Short Cationic Peptidomimetic Antimicrobials





Abstract
1. Antimicrobial Peptides
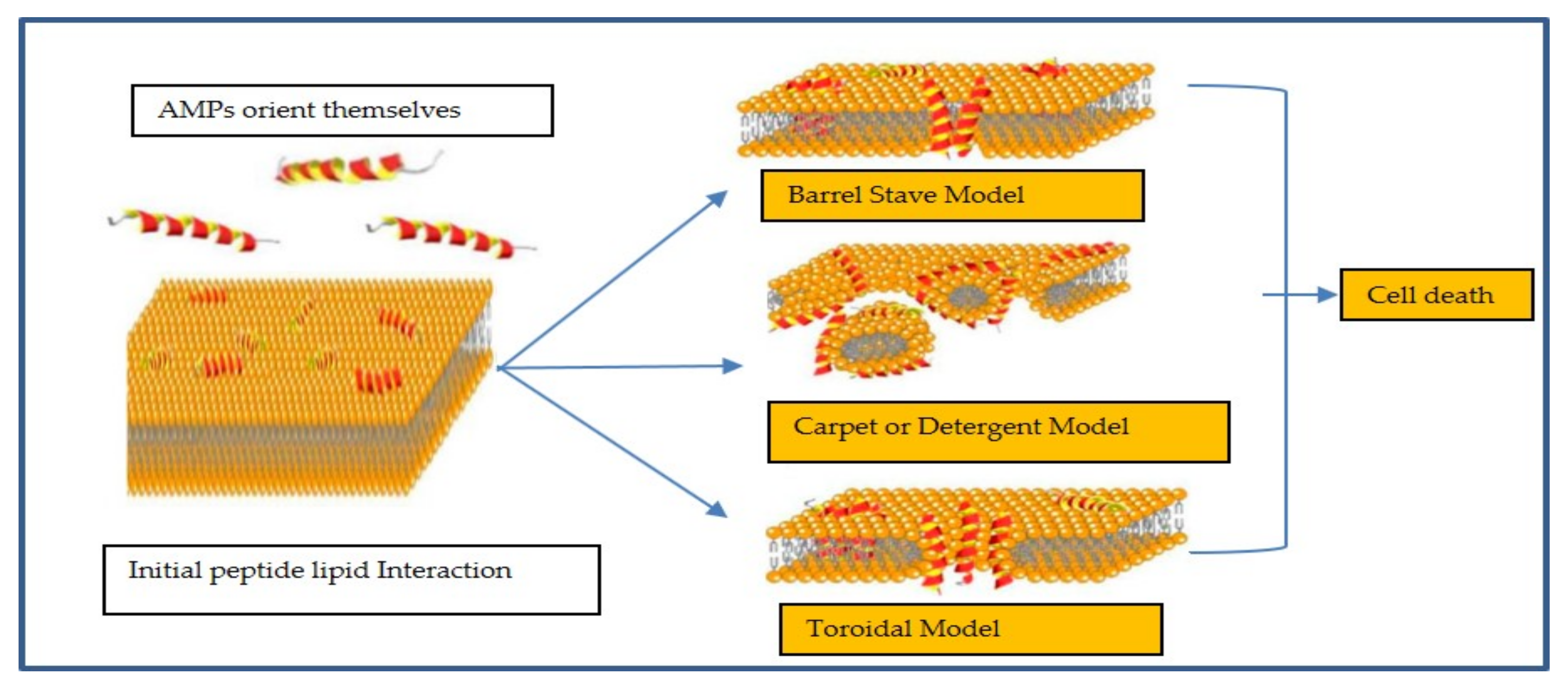
2. Mechanism of Action
3. Mechanism of Resistance
4. Development of Peptidomimetics
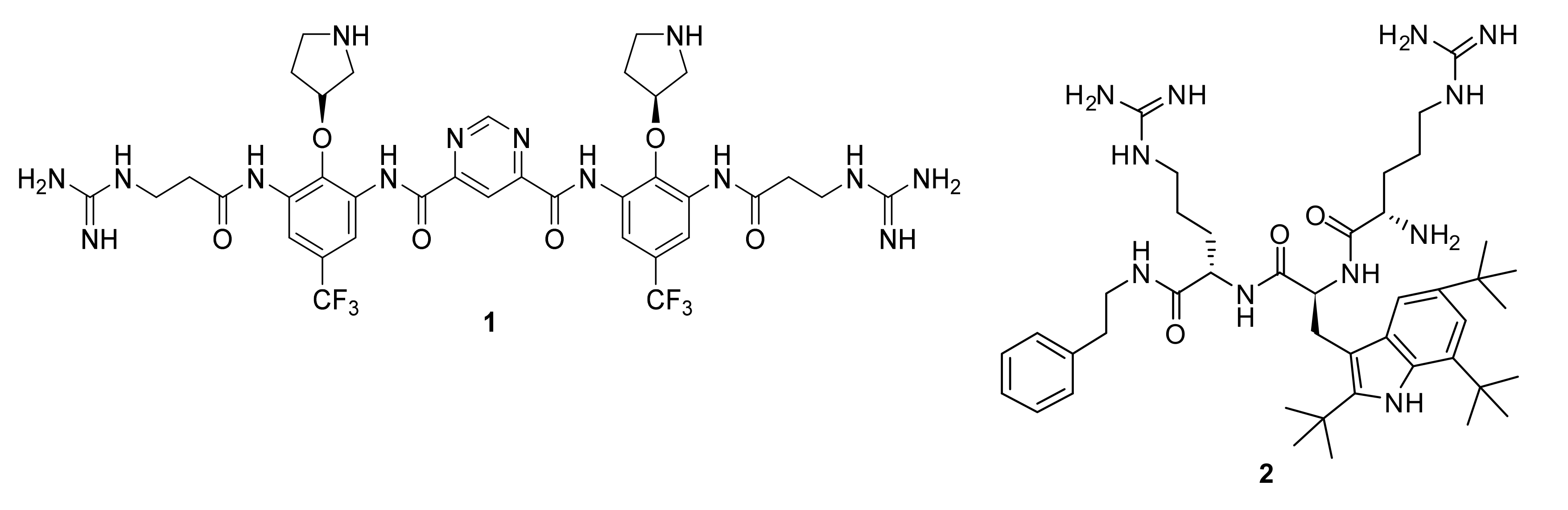
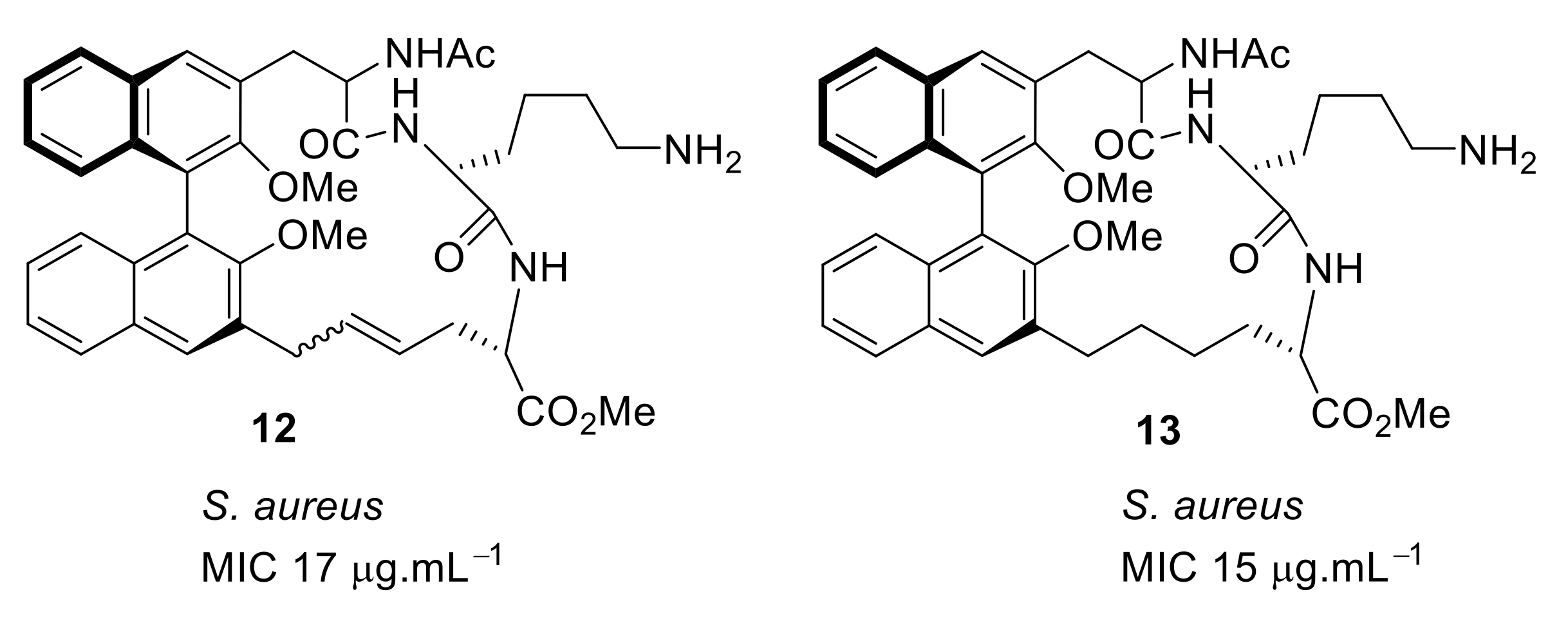
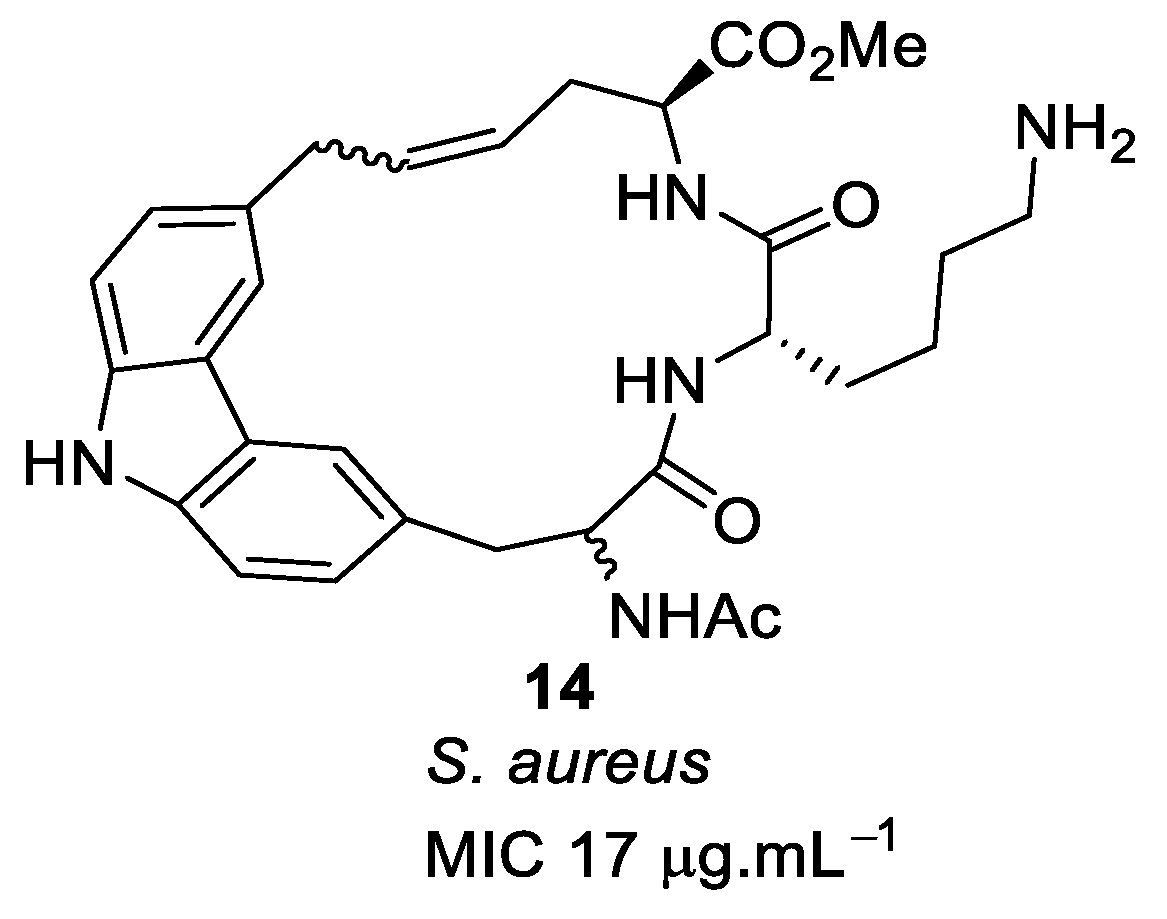
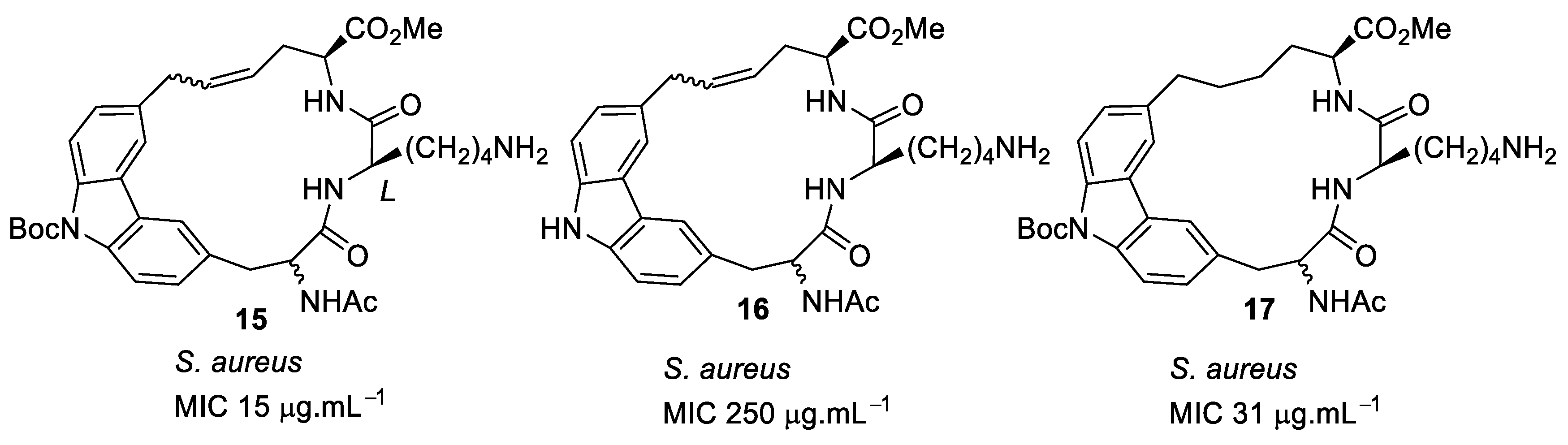
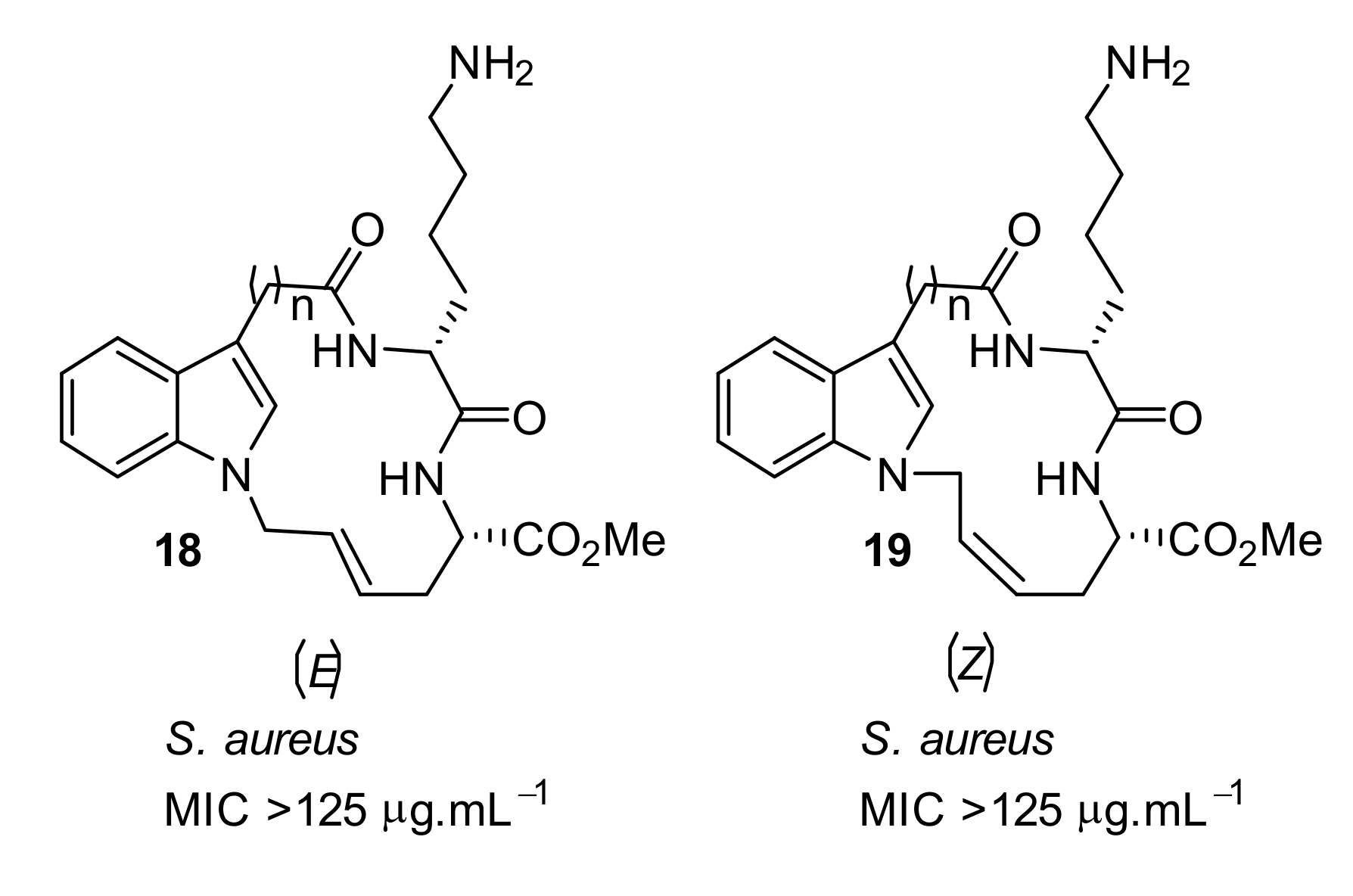
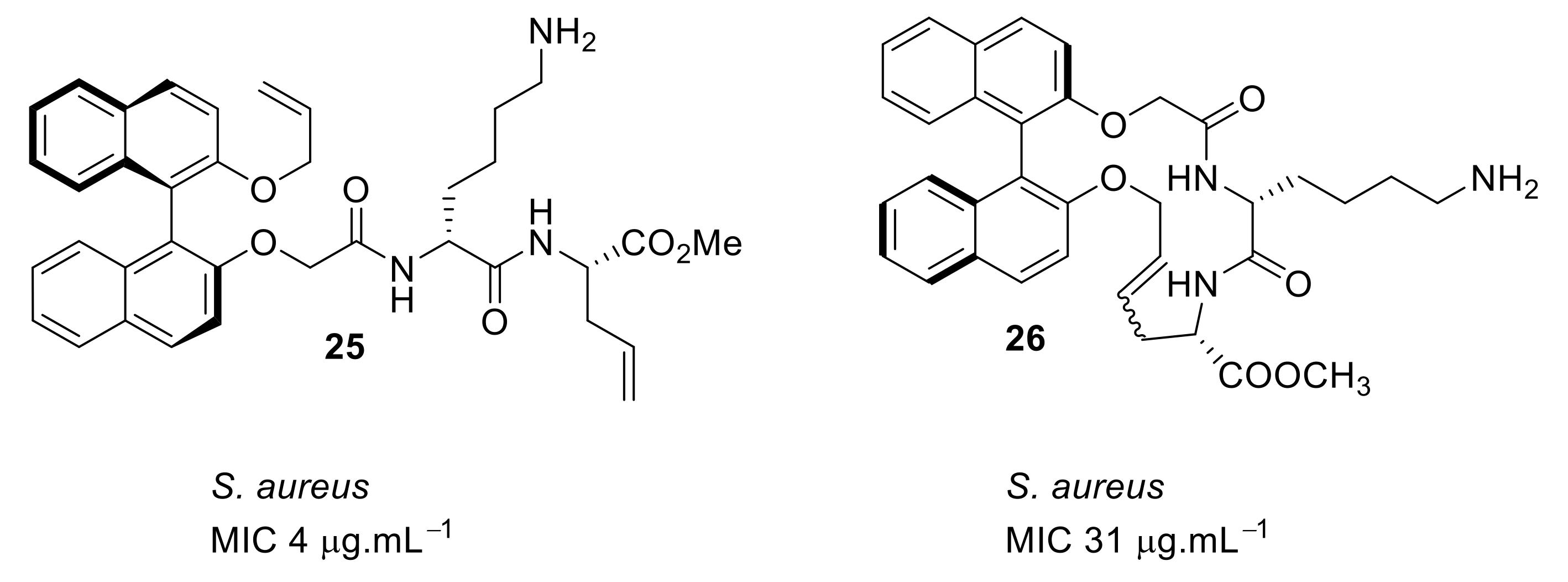
4.1. Binaphthyl Peptidomimetics
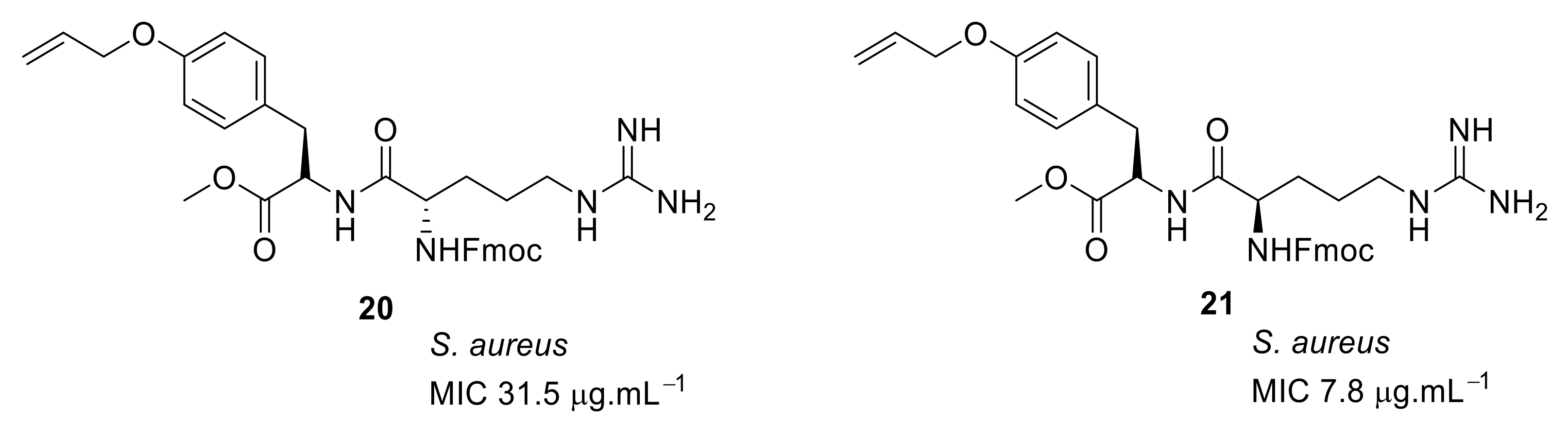
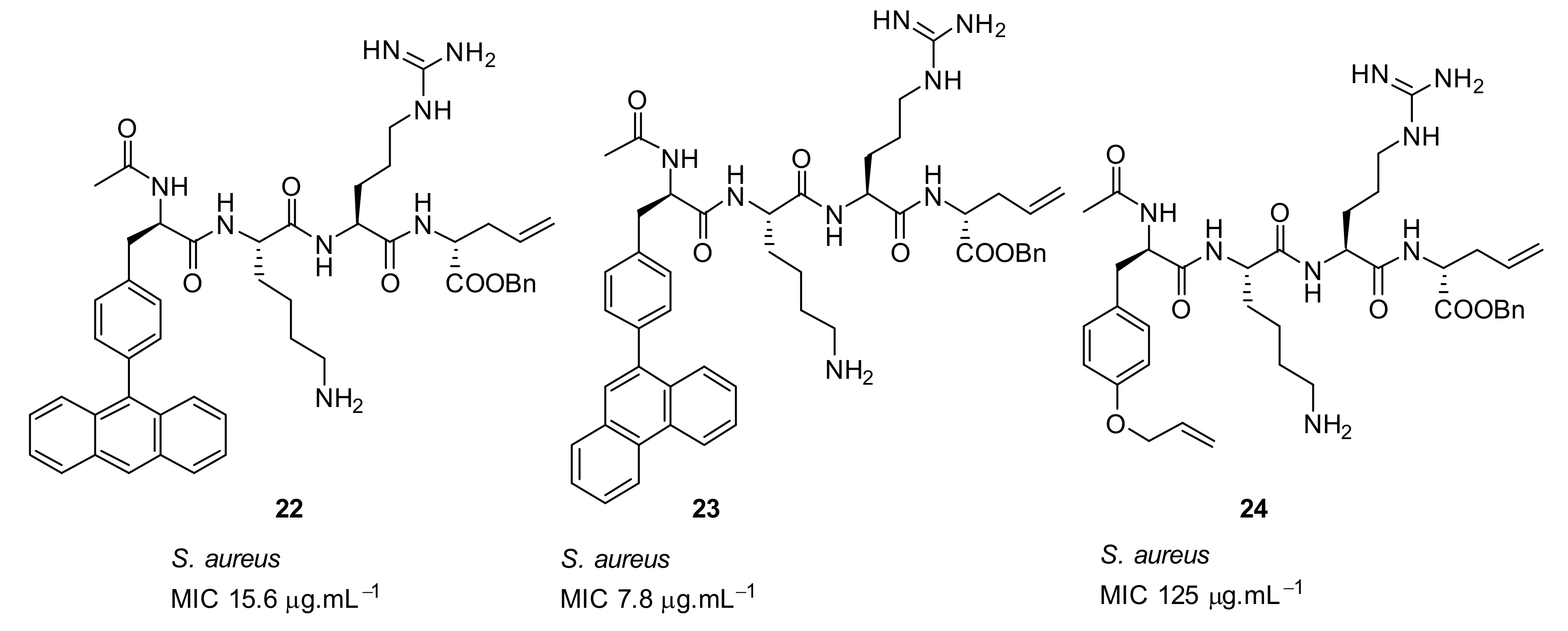
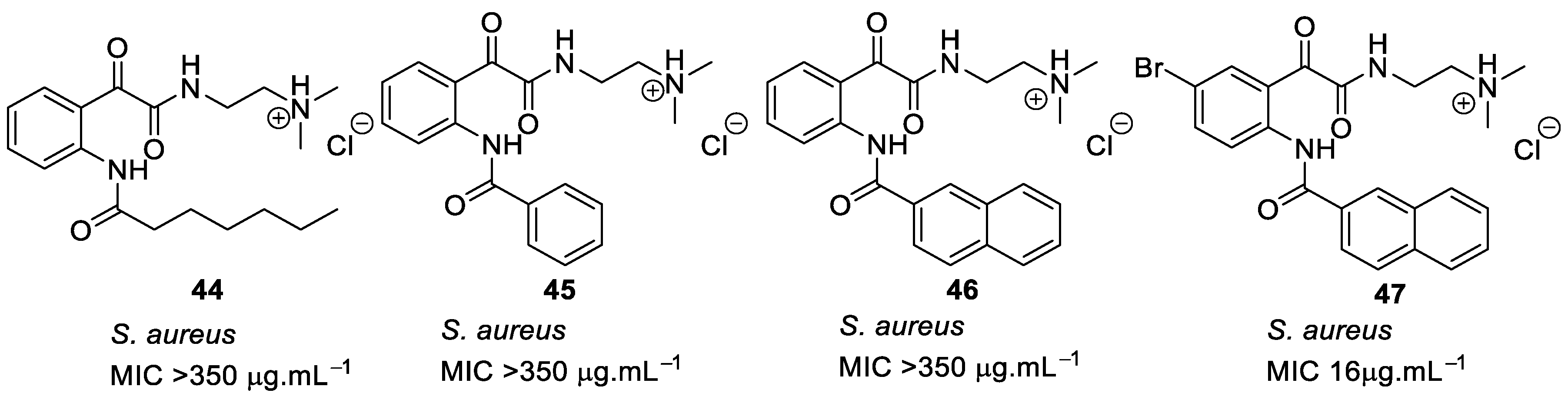
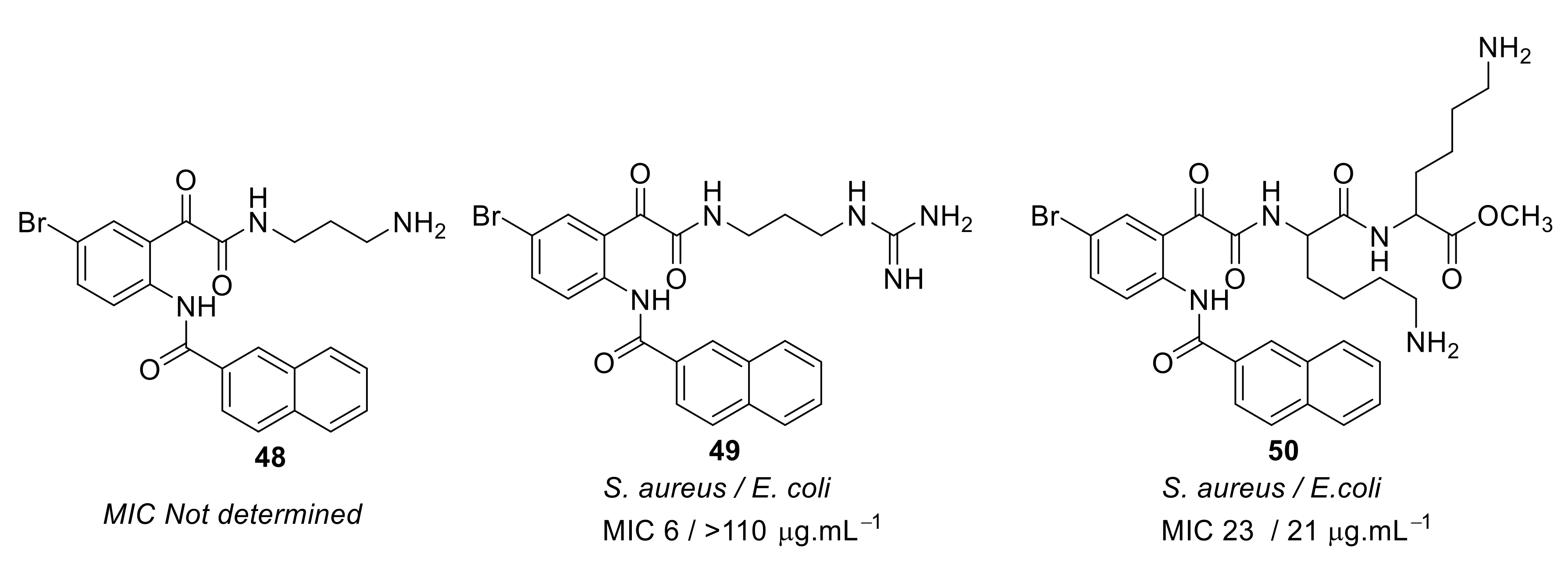
4.2. Glyoxamides and Carboxamide Peptidomimetics
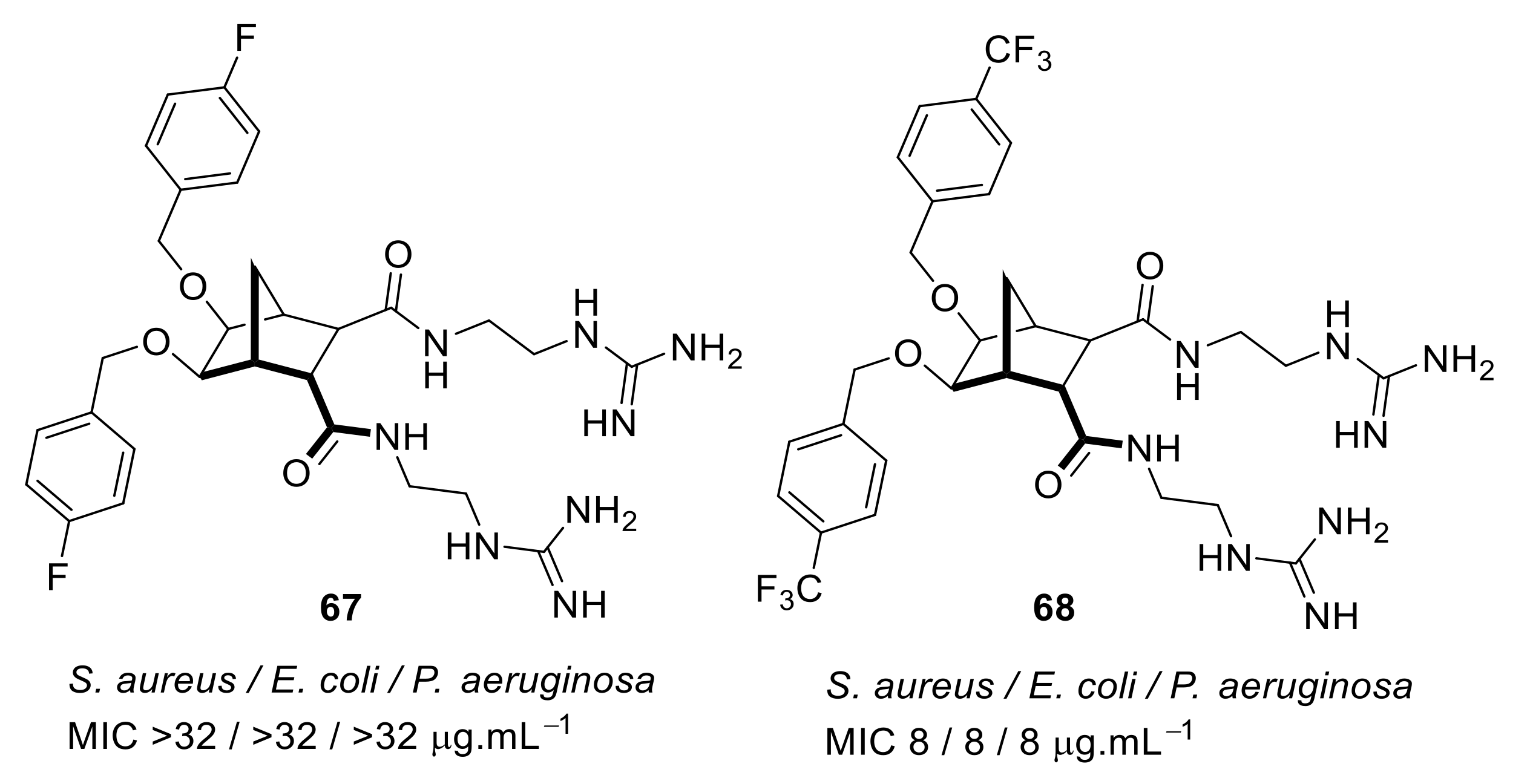
4.3. Norbornane Peptidomimetics
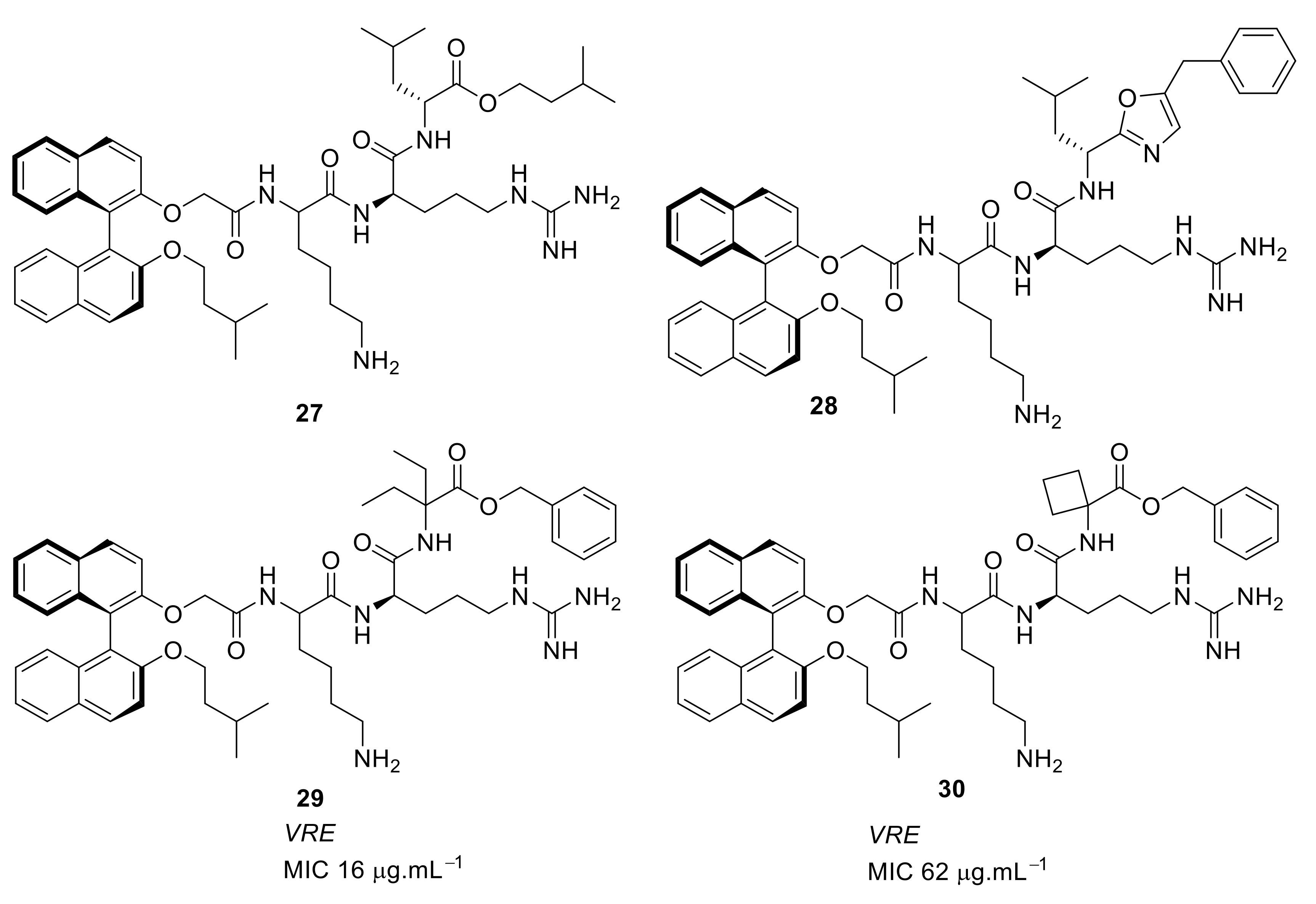
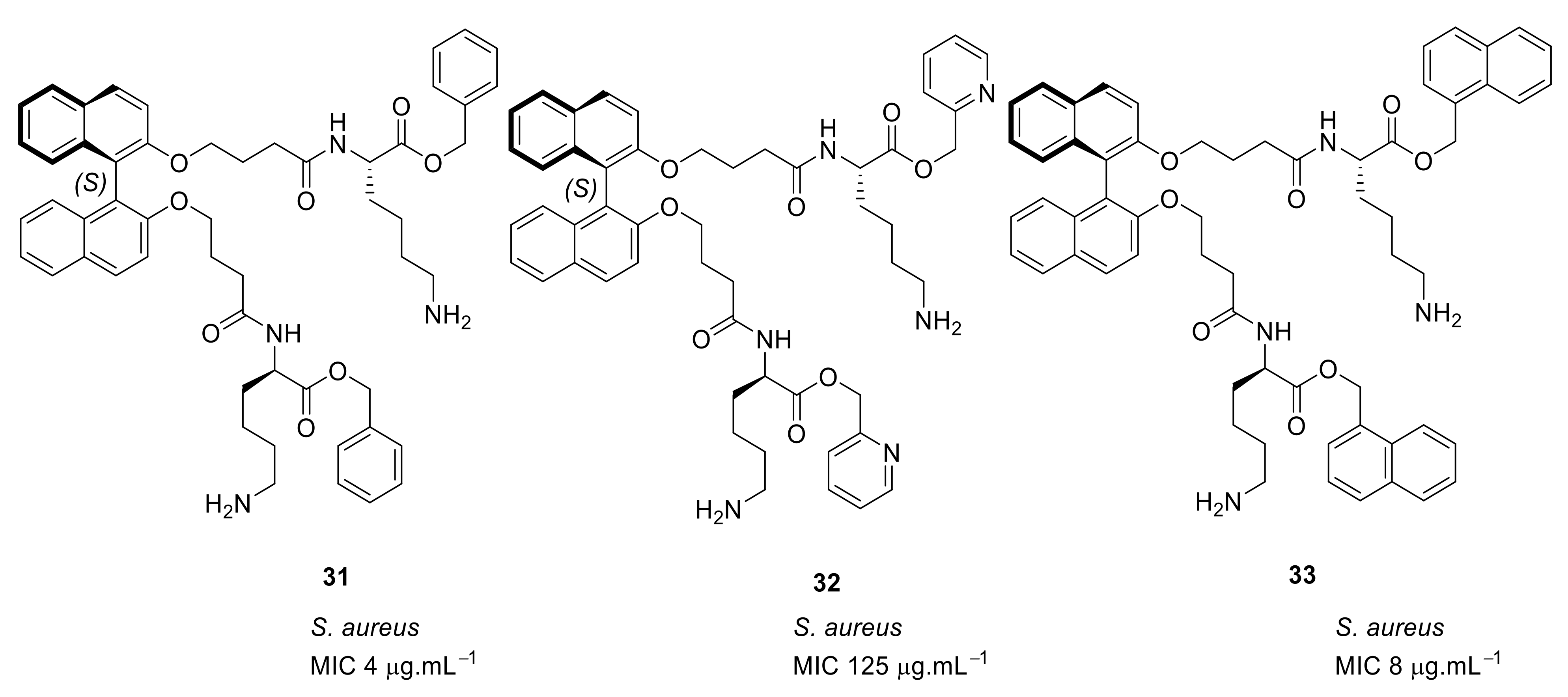
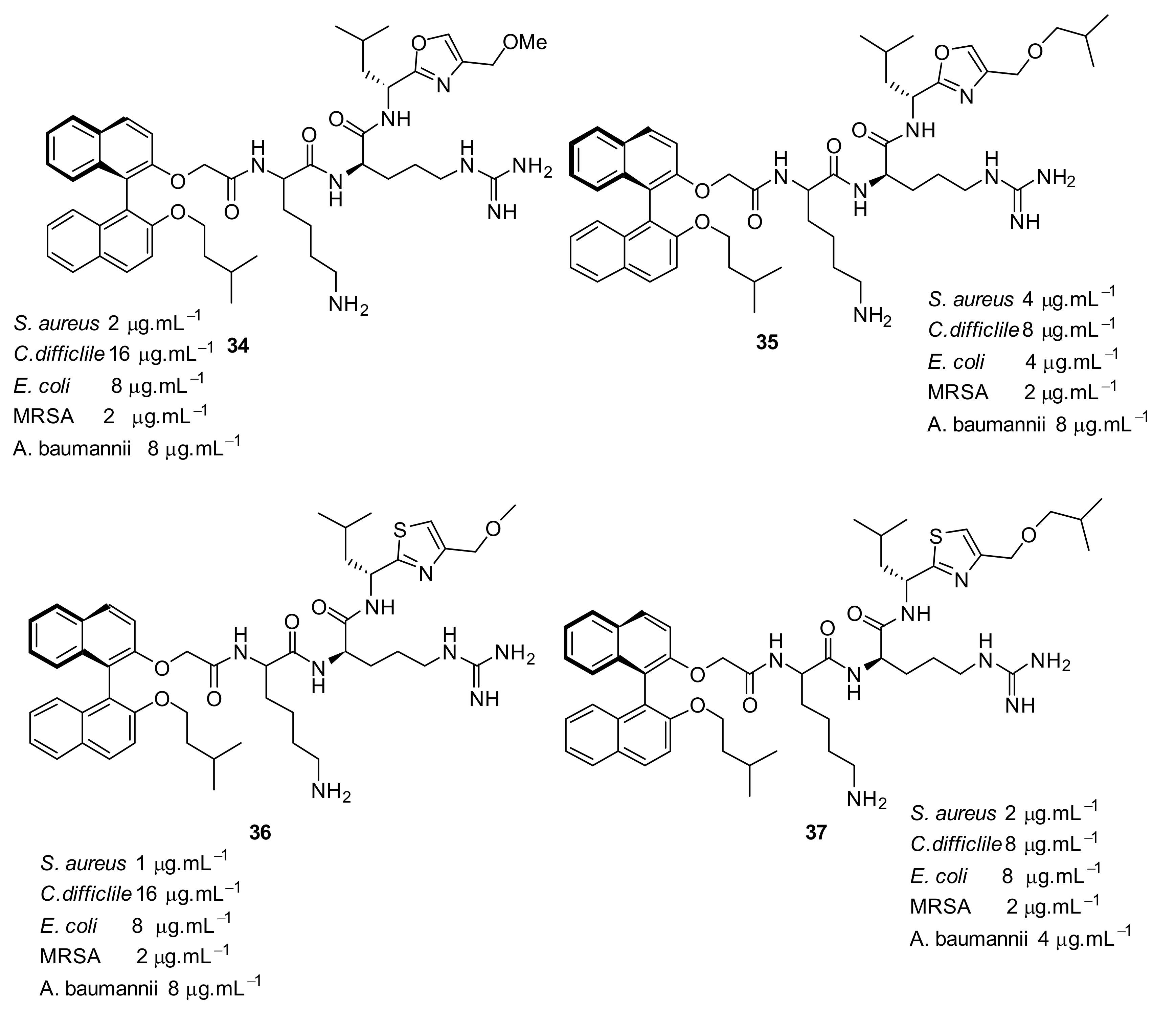
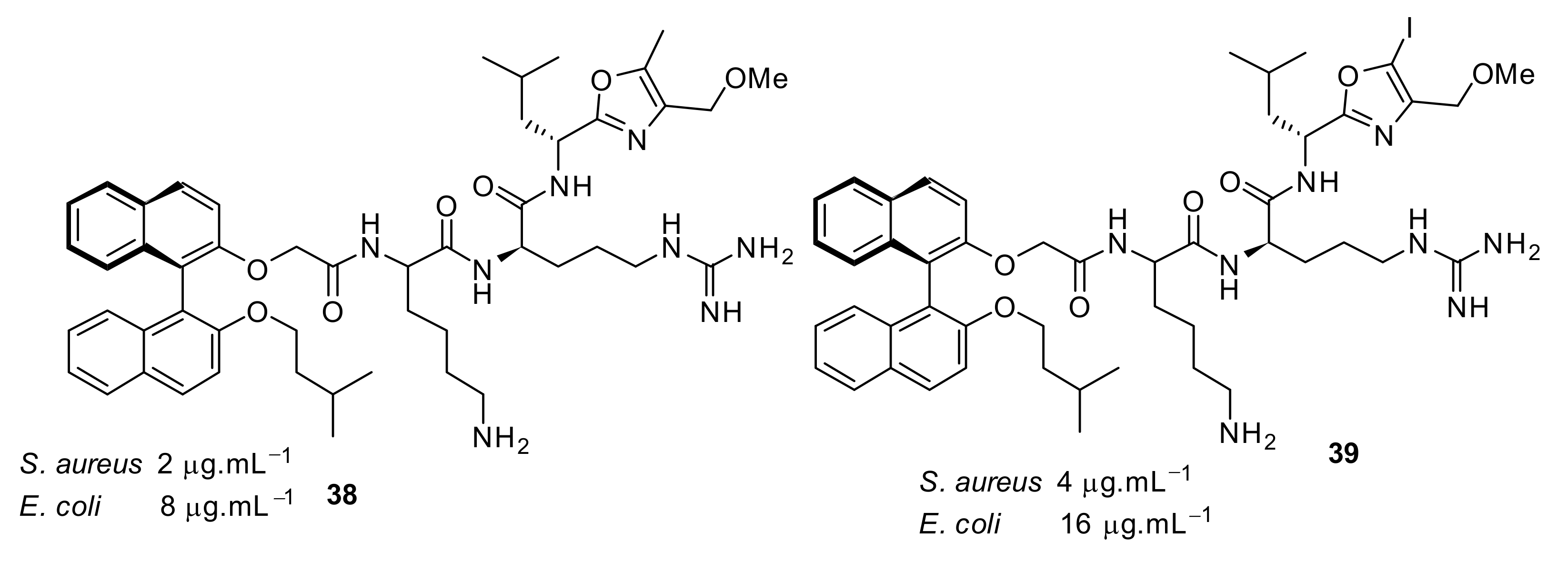
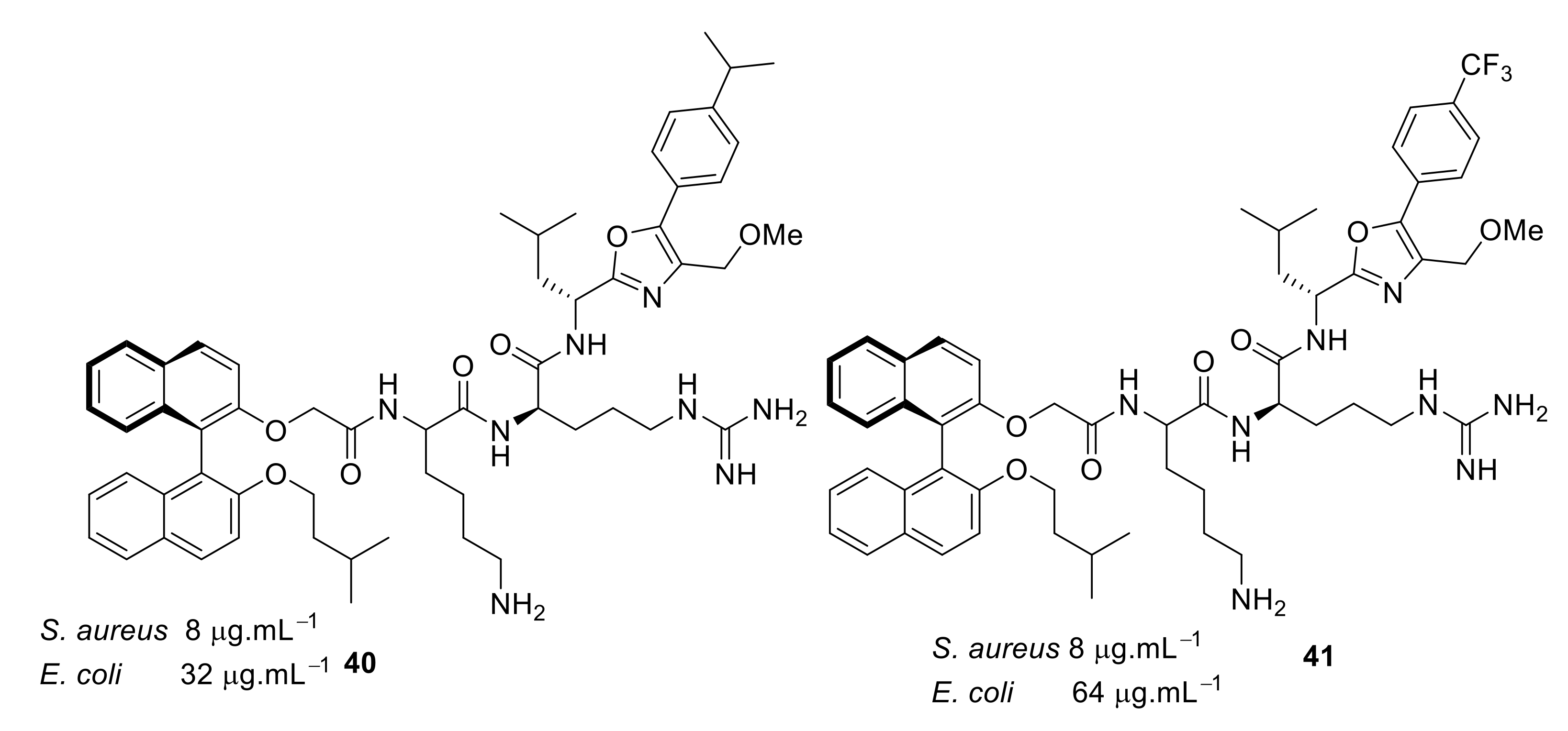
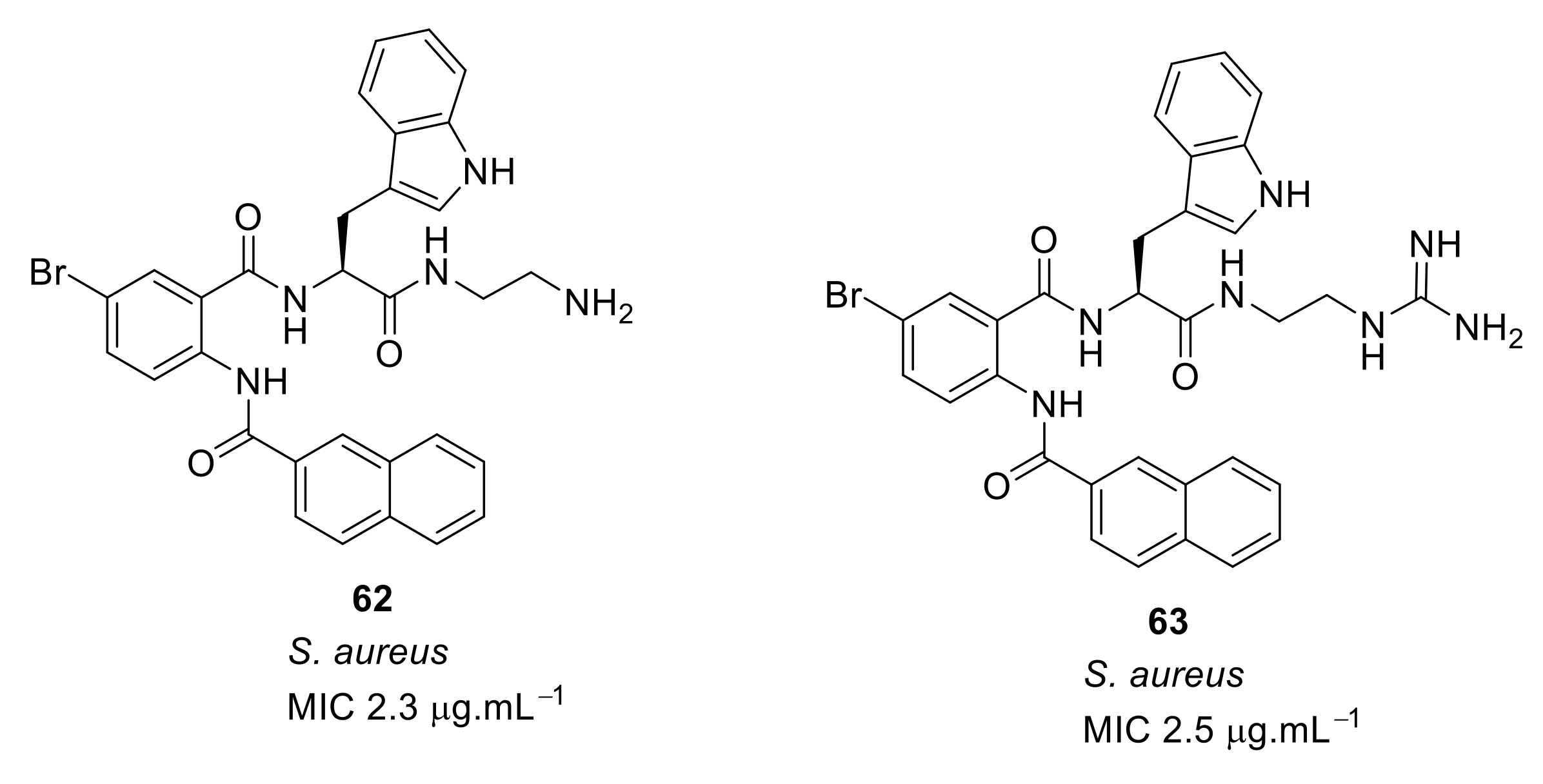
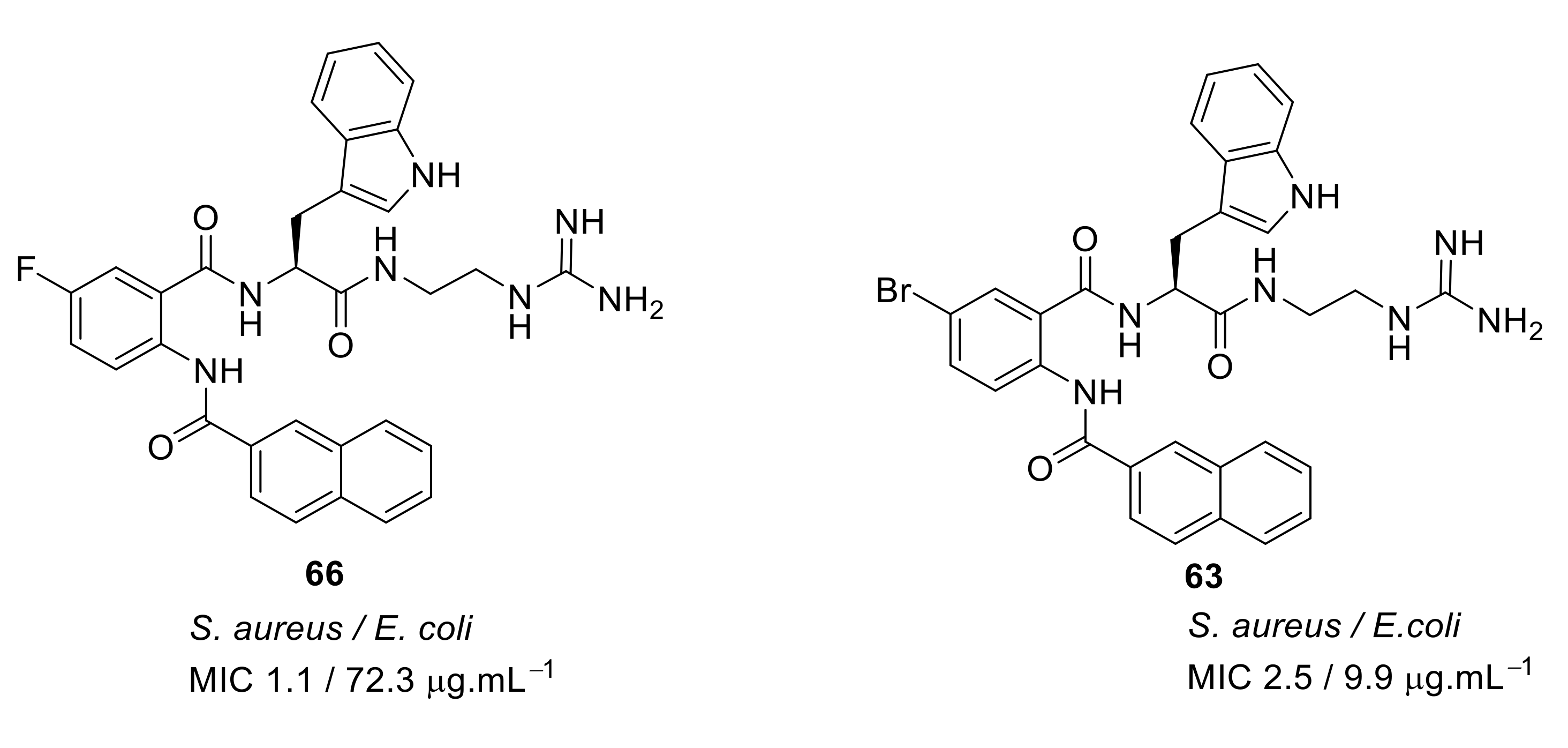
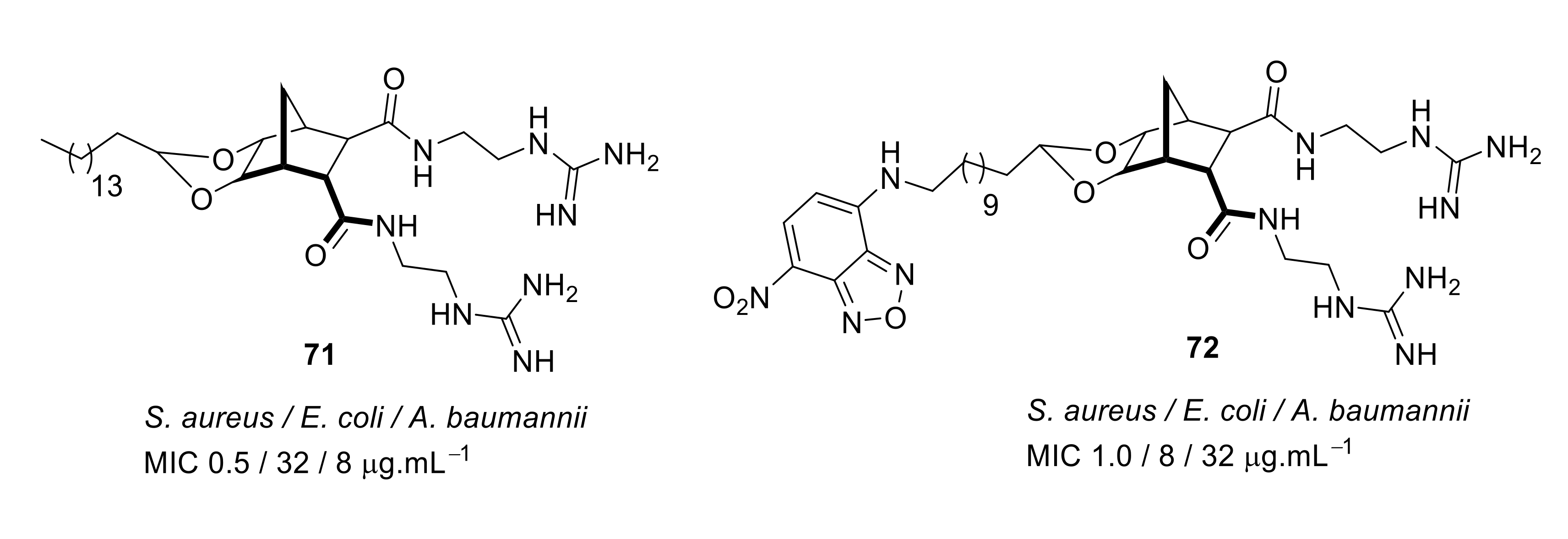
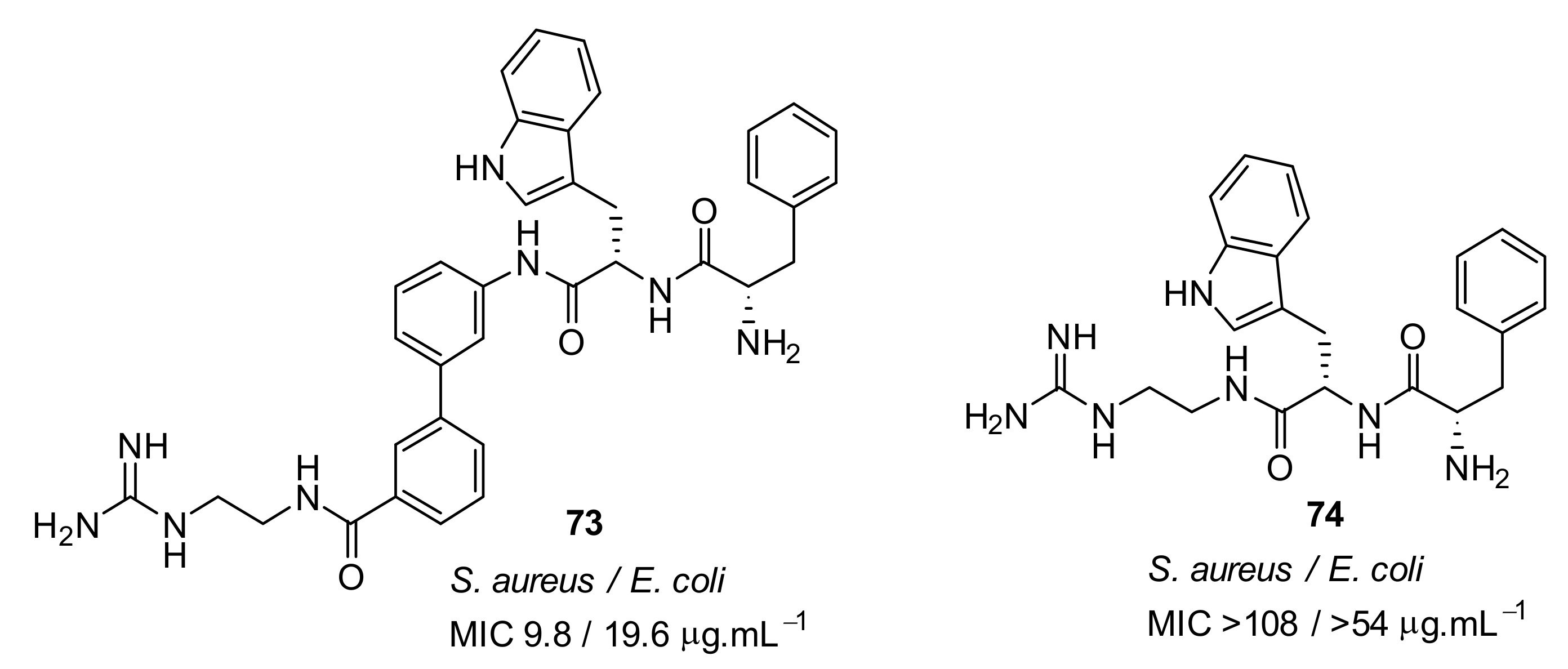
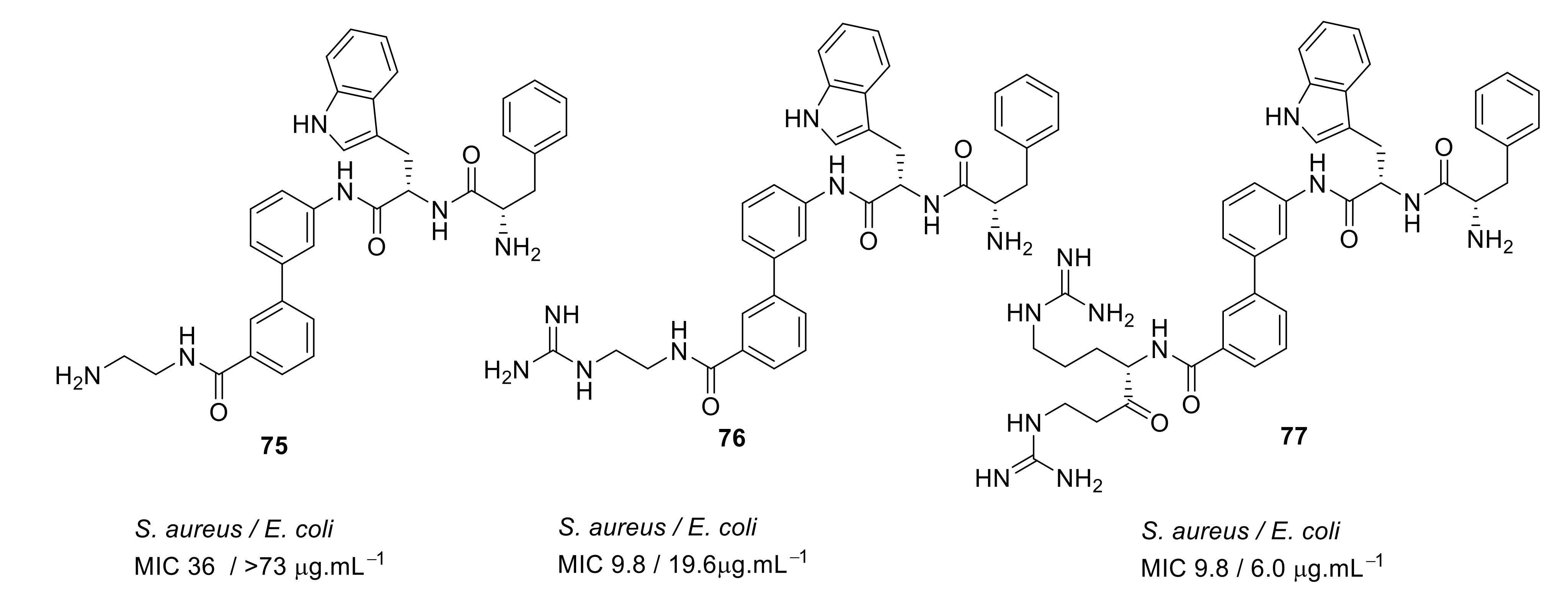
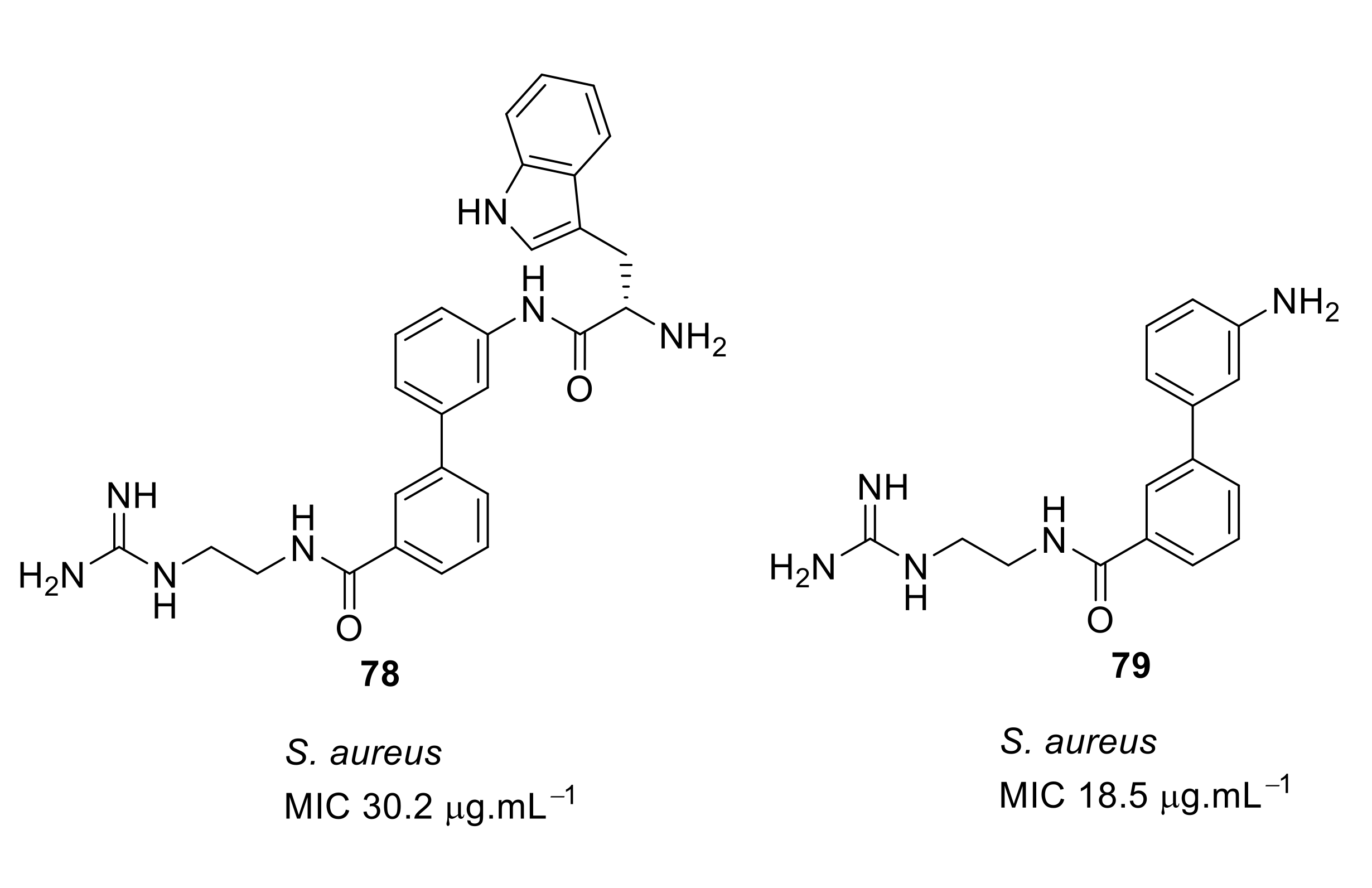
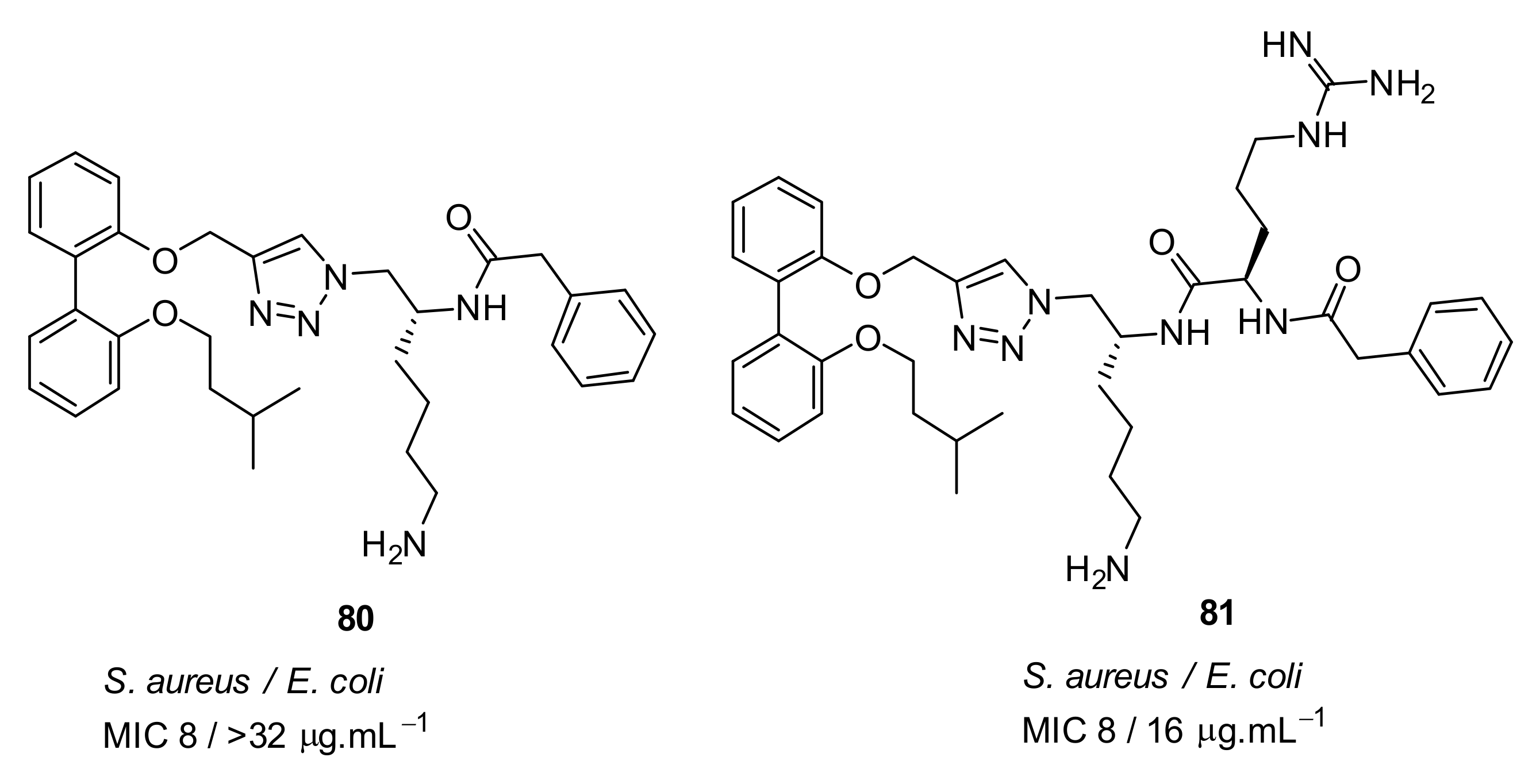
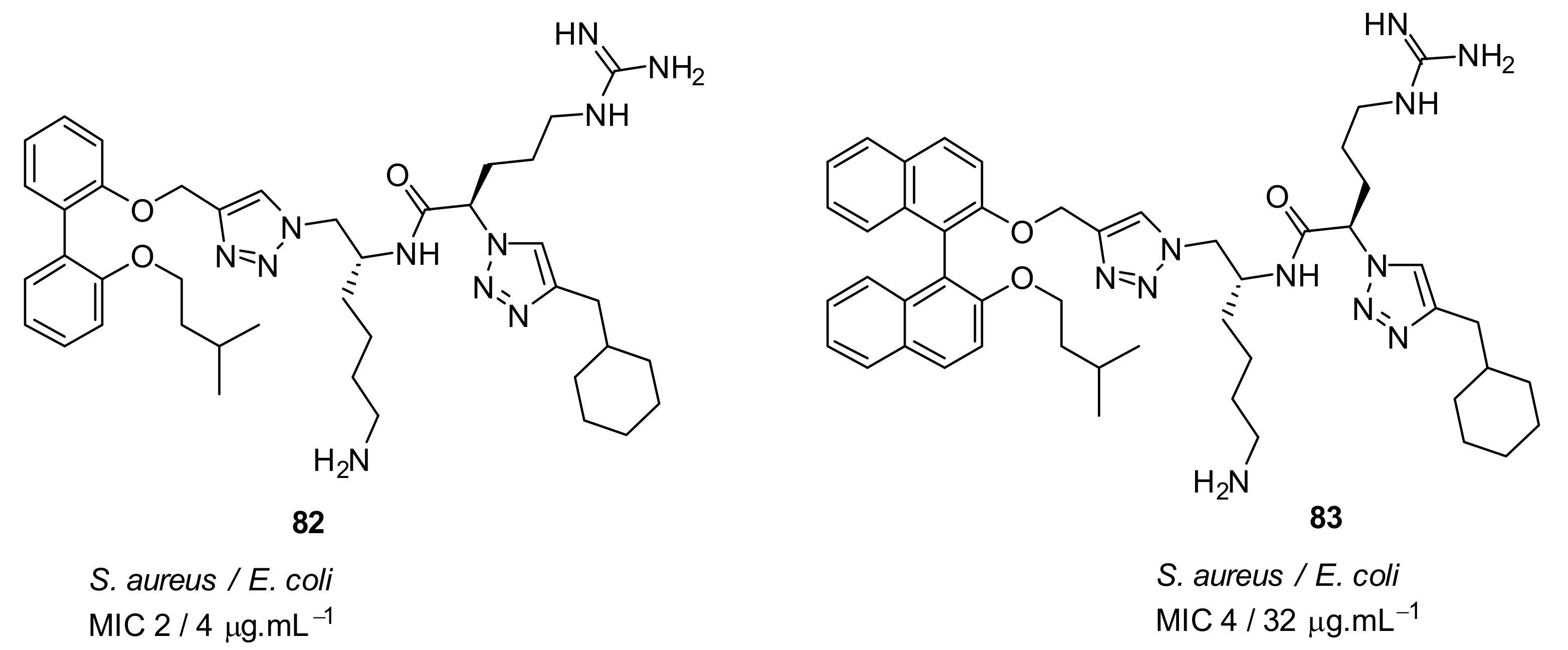
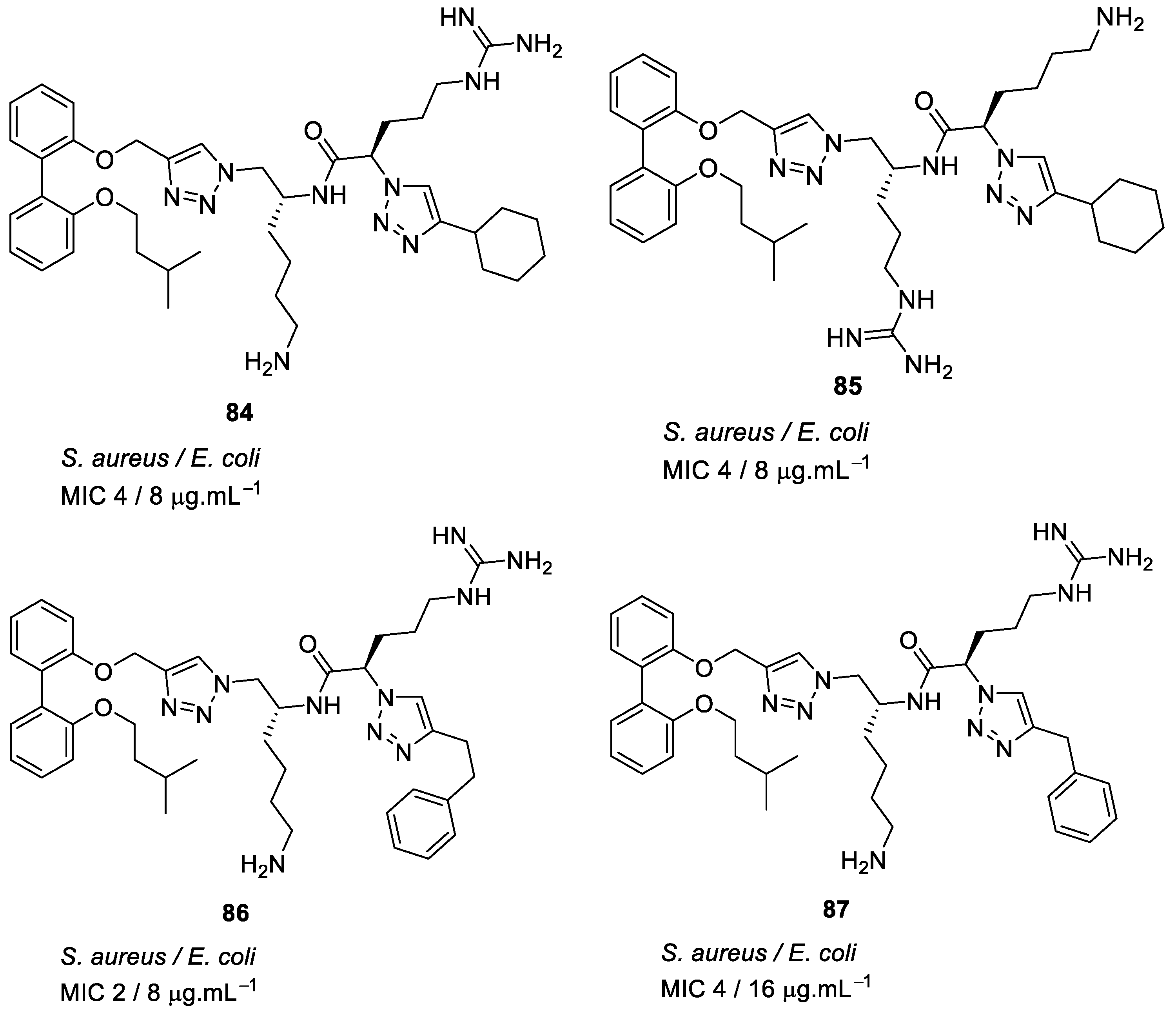
4.4. Biaryl (Biphenyl) Peptidomimetics
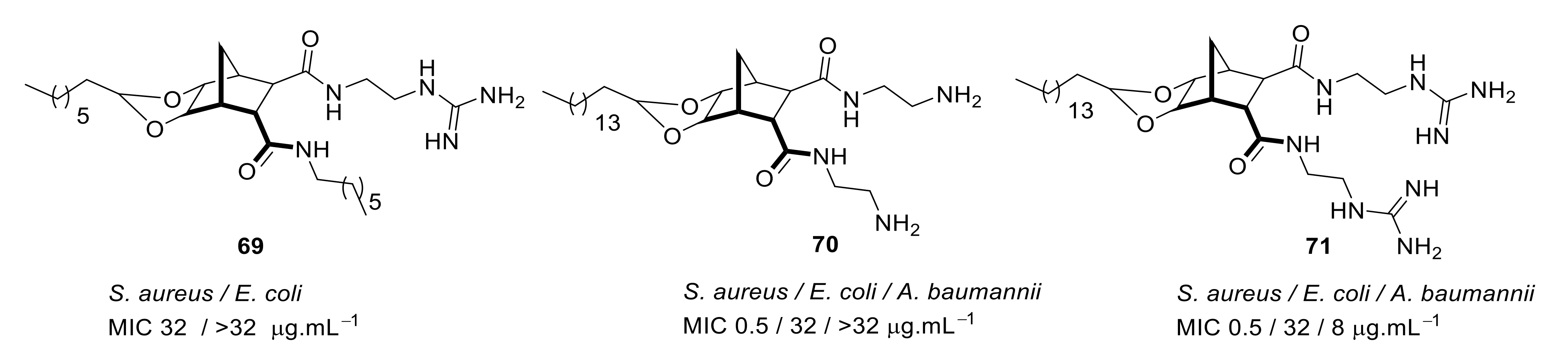
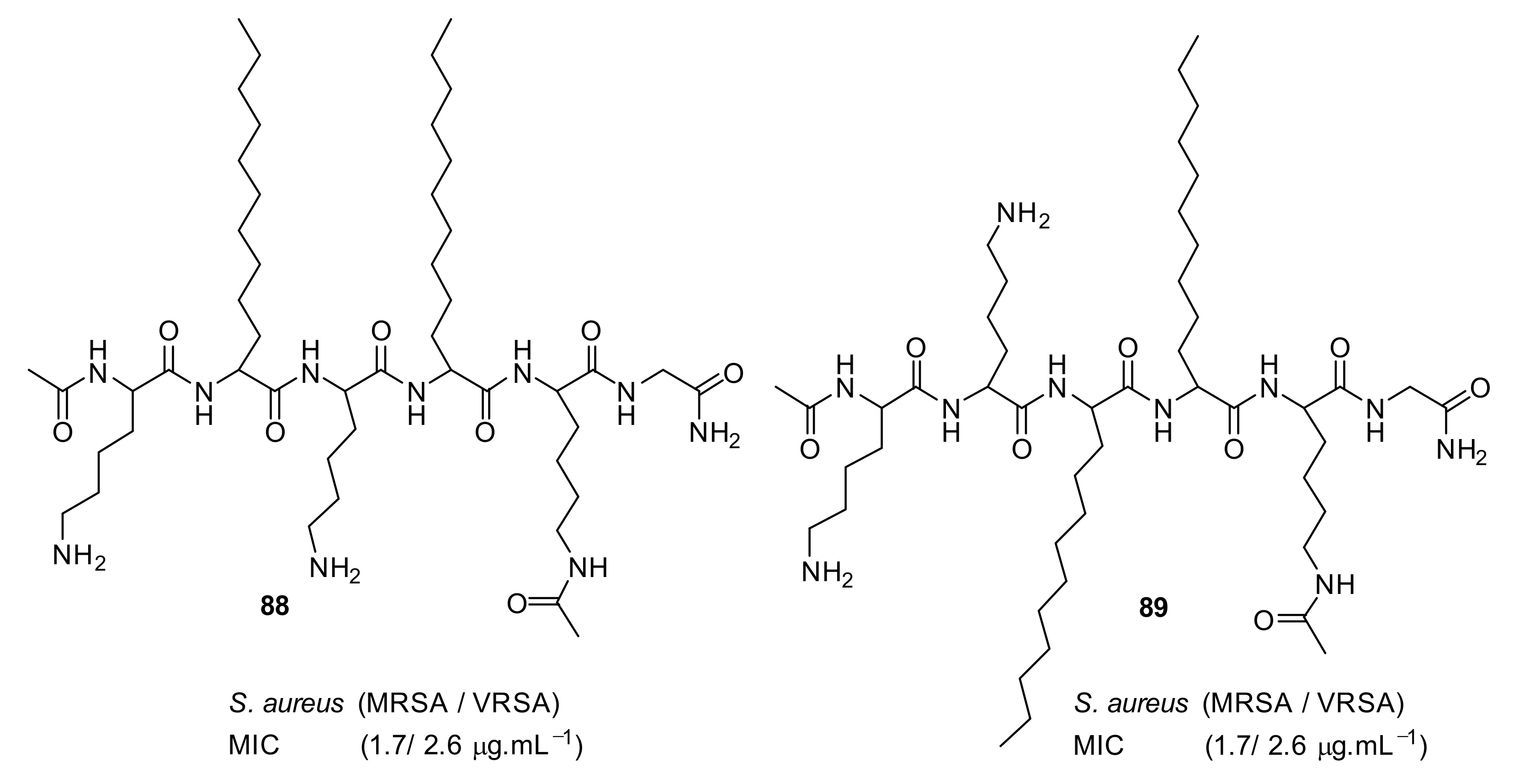
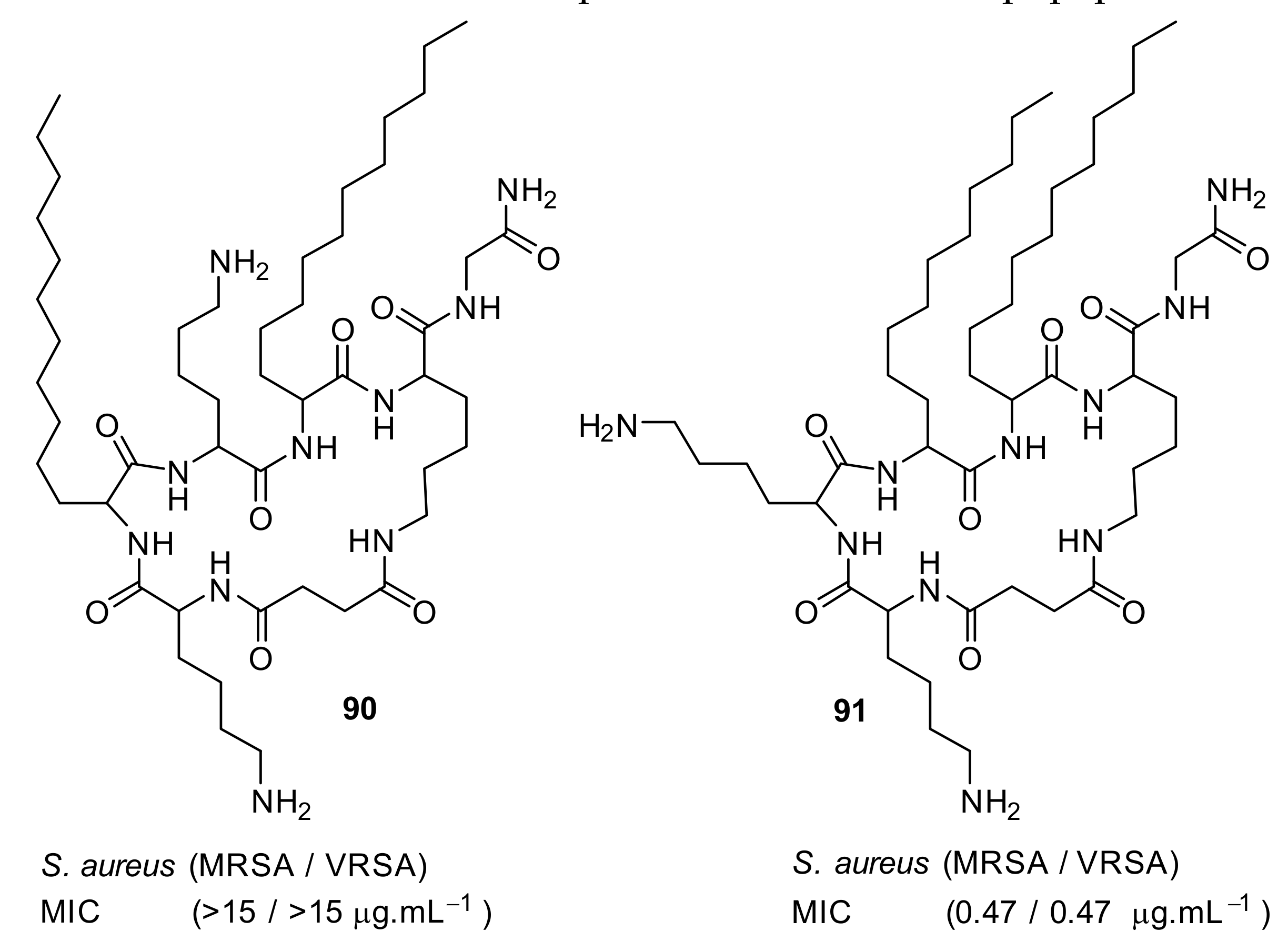
4.5. Short Cationic Lipopeptides
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lehrer, R.I.; Ganz, T. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol. 1999, 11, 23–27. [Google Scholar] [CrossRef]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, J. Cationic antimicrobial peptides: issues for potential clinical use. BioDrugs 2003, 17, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, α-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Park, C.B.; Yi, K.S.; Matsuzaki, K.; Kim, M.S.; Kim, S.C. Structure–activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. Proc. Natl. Acad. Sci. USA 2000, 97, 8245–8250. [Google Scholar] [CrossRef]
- Lay, F.T.; Anderson, M.A. Defensins—Components of the innate immune system in plants. Curr. Protein Pept. Sci. 2005, 6, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Molecular action mechanisms and membrane recognition of membrane-acting antimicrobial peptides. Yakugaku Zasshi 1997, 117, 253–264. [Google Scholar] [CrossRef][Green Version]
- Matsuzaki, K.; Yoneyama, S.; Fujii, N.; Miyajima, K.; Yamada, K.; Kirino, Y.; Anzai, K. Membrane permeabilization mechanisms of a cyclic antimicrobial peptide, tachyplesin I, and its linear analog. Biochemistry 1997, 36, 9799–9806. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [Google Scholar] [CrossRef]
- Oppenheim, F.G.; Xu, T.; McMillian, F.M.; Levitz, S.M.; Diamond, R.D.; Offner, G.D.; Troxler, R.F. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J. Biol. Chem. 1988, 263, 7472–7477. [Google Scholar]
- Huq, N.L.; Cross, K.J.; Ung, M.; Myroforidis, H.; Veith, P.D.; Chen, D.; Stanton, D.; He, H.; Ward, B.R.; Reynolds, E.C. A review of the salivary proteome and peptidome and saliva-derived peptide therapeutics. Int. J. Pept. Res. Ther. 2007, 13, 547–564. [Google Scholar] [CrossRef]
- Cabiaux, V.; Agerberth, B.; Johansson, J.; Homble, F.; Goormaghtigh, E.; Ruysschaert, J.M. Secondary structure and membrane interaction of PR-39, a Pro+Arg-rich antibacterial peptide. Eur. J. Biochem. 1994, 224, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Conti, S.; Radicioni, G.; Ciociola, T.; Longhi, R.; Polonelli, L.; Gatti, R.; Cabras, T.; Messana, I.; Castagnola, M.; Vitali, A. Structural and functional studies on a proline-rich peptide isolated from swine saliva endowed with antifungal activity towards Cryptococcus neoformans. Biochim. Biophys. Acta 2013, 1828, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhao, X.-Y.; Guo, S.; Yang, Q. Effect of loop structure of bovine lactoferricin on Escherichia Coli. AFSE 2018, 2. [Google Scholar] [CrossRef]
- Abdel Monaim, S.A.H.; Somboro, A.M.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Bacteria hunt bacteria through an intriguing cyclic peptide. Chem. Med. Chem. 2019, 14, 24–51. [Google Scholar] [CrossRef] [PubMed]
- Clifton, L.A.; Skoda, M.W.A.; Le Brun, A.P.; Ciesielski, F.; Kuzmenko, I.; Holt, S.A.; Lakey, J.H. Effect of divalent cation removal on the structure of Gram-negative bacterial outer membrane models. Langmuir 2015, 31, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.V.; Rao, S.S.; Atreya, C.D. Evaluation of antimicrobial peptides as novel bactericidal agents for room temperature-stored platelets. Transfusion 2010, 50, 166–173. [Google Scholar] [CrossRef]
- Straus, S.K.; Hancock, R.E. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 2006, 1758, 1215–1223. [Google Scholar] [CrossRef]
- Sang, Y.; Blecha, F. Antimicrobial peptides and bacteriocins: alternatives to traditional antibiotics. Anim. Health Res. Rev. 2008, 9, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta 2016, 1858, 936–946. [Google Scholar] [CrossRef]
- Biswas, R.; Martinez, R.E.; Gohring, N.; Schlag, M.; Josten, M.; Xia, G.; Hegler, F.; Gekeler, C.; Gleske, A.K.; Gotz, F.; et al. Proton-binding capacity of Staphylococcus aureus wall teichoic acid and its role in controlling autolysin activity. PLoS ONE 2012, 7, e41415. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Mensa, B.; Howell, G.L.; Scott, R.; DeGrado, W.F. Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Toke, O. Antimicrobial peptides: New candidates in the fight against bacterial infections. Biopolymers 2005, 80, 717–735. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Zhang, L.; Rozek, A.; Hancock, R.E. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar] [CrossRef]
- Shimazaki, K.; Tazume, T.; Uji, K.; Tanaka, M.; Kumura, H.; Mikawa, K.; Shimo-Oka, T. Properties of a heparin-binding peptide derived from bovine lactoferrin. J. Dairy Sci. 1998, 81, 2841–2849. [Google Scholar] [CrossRef]
- Epand, R.M.; Shai, Y.; Segrest, J.P.; Anantharamaiah, G.M. Mechanisms for the modulation of membrane bilayer properties by amphipathic helical peptides. Biopolymers 1995, 37, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Spaar, A.; Munster, C.; Salditt, T. Conformation of peptides in lipid membranes studied by x-ray grazing incidence scattering. Biophys. J. 2004, 87, 396–407. [Google Scholar] [CrossRef]
- Vad, B.S.; Bertelsen, K.; Johansen, C.H.; Pedersen, J.M.; Skrydstrup, T.; Nielsen, N.C.; Otzen, D.E. Pardaxin permeabilizes vesicles more efficiently by pore formation than by disruption. Biophys. J. 2010, 98, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Jenssen, H. Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid-membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Bechinger, B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta 1999, 1462, 157–183. [Google Scholar] [CrossRef]
- Sitaram, N.; Nagaraj, R. Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim. Biophys. Acta 1999, 1462, 29–54. [Google Scholar] [CrossRef]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.; Hale, J.D.; Elliot, M.; Hancock, R.E.; Straus, S.K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2009, 96, 552–565. [Google Scholar] [CrossRef]
- Guan, Q.; Huang, S.; Jin, Y.; Campagne, R.; Alezra, V.; Wan, Y. Recent advances in the exploration of therapeutic analogues of Gramicidin S, an old but still potent antimicrobial peptide. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Katsu, T.; Imamura, T.; Komagoe, K.; Masuda, K.; Mizushima, T. Simultaneous measurements of K+ and calcein release from liposomes and the determination of pore size formed in a membrane. Anal. Sci. 2007, 23, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Rautenbach, M.; Vosloo, J.A.; Siersma, T.; Aisenbrey, C.H.M.; Zaitseva, E.; Laubscher, W.E.; van Rensburg, W.; Behrends, J.C.; Bechinger, B.; et al. The multifaceted antibacterial mechanisms of the pioneering peptide antibiotics tyrocidine and gramicidin S. mBio 2018, 9, e00802–e00818. [Google Scholar] [CrossRef]
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane active antimicrobial peptides: Translating mechanistic insights to design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef]
- Lewis, L.A.; Choudhury, B.; Balthazar, J.T.; Martin, L.E.; Ram, S.; Rice, P.A.; Stephens, D.S.; Carlson, R.; Shafer, W.M. Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect. Immun. 2009, 77, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S. Bacterial modification of LPS and resistance to antimicrobial peptides. J. Endotoxin Res. 2001, 7, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lim, K.B.; Poduje, C.M.; Daniel, M.; Gunn, J.S.; Hackett, M.; Miller, S.I. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar] [CrossRef]
- Guina, T.; Yi, E.C.; Wang, H.; Hackett, M.; Miller, S.I. A PhoP-regulated outer membrane protease of Salmonella enterica serovar typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 2000, 182, 4077–4086. [Google Scholar] [CrossRef]
- Shafer, W.M.; Qu, X.D.; Waring, A.J.; Lehrer, R.I. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a, member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, F.J.; del Castillo, I.; Moreno, F. Construction and characterization of mutations at codon 751 of the Escherichia coli gyrB gene that confer resistance to the antimicrobial peptide microcin B17 and alter the activity of DNA gyrase. J. Bacteriol. 2001, 183, 2137–2140. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.; Scott, M.G.; Karunaratne, N.; Yan, H.; Hancock, R.E. Salt-resistant alpha-helical cationic antimicrobial peptides. Antimicrob. Agents Chemother. 1999, 43, 1542–1548. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Bayer, A.S.; Koo, S.P.; Foss, W.; Sullam, P.M. Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J. Clin. Investig. 1998, 101, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm formation mechanisms and targets for developing antibiofilm agents. Future Med. Chem. 2015, 7. [Google Scholar] [CrossRef]
- Van Hoek, A.H.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J. Acquired antibiotic resistance genes: an overview. Front. Microbiol. 2011, 2, 203. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, B.; Liang, B.; Xu, X.; Qiu, S.; Jia, L.; Li, P.; Yang, L.; Li, Y.; Xiang, Y.; et al. A novel mcr-1 variant carried by an IncI2-type plasmid identified from a multidrug resistant enterotoxigenic Escherichia coli. Front. Microbiol. 2018, 9, 815. [Google Scholar] [CrossRef]
- Dobias, J.; Poirel, L.; Nordmann, P. Cross-resistance to human cationic antimicrobial peptides and to polymyxins mediated by the plasmid-encoded MCR-1? Clin. Microbiol. Infect. 2017, 23. [Google Scholar] [CrossRef]
- Yeung, A.T.; Gellatly, S.L.; Hancock, R.E. Multifunctional cationic host defence peptides and their clinical applications. Cell Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef]
- Yang, Q.; Li, M.; Spiller, O.B.; Andrey, D.O.; Hinchliffe, P.; Li, H.; MacLean, C.; Niumsup, P.; Powell, L.; Pritchard, M.; et al. Balancing mcr-1 expression and bacterial survival is a delicate equilibrium between essential cellular defence mechanisms. Nat. Commun. 2017, 8, 2054. [Google Scholar] [CrossRef] [PubMed]
- Andres, E. Cationic antimicrobial peptides in clinical development, with special focus on thanatin and heliomicin. Eur J. Clin. Microbiol. Infect. Dis 2012, 31, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Ciumac, D.; Gong, H.; Hu, X.; Lu, J.R. Membrane targeting cationic antimicrobial peptides. J. Colloid Interface Sci. 2019, 537, 163–185. [Google Scholar] [CrossRef] [PubMed]
- Huttner, B.; Jones, M.; Rubin, M.A.; Neuhauser, M.M.; Gundlapalli, A.; Samore, M. Drugs of last resort? The use of polymyxins and tigecycline at US Veterans affairs medical centers, 2005-2010. PLoS ONE 2012, 7, e36649. [Google Scholar] [CrossRef]
- Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular mechanisms of colistin-induced nephrotoxicity. Molecules 2019, 24, 653. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef]
- Ghosh, C.; Haldar, J. Membrane-active small molecules: Designs inspired by antimicrobial peptides. Chem. Med. Chem. 2015, 10, 1606–1624. [Google Scholar] [CrossRef]
- Sarkar, P.; Yarlagadda, V.; Ghosh, C.; Haldar, J. A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. Medchemcomm 2017, 8, 516–533. [Google Scholar] [CrossRef]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to conventional antibiotics in the era of antimicrobial resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef]
- Isaksson, J.; Brandsdal, B.O.; Engqvist, M.; Flaten, G.E.; Svendsen, J.S.; Stensen, W. A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J. Med. Chem. 2011, 54, 5786–5795. [Google Scholar] [CrossRef]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar] [CrossRef]
- Koh, J.J.; Lin, S.; Aung, T.T.; Lim, F.; Zou, H.; Bai, Y.; Li, J.; Lin, H.; Pang, L.M.; Koh, W.L.; et al. Amino acid modified xanthone derivatives: novel, highly promising membrane-active antimicrobials for multidrug-resistant Gram-positive bacterial infections. J. Med. Chem. 2015, 58, 739–752. [Google Scholar] [CrossRef]
- Bucki, R.; Niemirowicz, K.; Wnorowska, U.; Byfield, F.J.; Piktel, E.; Watek, M.; Janmey, P.A.; Savage, P.B. Bactericidal activity of ceragenin CSA-13 in cell culture and in an animal model of peritoneal infection. Antimicrob. Agents Chemother. 2015, 59, 6274–6282. [Google Scholar] [CrossRef]
- Konai, M.M.; Samaddar, S.; Bocchinfuso, G.; Santucci, V.; Stella, L.; Haldar, J. Selectively targeting bacteria by tuning the molecular design of membrane-active peptidomimetic amphiphiles. Chem. Commun. (Camb) 2018, 54, 4943–4946. [Google Scholar] [CrossRef]
- Gunasekaran, P.; Rajasekaran, G.; Han, E.H.; Chung, Y.H.; Choi, Y.J.; Yang, Y.J.; Lee, J.E.; Kim, H.N.; Lee, K.; Kim, J.S.; et al. Cationic amphipathic triazines with potent anti-bacterial, anti-inflammatory and anti-atopic dermatitis properties. Sci. Rep. 2019, 9, 1292. [Google Scholar] [CrossRef]
- Ahn, M.; Gunasekaran, P.; Rajasekaran, G.; Kim, E.Y.; Lee, S.J.; Bang, G.; Cho, K.; Hyun, J.K.; Lee, H.J.; Jeon, Y.H.; et al. Pyrazole derived ultra-short antimicrobial peptidomimetics with potent anti-biofilm activity. Eur. J. Med. Chem. 2017, 125, 551–564. [Google Scholar] [CrossRef]
- Vooturi, S.K.; Dewal, M.B.; Firestine, S.M. Examination of a synthetic benzophenone membrane-targeted antibiotic. Org. Biomol. Chem. 2011, 9, 6367–6372. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Huo, D.; Nimmagadda, A.; Wu, J.; She, F.; Su, M.; Lin, X.; Yan, J.; Cao, A.; Xi, C.; et al. Small antimicrobial agents based on acylated reduced amide scaffold. J. Med. Chem. 2016, 59, 7877–7887. [Google Scholar] [CrossRef]
- Bremner, J.B.; Coates, J.A.; Coghlan, D.R.; David, D.M.; Keller, P.A.; Pyne, S.G. The synthesis of a novel binaphthyl-based cyclic peptoid with anti-bacterial activity. New J. Chem. 2002, 26, 1549–1551. [Google Scholar] [CrossRef]
- Williams, D.H.; Bardsley, B. The vancomycin group of antibiotics and the fight against resistant bacteria. Angew. Chem. Int. Ed. Engl. 1999, 38, 1172–1193. [Google Scholar] [CrossRef]
- Bremner, J.B.; Coates, J.A.; Keller, P.A.; Pyne, S.G.; Witchard, H.M. The synthesis of a novel carbazole-linked cyclic peptoid with antibacterial activity. Synlett 2002, 219–222. [Google Scholar] [CrossRef]
- Bremner, J.B.; Coates, J.A.; Keller, P.A.; Pyne, S.G.; Witchard, H.M. Synthesis of carbazole-linked cyclic and acyclic peptoids with antibacterial activity. Tetrahedron 2003, 59, 8741–8755. [Google Scholar] [CrossRef]
- Boyle, T.P.; Bremner, J.B.; Coates, J.; Deadman, J.; Keller, P.A.; Pyne, S.G.; Rhodes, D.I. New cyclic peptides via ring-closing metathesis reactions and their anti-bacterial activities. Tetrahedron 2008, 64, 11270–11290. [Google Scholar] [CrossRef]
- Boyle, T.P.; Bremner, J.B.; Coates, J.A.; Deadman, J.; Keller, P.A.; Pyne, S.G.; Somphol, K. Synthesis of novel N-protected hydrophobic phenylalanines and their application in potential antibacterials. Eur. J. Med. Chem. 2009, 44, 1001–1009. [Google Scholar] [CrossRef]
- Garas, A.; Bremner, J.B.; Coates, J.; Deadman, J.; Keller, P.A.; Pyne, S.G.; Rhodes, D.I. Binaphthyl scaffolded peptoids via ring-closing metathesis reactions and their anti-bacterial activities. Bioorg. Med. Chem. Lett. 2009, 19, 3010–3013. [Google Scholar] [CrossRef]
- Au, V.S.; Bremner, J.B.; Coates, J.; Keller, P.A.; Pyne, S.G. Synthesis of some cyclic indolic peptoids as potential antibacterials. Tetrahedron 2006, 62, 9373–9382. [Google Scholar] [CrossRef][Green Version]
- Bremner, J.B.; Keller, P.A.; Pyne, S.G.; Boyle, T.P.; Brkic, Z.; David, D.M.; Garas, A.; Morgan, J.; Robertson, M.; Somphol, K.; et al. Binaphthyl-based dicationic peptoids with therapeutic potential. Angew. Chem. Int. Ed. 2010, 49, 537. [Google Scholar] [CrossRef]
- Bremner, J.B.; Keller, P.A.; Pyne, S.G.; Robertson, M.J.; Sakthivel, K.; Somphol, K.; Baylis, D.; Coates, J.A.; Deadman, J.; Jeevarajah, D.; et al. Binaphthyl-anchored antibacterial tripeptide derivatives with hydrophobic C-terminal amino acid variations. Beilstein J. Org. Chem. 2012, 8, 1265–1270. [Google Scholar] [CrossRef]
- Robertson, M.; Bremner, J.B.; Coates, J.; Deadman, J.; Keller, P.A.; Pyne, S.G.; Somphol, K.; Rhodes, D.I. Synthesis and antibacterial activity of C2-symmetric binaphthyl scaffolded amino acid derivatives. Eur. J. Med. Chem. 2011, 46, 46–4201. [Google Scholar] [CrossRef]
- Wales, S.M.; Hammer, K.A.; Somphol, K.; Kemker, I.; Schroder, D.C.; Tague, A.J.; Brkic, Z.; King, A.M.; Lyras, D.; Riley, T.V.; et al. Synthesis and antimicrobial activity of binaphthyl-based, functionalized oxazole and thiazole peptidomimetics. Org. Biomol. Chem. 2015, 13, 10813–10824. [Google Scholar] [CrossRef]
- Cheah, W.C.; Black, D.S.; Goh, W.K.; Kumar, N. Synthesis of anti-bacterial peptidomimetics derived from N-acylisatins. Tetrahedron Lett. 2008, 49, 2965–2968. [Google Scholar] [CrossRef]
- Le, T.; Cheah, W.C.; Wood, K.; Black, D.S.C.; Willcox, M.D.; Kumar, N. Synthesis of dendrimeric N-glyoxylamide peptide mimics. Tetrahedron Lett. 2011, 52, 3645–3647. [Google Scholar] [CrossRef]
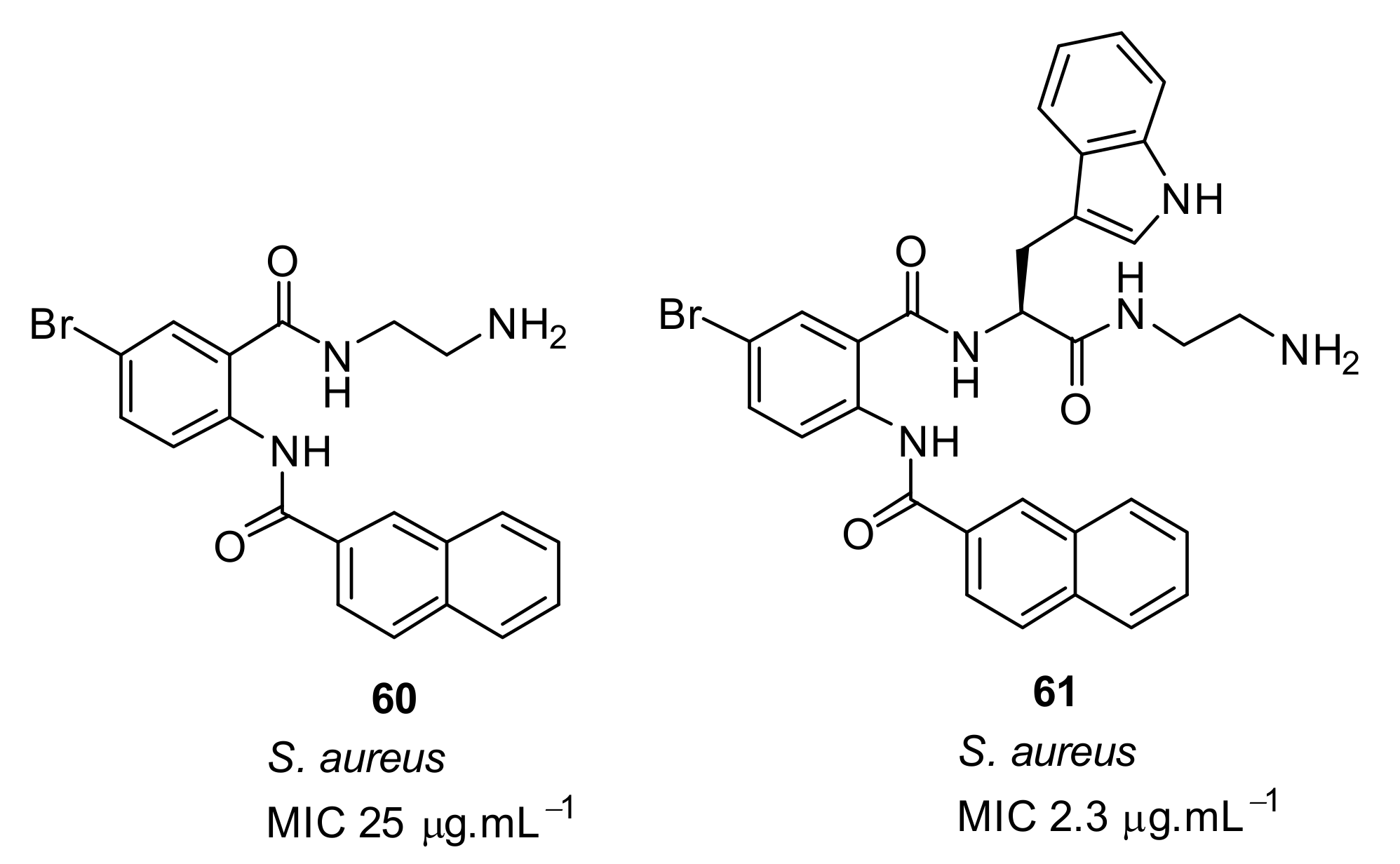
- Nizalapur, S.; Ho, K.K.K.; Kimyon, O.; Yee, E.; Berry, T.; Manefield, M.; Cranfield, C.G.; Willcox, M.; Black, D.S.; Kumar, N. Synthesis and biological evaluation of N-naphthoyl-phenylglyoxamide-based small molecular antimicrobial peptide mimics as novel antimicrobial agents and biofilm inhibitors. Org. Biomol. Chem. 2016, 14, 3623–3637. [Google Scholar] [CrossRef]
- Nizalapur, S.; Kimyon, O.; Yee, E.; Ho, K.; Berry, T.; Manefield, M.; Cranfield, C.G.; Willcox, M.; Black, D.S.; Kumar, N. Amphipathic guanidine-embedded glyoxamide-based peptidomimetics as novel antibacterial agents and biofilm disruptors. Org. Biomol. Chem. 2017, 15, 2033–2051. [Google Scholar] [CrossRef]
- Yu, T.T.; Nizalapur, S.; Ho, K.K.K.; Yee, E.; Berry, T.; Cranfield, C.G.; Willcox, M.; Black, D.S.; Kumar, N. Design, synthesis and biological evaluation of N-Sulfonylphenyl glyoxamide-based antimicrobial peptide mimics as novel antimicrobial agents. ChemistrySelect 2017, 2, 3452–3461. [Google Scholar] [CrossRef]
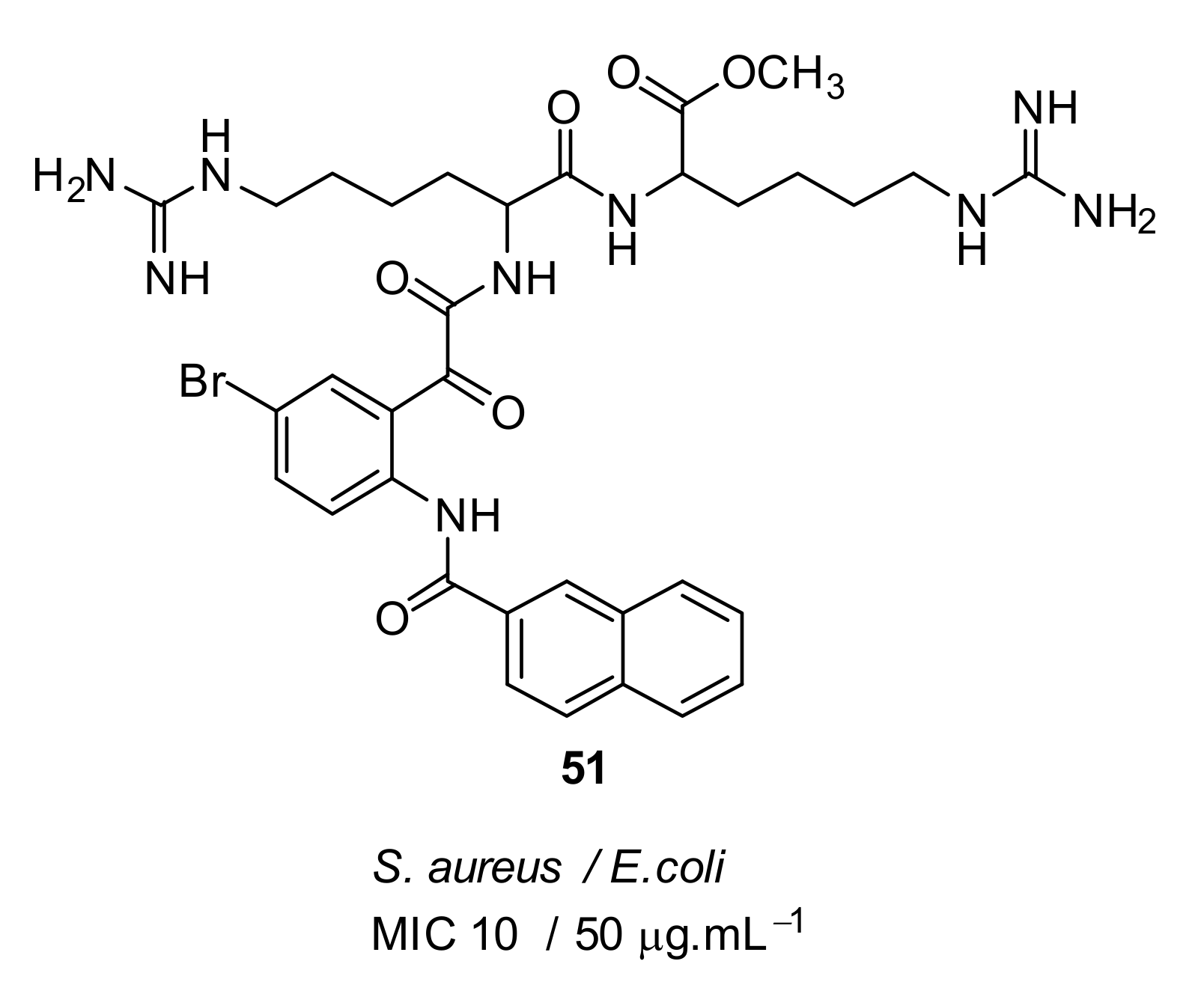
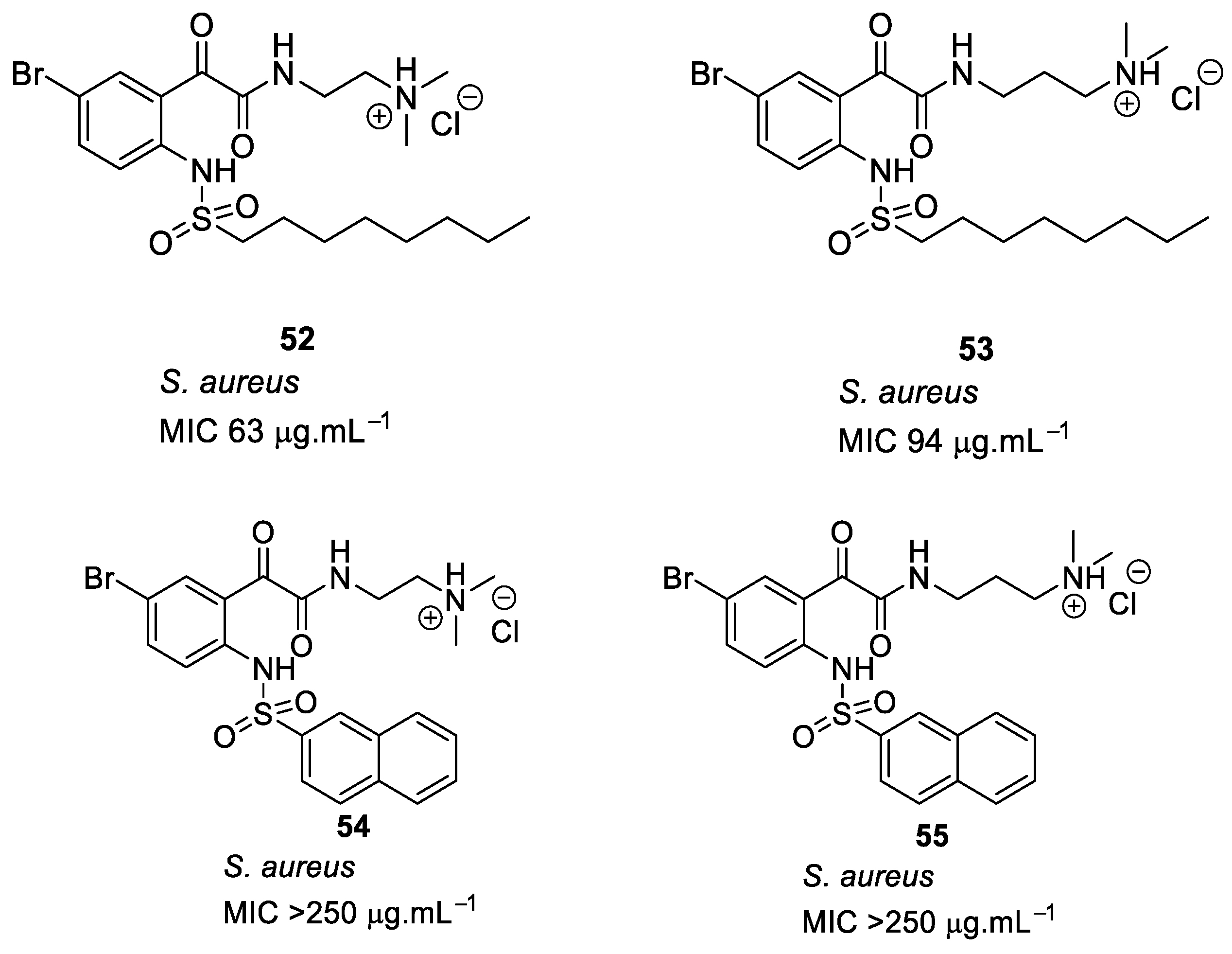
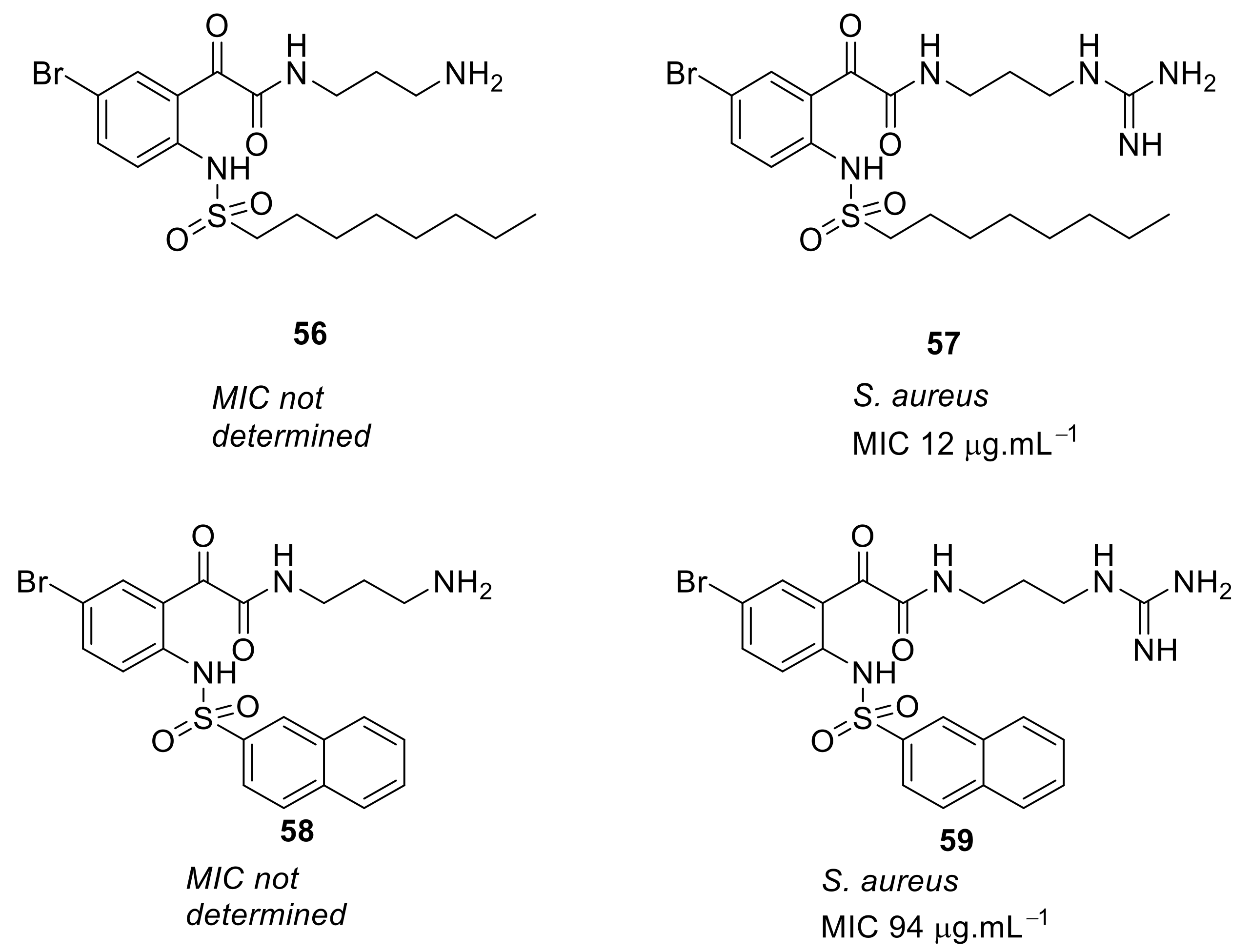
- Kuppusamy, R.; Yasir, M.; Yee, E.; Willcox, M.; Black, D.S.; Kumar, N. Guanidine functionalized anthranilamides as effective antibacterials with biofilm disruption activity. Org. Biomol. Chem. 2018, 16, 5871–5888. [Google Scholar] [CrossRef]
- Henderson, L.C.; Li, J.; Nation, R.L.; Velkov, T.; Pfeffer, F.M. Developing an anion host for lipid A binding and antibacterial activity. Chem. Commun. 2010, 46, 3197–3199. [Google Scholar] [CrossRef]
- Hickey, S.M.; Ashton, T.D.; Khosa, S.K.; Robson, R.N.; White, J.M.; Li, J.; Nation, R.L.; Yu, H.Y.; Elliott, A.G.; Butler, M.S.; et al. Synthesis and evaluation of cationic norbornanes as peptidomimetic antibacterial agents. Org. Biomol. Chem. 2015, 13, 6225–6241. [Google Scholar] [CrossRef]
- Hickey, S.M.; Ashton, T.D.; White, J.M.; Li, J.; Nation, R.L.; Yu, H.Y.; Elliott, A.G.; Butler, M.S.; Huang, J.X.; Cooper, M.A.; et al. Synthesis of norbornane bisether antibiotics via silver-mediated alkylation. RSC Adv. 2015, 5, 28582–28596. [Google Scholar] [CrossRef]
- Hickey, S.M.; Ashton, T.D.; Boer, G.; Bader, C.A.; Thomas, M.; Elliott, A.G.; Schmuck, C.; Yu, H.Y.; Li, J.; Nation, R.L.; et al. Norbornane-based cationic antimicrobial peptidomimetics targeting the bacterial membrane. Eur. J. Med. Chem. 2018, 160, 9–22. [Google Scholar] [CrossRef]
- Kuppusamy, R.; Yasir, M.; Berry, T.; Cranfield, C.G.; Nizalapur, S.; Yee, E.; Kimyon, O.; Taunk, A.; Ho, K.K.K.; Cornell, B.; et al. Design and synthesis of short amphiphilic cationic peptidomimetics based on biphenyl backbone as antibacterial agents. Eur. J. Med. Chem. 2018, 143, 1702–1722. [Google Scholar] [CrossRef]
- Cranfield, C.G.; Cornell, B.A.; Grage, S.L.; Duckworth, P.; Carne, S.; Ulrich, A.S.; Martinac, B. Transient potential gradients and impedance measures of tethered bilayer lipid membranes: Pore-forming peptide insertion and the effect of electroporation. Biophys. J. 2014, 106, 182–189. [Google Scholar] [CrossRef]
- Alghalayini, A.; Garcia, A.; Berry, T.; Cranfield, C.G. The use of tethered bilayer lipid membranes to identify the mechanisms of antimicrobial peptide Iinteractions with lipid bilayers. Antibiotics (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Tague, A.J.; Putsathit, P.; Hammer, K.A.; Wales, S.M.; Knight, D.R.; Riley, T.V.; Keller, P.A.; Pyne, S.G. Cationic biaryl 1,2,3-triazolyl peptidomimetic amphiphiles: synthesis, antibacterial evaluation and preliminary mechanism of action studies. Eur. J. Med. Chem. 2019, 168, 386–404. [Google Scholar] [CrossRef]
- Wales, S.M.; Hammer, K.A.; King, A.M.; Tague, A.J.; Lyras, D.; Riley, T.V.; Keller, P.A.; Pyne, S.G. Binaphthyl-1,2,3-triazole peptidomimetics with activity against Clostridium difficile and other pathogenic bacteria. Org. Biomol. Chem. 2015, 13, 5743–5756. [Google Scholar] [CrossRef] [PubMed]
- Azmi, F.; Elliott, A.G.; Khalil, Z.G.; Hussein, W.M.; Kavanagh, A.; Huang, J.X.; Quezada, M.; Blaskovich, M.A.T.; Capon, R.J.; Cooper, M.A.; et al. Self-assembling lipopeptides with a potent activity against Gram-positive bacteria, including multidrug resistant strains. Nanomedicine 2015, 10, 3359–3371. [Google Scholar] [CrossRef]
- Liu, L.H.; Xu, K.J.; Wang, H.Y.; Tan, P.K.J.; Fan, W.M.; Venkatraman, S.S.; Li, L.J.; Yang, Y.Y. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat. Nanotechnol. 2009, 4, 457–463. [Google Scholar] [CrossRef]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Class | Number of AMPs | Percentage |
|---|---|---|
| α-helix | 465 | 15.2% |
| β-structure | 82 | 2.68% |
| Mix of α-helix and β-sheet | 106 | 3.46% |
| Extended | 100 | 3.27% |
| Disulfide bridge | 493 | 16.12% |
| Unknown | 1789 | 58.5% |
| Structure | MIC µg·mL−1 | Ref |
|---|---|---|
 | S. aureus (3) E. coli (8) P. aeruginosa (5) Methicillin-resistant S. aureus (3) Methicillin-resistant S. epidermidis (1) glycopeptide-intermediate S. aureus (3) | Isaksson et al. [70] |
 | S. aureus (0.045) E. coli (0.4) K. pneumoniae (2) P. aeruginosa (2) | Choi et al. [71] |
 | S. aureus (0.5) E. coli (1) | Isaksson et al. [70] |
 | S. aureus (0.5) Bacillus cereus (2) Methicillin-resistant S. aureus (2) | Koh et al. [72] |
 | S. aureus (0.5) E. coli (2) | Bucki et al. [73] |
 | S. aureus (2) E. coli (3) P. aeruginosa (3) Methicillin-resistant S. aureus (3) | Konai et al. [74] |
 | S. aureus (3) E. coli (5) P. aeruginosa (5) | Gunasekaran et al. [75] |
 | S. aureus (4) E. coli (32) P. aeruginosa (8) Methicillin-resistant S. aureus (8) Vancomycin-resistant E. faecalis (8) | Ahn et al. [76] |
 | Methicillin-resistant S. aureus (1) Vancomycin-resistant S. aureus (0.5) S. aureus (4) | Vooturi et al. [77] |
 | Methicillin-resistant S. aureus (2) Vancomycin-resistant enterococci (6) Methicillin-resistant S. epidermidis (3) E. coli (3) P. aeruginosa (6) | Teng et al. [78] |
| Structure | MIC µg·mL−1 | Ref |
|---|---|---|
 | S. aureus (17) | Bremner et al. [79] |
 | S. aureus (15) | Bremner et al. [81] |
 | S. aureus (>125) | Au et al. [86] |
 | S. aureus (8) | Boyle et al. [84] |
 | Methicillin-resistant S. aureus (2–4) Methicillin-sensitive S. aureus (4) Vancomycin-resistant S. aureus (2) S. epidermidis (4) Vancomyin-resistant enterococci (4) A. baumannii (4) E. coli (16) | Bremner et al. [87] |
 | Methicillin-resistant S. aureus (4) Methicillin-sensitive S. aureus (4) Vancomycin-resistant S. aureus (4) S. epidermidis (4) Vancomyin-resistant enterococci (4) A. baumannii (8) E. coli (32) | Bremner et al. [87] |
 | Methicillin-sensitive S. aureus (4) Methicillin-resistant S. aureus (4) Vancomycin-intermediate S. aureus (4) S. epidermidis (2) | Bremner et al. [88] |
 | S. aureus (4/8) (Bn (31)/Nap (33)) Vancomycin-resistant S. aureus (83/>125) (Bn (31)/Nap (33)) | Robertson et al. [89] |
 | S. aureus (1) MRSA (2) E. faecalis (2) S. pneumoniae (2) E. coli (4) A. baumannii (4) C. difficile (8) | Wales et al. [90] |
| Structure | MIC µg·mL−1 | Ref |
|---|---|---|
 | S. aureus (6) E. coli (>110) | Nizalapur et al. [94] |
 | S. aureus (12) | Yu et al. [95] |
 | S. aureus (3) E. coli (10) | Kuppusamy et al. [96] |
| Structure | MIC µg mL−1 | Ref |
|---|---|---|
 | S. aureus (0.5) E. coli (32) | Henderson et al. Hickey et al. [97,98] |
 | S. aureus (1) E. coli (8) S. pneumoniae (1) | Hickey et al. [100] |
| Structure | MIC µg mL−1 | Ref |
|---|---|---|
 | S. aureus (10) E. coli (6) | Kuppusamy et al. [101] |
 | S. aureus (4) E. coli (32) Vancomyin-resistant enterococci (4) C. difficile (4) A. baumannii (4) | Wales et al. [105] |
 | MRSA (2) E. coli (4) C. difficile (4) A. baumannii (8) | Tague et al. [104] |
| Structure | MIC µg·mL−1 | Ref |
|---|---|---|
 | methicillin-resistant S. aureus (1.7) Glycopeptide-intermediate S. aureus (7.5) Vancomycin-resistant S. aureus (2.6) S. pneumoniae (5.6) | Azmi et al. [106] |
 | methicillin-resistant S. aureus (0.47) Glycopeptide-intermediate S. aureus (7.5) Vancomycin-resistant S. aureus (0.47) S. pneumoniae (2.8) | Azmi et al. [106] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuppusamy, R.; Willcox, M.; Black, D.S.; Kumar, N. Short Cationic Peptidomimetic Antimicrobials. Antibiotics 2019, 8, 44. https://doi.org/10.3390/antibiotics8020044
Kuppusamy R, Willcox M, Black DS, Kumar N. Short Cationic Peptidomimetic Antimicrobials. Antibiotics. 2019; 8(2):44. https://doi.org/10.3390/antibiotics8020044
Chicago/Turabian StyleKuppusamy, Rajesh, Mark Willcox, David StC. Black, and Naresh Kumar. 2019. "Short Cationic Peptidomimetic Antimicrobials" Antibiotics 8, no. 2: 44. https://doi.org/10.3390/antibiotics8020044
APA StyleKuppusamy, R., Willcox, M., Black, D. S., & Kumar, N. (2019). Short Cationic Peptidomimetic Antimicrobials. Antibiotics, 8(2), 44. https://doi.org/10.3390/antibiotics8020044