholar] [CrossRef]
- Speck, P.; Smithyman, A. Safety and efficacy of phage therapy via the intravenous route. FEMS Microbiol. Lett. 2016, 363, 242. [Google Scholar] [CrossRef]
- Jennes, S.; Merabishvili, M.; Soentjens, P.; Pang, K.W.; Rose, T.; Keersebilck, E.; Soete, O.; François, P.M.; Teodorescu, S.; Verween, G.; et al. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury—A case report. Crit. Care 2017, 21, 129. [Google Scholar] [CrossRef] [PubMed]
- Sula, L.; Sulova, J.; Stolcpartova, M. Therapy of experimental tuberculosis in guinea pigs with mycobacterial phages DS-6A, GR-21 T, My-327. Czech. Med. 1981, 4, 209–214. [Google Scholar] [PubMed]
- Trigo, G.; Martins, T.G.; Fraga, A.G.; Longatto-Filho, A.; Castro, A.G.; Azeredo, J.; Pedrosa, J. Phage Therapy Is Effective against Infection by Mycobacterium ulcerans in a Murine Footpad Model. PLoS Negl. Trop. Dis. 2013, 7, 2183. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Pabary, R.; Singh, C.; Morales, S.; Bush, A.; Alshafi, K.; Bilton, D.; Alton, E.; Smithyman, A.; Davies, J. Anti pseudomonal bacteriophage reduces infective burden and inflammatory response in the murine lung. Antimicrob. Agents Chemother. 2016, 60, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mi, Z.; Niu, W.; An, X.; Yuan, X.; Liu, H.; Li, L.; Liu, Y.; Feng, Y.; Huang, Y.; et al. Intranasal treatment with bacteriophage rescues mice from Acinetobacter baumannii-mediated pneumonia. Future Microbiol. 2016, 11, 631–641. [Google Scholar] [CrossRef]
- Waters, E.M.; Neill, D.R.; Kaman, B.; Sahota, J.S.; Clokie, M.R.J.; Winstanley, C.; Kadioglu, A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 2017, 72, 666–667. [Google Scholar] [CrossRef]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Médecine et Maladies Infectieuses 2008, 38, 426–430. [Google Scholar] [CrossRef]
- Matinkhoo, S.; Lynch, K.; Dennis, J.; Finlay, W.; Vehring, R. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J. Pharm. Sci. 2011, 100, 5197–5205. [Google Scholar] [CrossRef]
- Saussereau, E.; Vachier, I.; Chiron, R.; Godbert, B.; Sermet, I.; Dufour, N.; Pirnay, J.D.; De Vos, F.; Carrié, N.; Debarbieux, L. Effectiveness of bacteriophages in the sputum of cystic fibrosis patients. Clin. Microbiol. Infect. 2014, 20, 983–990. [Google Scholar] [CrossRef]
- Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Leitner, L.; Mehnert, U.; Chkhotua, A.; Kessler, K.; Sybesma, W. Adapted Bacteriophages for Treating Urinary Tract Infections. Front. Microbiol. 2018, 9, 1832. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; Berger, B.; Deng, Y.; Kieser, S.; Foata, F.; Moine, D.; Descombes, P.; Sultana, S.; Huq, S.; Kumar-Bardhan, P.; et al. Oral application of Escherichia coli bacteriophage: Safety tests in healthy and diarrheal children from Bangladesh. Environ. Microbiol. 2016, 19, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; Brüssow, H. From bench to bed and back again: Phage therapy of childhood Escherichia coli diarrhea. Ann. N. Y. Acad. Sci. 2016, 1372, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Ferry, T.; Leboucher, G.; Fevre, C.; Herry, Y.; Conrad, A.; Josse, J.; Batailler, C.; Chidiac, C.; Medina, M.; Lustig, S.; et al. Salvage Debridement, Antibiotics and Implant Retention (“DAIR”) with local injection of a selected cocktail of bacteriophages: Is it an option for an elderly patient with relapsing Staphylococcus aureus prosthetic-joint infection? Open Forum Infect. Dis. 2018, 24, 5. [Google Scholar] [CrossRef] [PubMed]
- Akanda, Z.Z.; Taha, M.; Abdelbary, H. Current review-The rise of bacteriophage as a unique therapeutic platform in treating peri-prosthetic joint infections. J. Orthop. Res. 2017, 36, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.D.; Wolcott, R.D.; Kuskowski, M.A.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care 2009, 238, 240–243. [Google Scholar] [CrossRef]
- Rose, T.; Verbeken, G.; De Vos, D.; Merabishvili, M.; Vaneechoutte, M.; Lavigne, R.; Jennes, S.; Zizi, M.; Pirnay, J.P. Experimental phage therapy of burn wound infection: difficult first steps. Int. J. Burns Trauma 2014, 4, 66–73. [Google Scholar] [PubMed]
- Wright, A.; Hawkins, C.H.; Anggard, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic phage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Yoichi, M.; Abe, M.; Miyanaga, K.; Unno, H.; Tanji, Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157: H7. J. Biotechnol. 2005, 115, 101–107. [Google Scholar] [CrossRef]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol. Lett. 2009, 295, 211–217. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Lo, Y.-H.; Tseng, P.-W.; Chang, S.-F.; Lin, Y.-T.; Chen, T.-S. A T3 and T7 recombinant phage acquires efficient adsorption and a broader host range. PLoS ONE 2012, 7, e30954. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nature Biotechnol. 2014, 32, 1146. [Google Scholar] [CrossRef]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nature Biotechnol. 2014, 32, 1141. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, C.L.; Merril, C.R.; Adhya, S. An amino acid substitution in a capsid protein enhances phage survival in the mouse circulatory system more than a 1000-fold. Virus Res. 2005, 114, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Hagens, S.; Habel, A.; Von Ahsen, U.; Von Gabain, A.; Bläsi, U. Therapy of experimental Pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob. Agents Chemother. 2004, 48, 3817–3822. [Google Scholar] [CrossRef]
- Matsuda, T.; Freeman, T.A.; Hilbert, D.W.; Duff, M.; Fuortes, M.; Stapleton, P.P.; Daly, J.M. Lysis-deficient bacteriophage therapy decreases endotoxin and inflammatory mediator release and improves survival in a murine peritonitis model. Surgery 2005, 137, 639–646. [Google Scholar] [CrossRef]
- Fauconnier, A. Regulating phage therapy: The biological master file concept could help to overcome the regulatory challenge of personalized medicines. EMBO Rep. 2017, 18, 198–200. [Google Scholar] [CrossRef]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Commission de la santé publique, de l’environnement et du renouveau de la société. Questions jointes de Mme Muriel Gerkenset, M. Philippe Blanchart àlaministredes Affaires sociales et de la Santé publiques ur’la phagothérapie’ àla ministre des Affaires sociales et de la Santé publique’ (N 11955 and N 12911). 2016. Available online: https://www.dekamer.be/doc/CCRA/pdf/54/ac464.pdf (accessed on 1 August 2019).
- El-Shibiny, A.; El-Sahhar, S. Bacteriophages: The possible solution to treat infections caused by pathogenic bacteria. Can. J. Microbiol. 2017, 63, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage therapy: A potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries. 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; De Roock, S.; Opal, S.M. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, J.; Sutherland, I.W. The use of phages for the removal of infectious biofilms. Cur. Pharm. Biotechnol. 2008, 9, 261–266. [Google Scholar] [CrossRef]
- Anon. When is an allergy to an antibiotic really an allergy? Best Pract. J. 2015, 68, 22. [Google Scholar]
- Terico, A.T.; Gallagher, J.C. Beta-lactam hypersensitivity and cross-reactivity. J. Pharm. Pract. 2014, 27, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Ligonenko, O.V.; Borysenko, M.M.; Digtyar, I.I.; Ivashchenko, D.M.; Zubakha, A.B.; Chorna, I.O.; Shumeyko, I.A.; Storozhenko, O.V.; Gorb, L.I.; Ligonenko, O.O. Application of bacteriophages in complex of treatment of a shot-gun wounds of soft tissues in the patients, suffering multiple allergy for antibiotics. Klin. Khir. 2015, 10, 65–66. [Google Scholar]
- Ghannad, M.S.; Mohammadi, A. Bacteriophage: Time to re-evaluate the potential of phage therapy as a promising agent to control multidrug-resistant bacteria. Iran. J. Basic Med. Sci. 2012, 15, 693–701. [Google Scholar]
- Kucharewicz-Krukowsk, A.; Slopek, S. Immunogenic effect of bacteriophage in patients subjected to phage therapy. Arch. Immunol. Ther. Exp. (Warsz.) 1987, 35, 553–561. [Google Scholar]



{kind=link}
{kind=link}
| Delivery Route | Advantages | Disadvantages | Mitigations to Hurdles |
|---|---|---|---|
| Intraperitoneal | Higher dosage volumes possible. Diffusion to other sites. | Extent of diffusion to other sites may be overestimated in humans (most data from small animals). | Multiple delivery sites. |
| Intramuscular | Phages delivered at infection site. | Slower diffusion of phages (possibly). Lower dosage volumes. | Multi-dose courses. Multi-dose courses. |
| Subcutaneous | Localized and systemic diffusion. | Lower dosage volumes. | Multi-dose courses. |
| Intravenous | Rapid systemic diffusion. | Rapid clearing of phages by the immune system. | In vivo selection of low-immunogenic phages may be possible. |
| Topical | High dose of phages delivered at infection site. | Run-off from target site if phages suspended in liquid. | Incorporate phages into gels and dressings. |
| Suppository | Slow, stable release of phages over long time. | Limited applications/sites. Risk of insufficient dosing. Technically challenging to manufacture. | Careful consideration of phage kinetics required. |
| Oral | Ease of delivery. Higher dosage volumes possible. | Stomach acid reduces phage titer. Non-specific adherence of phages to stomach contents and other microflora. | Add calcium carbonate to buffer pH. Microencapsulation to deliver phages to target area. |
| Aerosol | Relative ease of delivery. Can reach poorly perfused regions of infected lungs. | High proportion of phages lost. Delivery can be impaired by mucus and biofilms | Use of depolymerases to reduce mucus. |
| Consideration | Antibiotic Therapy | Phage Therapy |
|---|---|---|
| Specificity | Low | High |
| Development costs | High | Low-moderate |
| Side effects | Moderate-high | Usually low, but yet to be fully established |
| Resistance | Increasing incidence of multi-drug resistant isolates. | Can treat multi-drug-resistant isolates. Phage resistant isolates generally lack fitness. |
| Delivery to target | Moderate | Moderate to good. Can penetrate the blood-brain barrier. |
| Formulation | Fixed | Fixed or variable |
| Regulation | Well established | Underdeveloped |
| Kinetics | Single hit | Single hit or self-amplifying |
| Immunogenicity | Variable | Likely low, but not well established |
| Clinical validation | Many trial studies | Relatively few trial studies |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Calle, D.; Guimarães Benevides, R.; Góes-Neto, A.; Billington, C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics 2019, 8, 138. https://doi.org/10.3390/antibiotics8030138
Romero-Calle D, Guimarães Benevides R, Góes-Neto A, Billington C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics. 2019; 8(3):138. https://doi.org/10.3390/antibiotics8030138
Chicago/Turabian StyleRomero-Calle, Danitza, Raquel Guimarães Benevides, Aristóteles Góes-Neto, and Craig Billington. 2019. "Bacteriophages as Alternatives to Antibiotics in Clinical Care" Antibiotics 8, no. 3: 138. https://doi.org/10.3390/antibiotics8030138
APA StyleRomero-Calle, D., Guimarães Benevides, R., Góes-Neto, A., & Billington, C. (2019). Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics, 8(3), 138. https://doi.org/10.3390/antibiotics8030138