Antibiotic Discovery: Where Have We Come from, Where Do We Go?




Abstract
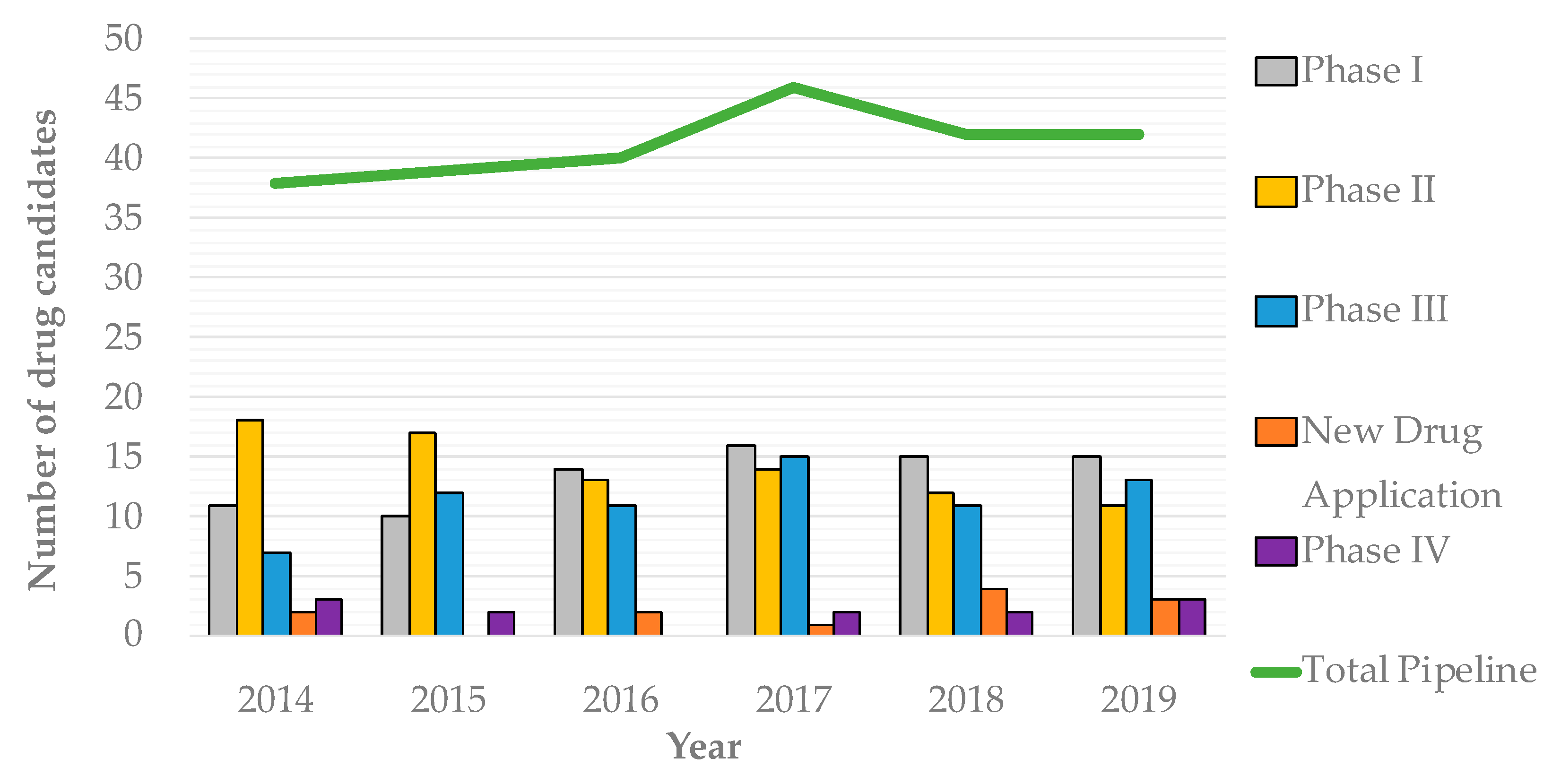
1. Introduction—The Desperate Need for New Antibiotics
2. The Birth of Antimicrobial Chemotherapy
3. Towards the Golden Era: Waksman Platform
4. Onto the Medicinal Chemistry Era: Semi-Synthesis
5. From the Ground Up: Fully Synthetic Antibiotics
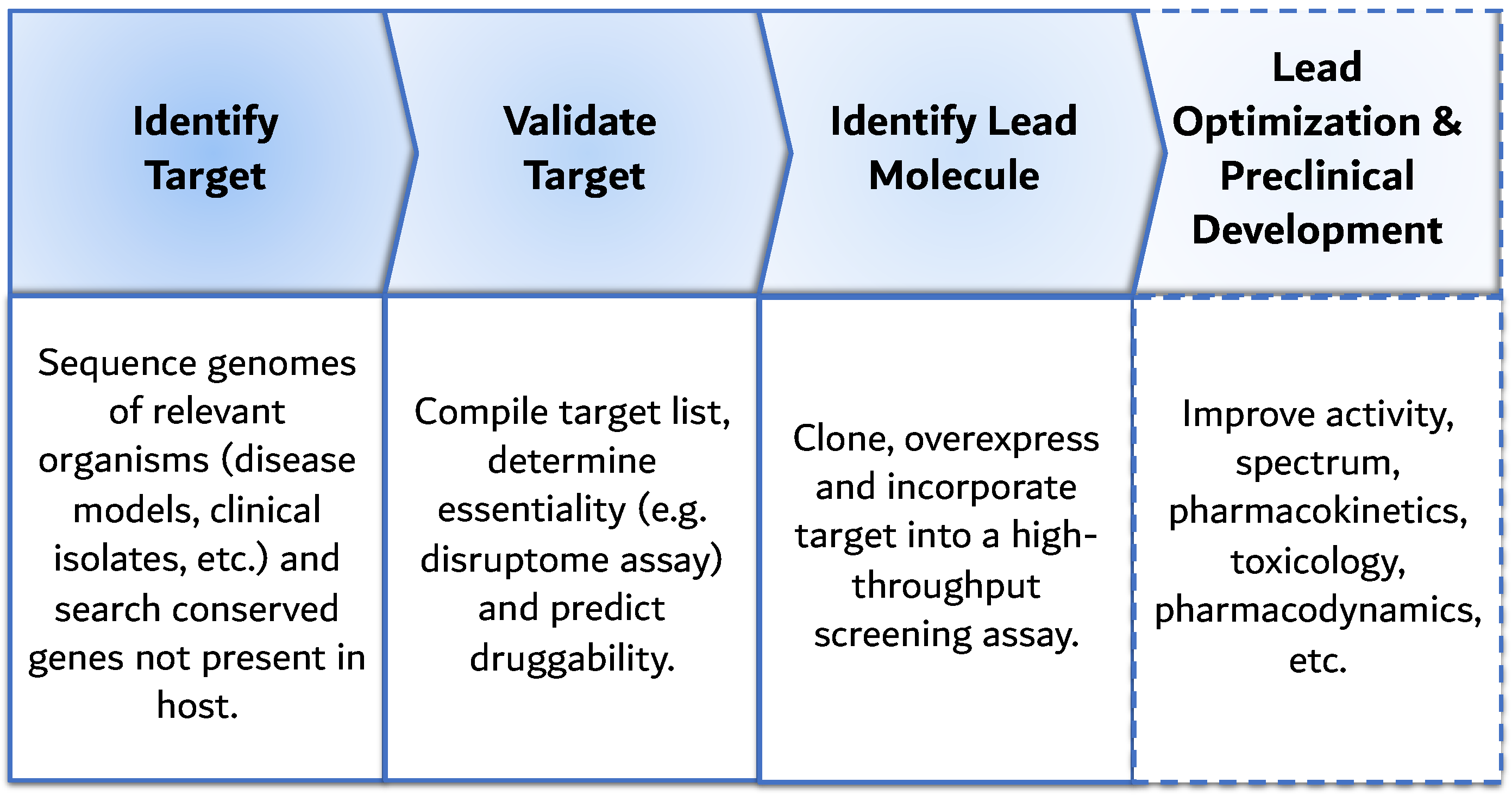
6. Advent of Genomics: Target-Based Screening
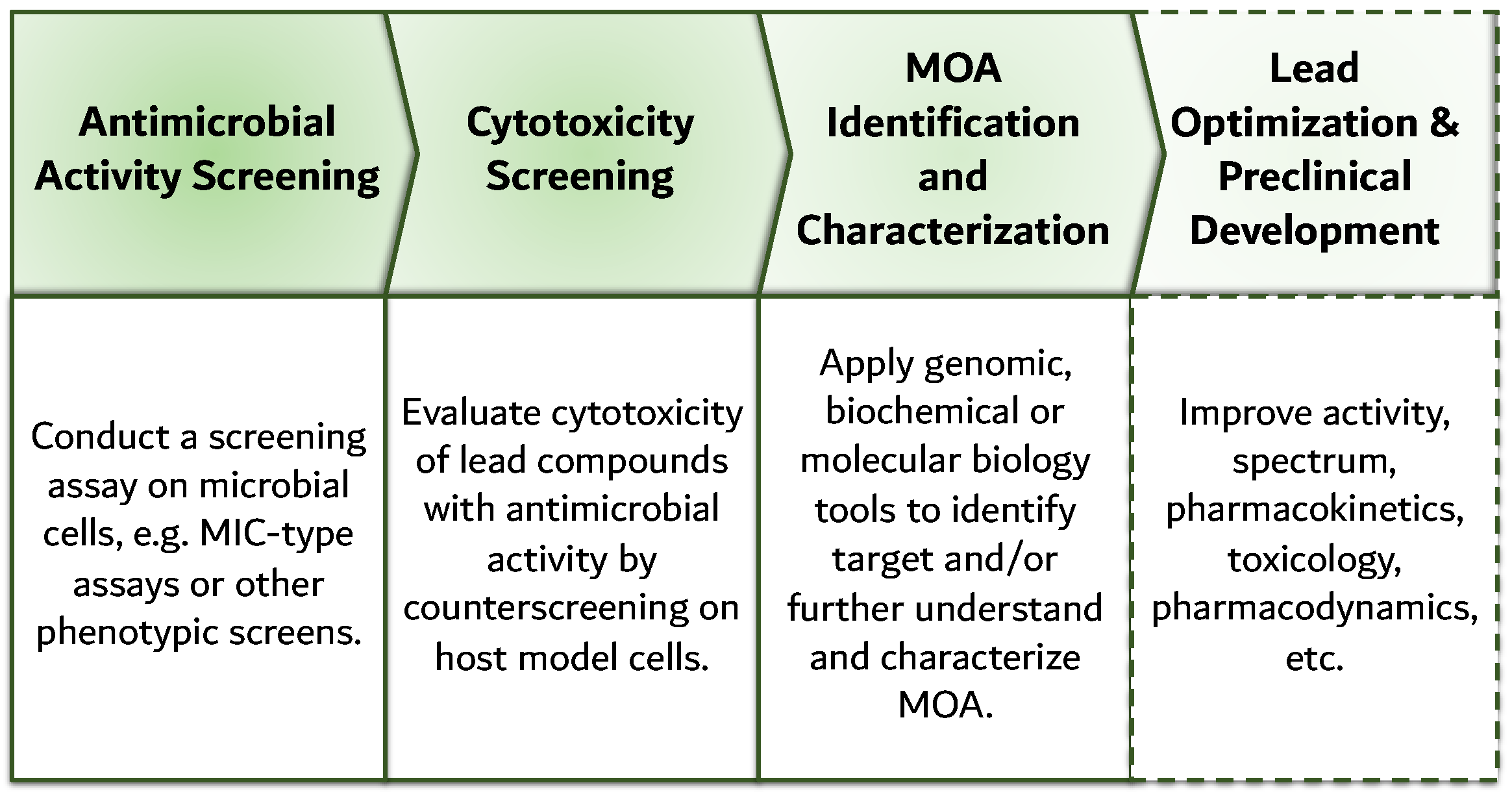
7. Reverse Genomics: Revival of Cell-Based Screening
8. Post-Genomics: Transcriptomics, Proteomics and Lipidomics
9. Post-Genomics: From Metabolomics towards Meta-omics
10. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, P.W.; Watkins, K.; Hewlett, A. Infection control through the ages. Am. J. Infect. Control 2012, 40, 35–42. [Google Scholar] [CrossRef]
- Clardy, J.; Fischbach, M.A.; Currie, C.R. The natural history of antibiotics. Curr. Biol. 2009, 19, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Discovery research: The scientific challenge of finding new antibiotics. J. Antimicrob. Chemother. 2011, 66, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Saga, T.; Yamaguchi, K. History of antimicrobial agents and resistant. Jpn. Med. Assoc. J. 2009, 137, 103–108. [Google Scholar]
- WHO Report on Global Surveillance of Epidemic-prone Infectious Diseases—Introduction. Available online: http://www.who.int/csr/resources/publications/introduction/en/index1.html (accessed on 12 October 2018).
- Center for Disease Control and Prevention. Antibiotic Use in the United States, 2017: Progress and Opportunities; US Department of Health and Human Service: Atlanta, GA, USA, 2017; pp. 1–40.
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2018, 3099, 1–11. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control; European Medicines Agency. The Bacterial Challenge: Time to React; European Centre for Disease Prevention and Control: Stockholm, Sweden, 2009; Volume 6, ISBN 9789291931934. [Google Scholar]
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef]
- Martens, E.; Demain, A.L. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 2017, 70, 520–526. [Google Scholar] [CrossRef]
- Akova, M. Epidemiology of antimicrobial resistance in bloodstream infections. Virulence 2016, 7, 252–266. [Google Scholar] [CrossRef]
- McFee, R.B. Nosocomial or hospital-acquired infections: An overview. Dis. Mon. 2009, 55, 422–438. [Google Scholar] [CrossRef]
- Theuretzbacher, U. Accelerating resistance, inadequate antibacterial drug pipelines and international responses. Int. J. Antimicrob. Agents 2012, 39, 295–299. [Google Scholar] [CrossRef]
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Available online: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (accessed on 24 March 2019).
- Rather, I.A.; Kim, B.C.; Bajpai, V.K.; Park, Y.H. Self-medication and antibiotic resistance: Crisis, current challenges, and prevention. Saudi J. Biol. Sci. 2017, 24, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Dodds, D.R. Antibiotic resistance: A current epilogue. Biochem. Pharmacol. 2017, 134, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Renwick, M.J.; Brogan, D.M.; Mossialos, E. A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics. J. Antibiot. 2016, 69, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Smith, D. Antibiotics Are Money-Losers For Big Pharma. How Can We Incentivize the Development of New Ones? Available online: https://www.forbes.com/sites/quora/2018/01/02/antibiotics-are-money-losers-for-big-pharma-how-can-we-incentivize-the-development-of-new-ones/#9318630487f3 (accessed on 23 March 2019).
- Kostyanev, T.; Bonten, M.J.M.; Steel, H.; Ross, S.; Franc, B.; Tacconelli, E.; Winterhalter, M.; Stavenger, R.A.; Harbarth, S.; Hackett, J.; et al. The innovative medicines initiative’s new drugs for bad bugs programme: European public—Private partnerships for the development of new strategies to tackle antibiotic resistance. J. Antimicrob. Chemother. 2016, 71, 290–295. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the infectious diseases society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Brown, D.G. New drugs and emerging leads in antibacterial drug discovery. Ref. Modul. Chem. Mol. Sci. Chem. Eng. 2016. [Google Scholar] [CrossRef]
- The Pew Charitable Trusts Antibiotics Currently in Global Clinical Development. Available online: https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development (accessed on 23 March 2019).
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 2015, 14, 529–542. [Google Scholar] [CrossRef]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Herrmann, M.; Nkuiya, B.; Dussault, A.R. Innovation and antibiotic use within antibiotic classes: Market incentives and economic instruments. Resour. Energy Econ. 2013, 35, 582–598. [Google Scholar] [CrossRef]
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef]
- Cui, L.; Su, X. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti-infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Fetzner, S.; Drees, S.L. Old molecules, new biochemistry. Chem. Biol. 2013, 20, 1438–1440. [Google Scholar] [CrossRef]
- Nicholson, D.J. Biological atomism and cell theory. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2010, 41, 202–211. [Google Scholar] [CrossRef]
- Levine, R.; Evers, C. The Slow Death of Spontaneous Generation; North Carolina State University: Raleigh, NC, USA, 2000; pp. 7–8. [Google Scholar]
- Brack, A. The Molecular Origins of Life: Assembling Pieces of the Puzzle; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Newsom, S.W.B. Pioneers in infection control—Joseph Lister. J. Hosp. Infect. 2003, 55, 246–253. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 Years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J. The introduction of “chemotherapy” using arsphenamine—The first magic bullet. J. R. Soc. Med. 2009, 102, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Ferrie, J.E. Arsenic, antibiotics and interventions. Int. J. Epidemiol. 2014, 43, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M.; Kristiansen, J.E. On the 75th anniversary of Prontosil. Dyes Pigments 2011, 88, 231–234. [Google Scholar] [CrossRef]
- Ligon, B.L. Penicillin: its discovery and early development. Semin. Pediatr. Infect. Dis. 2004, 15, 52–57. [Google Scholar] [CrossRef]
- Fleming, A. Sir Alexander Fleming—Nobel Lecture. Available online: https://www.nobelprize.org/prizes/medicine/1945/fleming/lecture/ (accessed on 12 November 2018).
- Bynum, B. Rediscovering penicillin. Lancet 2018, 392, 1108–1109. [Google Scholar] [CrossRef]
- Sales, K.C.; Rosa, F.; Sampaio, P.N.; Fonseca, L.P.; Lopes, M.B.; Calado, C.R.C. In situ near-infrared (NIR) versus high-throughput mid-infrared (MIR) spectroscopy to monitor biopharmaceutical production. Appl. Spectrosc. 2015, 69, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R. Different roads to discovery; Prontosil (hence sulfa drugs) and penicillin (hence β-lactams). J. Ind. Microbiol. Biotechnol. 2009, 36, 775–786. [Google Scholar] [CrossRef]
- Woodruff, H.B.; Selman, A. Waksman, winner of the 1952 nobel prize for physiology or medicine. Appl. Environ. Microbiol. 2014, 80, 2–8. [Google Scholar] [CrossRef]
- Lewis, K. Recover the lost art of drug discovery. Nature 2012, 485, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Willey, J.; Sherwood, L.; Woolverton, C. Antimicrobial chemotherapy. In Prescott, Harley and Klein’s Microbiology; Willey, J., Sherwood, L., Woolverton, C., Eds.; Colin Wheatley/Janice Roerig-Blong: New York, NY, USA, 2008; pp. 835–837. ISBN 978-0–07–299291–5. [Google Scholar]
- Wainwright, M. Streptomycin: Discovery and resultant controversy. Hist. Philos. Life Sci. 1991, 13, 97–124. [Google Scholar] [PubMed]
- Wiest, D.B.; Cochran, J.B.; Tecklenburg, F.W. Chloramphenicol toxicity revisited: A 12-year-old patient with a brain abscess. J. Pediatr. Pharmacol. Ther. 2012, 17, 182–188. [Google Scholar] [CrossRef]
- Liu, F.; Myers, A.G. Development of a platform for the discovery and practical synthesis of new tetracycline antibiotics. Curr. Opin. Chem. Biol. 2016, 32, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, E.; Wallat, J.; Pokorski, J.; von Recum, H. Erythromycin modification that improves its acidic stability while optimizing it for local drug delivery. Antibiotics 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Mast, Y.; Wohlleben, W. Streptogramins—Two are better than one! Int. J. Med. Microbiol. 2014, 304, 44–50. [Google Scholar] [CrossRef]
- James, R.C.; Pierce, J.G.; Okano, A.; Xie, J.; Boger, D.L. Redesign of glycopeptide antibiotics: Back to the future. ACS Chem. Biol. 2012, 7, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Floss, H.G.; Yu, T. Rifamycin mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, A.S.; Livaditis, I.G.; Gougoutas, V. The revival of fosfomycin. Int. J. Infect. Dis. 2011, 15, e732–e739. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.S.; Barone, A.A.; Penço, J.; Santos, M.V.; Marinho, I.S.; Arruda, E.A.G.; Manrique, E.I.; Costa, S.F. Intravenous colistin as therapy for nosocomial infections caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Clin. Infect. Dis. 1999, 28, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, B.I.; Oleson, F.B., Jr.; Baltz, R.H. Daptomycin: From the mountain to the clinic, with essential help from Francis Tally, MD. Clin. Infect. Dis. 2010, 50, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.D.; Palmer, M. The action mechanism of daptomycin. Bioorg. Med. Chem. 2016, 24, 6253–6268. [Google Scholar] [CrossRef]
- Adeyemo, A.A.; Oluwatosin, O.; Omotade, O.O. Study of streptomycin-induced ototoxicity: Protocol for a longitudinal study. Springerplus 2016, 5, 758. [Google Scholar] [CrossRef]
- Townsend, C.A. Convergent biosynthetic pathways to β-lactam antibiotics. Curr. Opin. Chem. Biol. 2016, 35, 97–108. [Google Scholar] [CrossRef]
- Güngör, Ö.Ö.N.; Gürkan, P.; Özçelik, B.; Oyardi, Ö. Synthesis and antimicrobial activities of new higher amino acid Schiff base derivatives of 6-aminopenicillanic acid and 7-aminocephalosporanic acid. J. Mol. Struct. 2016, 1106, 181–191. [Google Scholar]
- Deng, S.; Ma, X.; Su, E.; Wei, D. Efficient cascade synthesis of ampicillin from penicillin G potassium salt using wild and mutant penicillin G acylase from Alcaligenes faecalis. J. Biotechnol. 2016, 219, 142–148. [Google Scholar] [CrossRef]
- Gröger, H.; Pieper, M.; König, B.; Bayer, T.; Schleich, H. Industrial landmarks in the development of sustainable production processes for the β-lactam antibiotic key intermediate 7-aminocephalosporanic acid (7-ACA). Sustain. Chem. Pharm. 2017, 5, 72–79. [Google Scholar] [CrossRef]
- Hu, Y.; Zhu, B. Study on genetic engineering of Acremonium chrysogenum, the cephalosporin C producer. Synth. Syst. Biotechnol. 2016, 1, 143–149. [Google Scholar] [CrossRef]
- Nelson, M.L.; Levy, S.B. The history of the tetracyclines. Ann. N. Y. Acad. Sci. 2011, 1241, 17–32. [Google Scholar] [CrossRef]
- Charest, M.G.; Lerner, C.D.; Brubaker, J.D.; Siegel, D.R.; Myers, A.G. A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 2005, 308, 395–398. [Google Scholar] [CrossRef]
- Perez, M.; Echeverria, P.G.; Martinez-Arripe, E.; Ez Zoubir, M.; Touati, R.; Zhang, Z.; Genet, J.P.; Phansavath, P.; Ayad, T.; Ratovelomanana-Vidal, V. An efficient stereoselective total synthesis of all stereoisomers of the antibiotic thiamphenicol through ruthenium-catalyzed asymmetric reduction by dynamic kinetic resolution. Eur. J. Org. Chem. 2015, 2015, 5949–5958. [Google Scholar] [CrossRef]
- Alauzet, C.; Lozniewski, A.; Marchandin, H. Metronidazole resistance and nim genes in anaerobes: A review. Anaerobe 2019, 55, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef] [PubMed]
- Caron, F.; Wehrle, V.; Etienne, M. Triméthoprime: Un antibiotique en voie de réhabilitation en France. Med. Mal. Infect. 2017, 47, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Bisacchi, G.S. Origins of the quinolone class of antibacterials: An expanded “discovery story”. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef]
- Lee, G.C.; Reveles, K.R.; Attridge, R.T.; Lawson, K.A.; Mansi, I.A.; Lewis, J.S.; Frei, C.R. Outpatient antibiotic prescribing in the United States: 2000 to 2010. BMC Med. 2014, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Carlsen, P.N.; et al. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Brahim, I.; Hisham, N.; Aladdin, R.; Mohammed, H.; Bahaaeldin, A. Recent updates of carbapenem antibiotics. Eur. J. Med. Chem. 2017, 131, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.-G.; Hu, X.-X.; Li, C.-R.; Li, Y.-H.; Wang, Y.-X.; Jiang, J.-D.; Bi, C.-W.; Tang, S.; You, X.-F.; Song, D.-Q. Design, synthesis and biological evaluation of monobactams as antibacterial agents against gram-negative bacteria. Eur. J. Med. Chem. 2016, 110, 151–163. [Google Scholar] [CrossRef]
- Aminov, R. History of antimicrobial drug discovery: Major classes and health impact. Biochem. Pharmacol. 2017, 133, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.L.; Brickner, S.J.; Noe, M.C.; Miller, P.F. Linezolid, the first oxazolidinone antibacterial agent. Ann. N. Y. Acad. Sci. 2011, 1222, 49–54. [Google Scholar] [CrossRef] [PubMed]
- De Souza Mendes, C.; de Souza Antunes, A. Pipeline of known chemical classes of antibiotics. Antibiotics 2013, 2, 500–534. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.D. When will the genomics investment pay off for antibacterial discovery? Biochem. Pharmacol. 2006, 71, 1096–1102. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Foulkes, J. Elitra pharmaceuticals: new paradigms for antimicrobial drug discovery. Drug Discov. Today 2002, 7, S12–S15. [Google Scholar] [CrossRef]
- Brinster, S.; Lamberet, G.; Staels, B.; Trieu-Cuot, P.; Gruss, A.; Poyart, C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 2009, 458, 83–86. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Sass, P. Postgenomic strategies in antibacterial drug discovery. Future Microbiol. 2010, 5, 1553–1579. [Google Scholar] [CrossRef] [PubMed]
- Fields, F.R.; Lee, S.W.; McConnell, M.J. Using bacterial genomes and essential genes for the development of new antibiotics. Biochem. Pharmacol. 2017, 134, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, F. Recent progress on the development of novel antitubercular agents from whole cell screening hits. J. Syn. Org. Chem. Jpn. 2014, 72, 1239–1249. [Google Scholar] [CrossRef][Green Version]
- Moloney, M.G. Natural products as a source for novel antibiotics. Trends Pharmacol. Sci. 2016, 37, 689–701. [Google Scholar] [CrossRef]
- Lee, J.A.; Berg, E.L. Neoclassic drug discovery: The case for lead generation using phenotypic and functional approaches. J. Biomol. Screen. 2013, 18, 1143–1155. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336. [Google Scholar] [CrossRef] [PubMed]
- Hutter, B.; Schaab, C.; Albrecht, S.; Borgmann, M.; Brunner, N.A.; Freiberg, C.; Ziegelbauer, K.; Rock, C.O.; Ivanov, I.; Loferer, H. Prediction of mechanisms of action of antibacterial compounds by gene expression profiling. Antimicrob. Agents Chemother. 2004, 48, 2838–2844. [Google Scholar] [CrossRef] [PubMed]
- Hutter, B.; Fischer, C.; Jacobi, A.; Schaab, C.; Loferer, H. Panel of Bacillus subtilis reporter strains indicative of various modes of action. Antimicrob. Agents Chemother. 2004, 48, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Michelini, E.; Cevenini, L.; Mezzanotte, L.; Coppa, A.; Roda, A. Cell-based assays: Fuelling drug discovery. Anal. Bioanal. Chem. 2010, 398, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.; Sedrani, R.; Wiesmann, C. The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 2014, 13, 577–587. [Google Scholar] [CrossRef]
- Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 2016, 70, 3. [Google Scholar] [CrossRef] [PubMed]
- Wencewicz, T.A. New antibiotics from Nature’s chemical inventory. Bioorg. Med. Chem. 2016, 24, 6227–6252. [Google Scholar] [CrossRef]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Sieron, A.; King, A.M.; De Pascale, G.; Pawlowski, A.C.; Koteva, K.; Wright, G.D. A common platform for antibiotic dereplication and adjuvant discovery. Cell Chem. Biol. 2017, 24, 98–109. [Google Scholar] [CrossRef]
- Lewis, K. New approaches to antimicrobial discovery. Biochem. Pharmacol. 2017, 134, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Wang, Z.; Deisboeck, T.S. Mathematical modeling in cancer drug discovery. Drug Discov. Today 2014, 19, 145–150. [Google Scholar] [CrossRef]
- Dersch, P.; Khan, M.A.; Mühlen, S.; Görke, B. Roles of regulatory RNAs for antibiotic resistance in bacteria and their potential value as novel drug targets. Front. Microbiol. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Woollard, P.M.; Mehta, N.A.L.; Vamathevan, J.J.; Van Horn, S.; Bonde, B.K.; Dow, D.J. The application of next-generation sequencing technologies to drug discovery and development. Drug Discov. Today 2011, 16, 512–519. [Google Scholar] [CrossRef]
- Mäder, U.; Nicolas, P.; Richard, H.; Bessières, P.; Aymerich, S. Comprehensive identification and quantification of microbial transcriptomes by genome-wide unbiased methods. Curr. Opin. Biotechnol. 2011, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mewalal, R.; Hu, R.; Tuskan, G.A.; Yang, X. New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic. Res. 2017, 4, 1–8. [Google Scholar] [CrossRef]
- Lloyd, N.C.; Morgan, H.W.; Nicholson, B.K.; Ronimus, R.S. The composition of Ehrlich’s Salvarsan: Resolution of a century-old debate. Angew. Chem. Int. Ed. 2005, 44, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Brazas, M.D.; Hancock, R.E.W. Using microarray gene signatures to elucidate mechanisms of antibiotic action and resistance. Drug Discov. Today 2005, 10, 1245–1252. [Google Scholar] [CrossRef]
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Prediction of antibiotic resistance by gene expression profiles. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Unlu, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis. A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef]
- Hannigan, A.; Burchmore, R.; Wilson, J.B. The optimization of protocols for proteome difference gel electrophoresis (DiGE) analysis of preneoplastic skin. J. Proteome Res. 2007, 6, 3422–3432. [Google Scholar] [CrossRef]
- Roe, M.R.; Griffin, T.J. Gel-free mass spectrometry-based high throughput proteomics: Tools for studying biological response of proteins and proteomes. Proteomics 2006, 6, 4678–4687. [Google Scholar] [CrossRef]
- Pulido, M.R.; García-Quintanilla, M.; Gil-Marqués, M.L.; McConnell, M.J. Identifying targets for antibiotic development using omics technologies. Drug Discov. Today 2016, 21, 465–472. [Google Scholar] [CrossRef]
- Vranakis, I.; Goniotakis, I.; Psaroulaki, A.; Sandalakis, V.; Tselentis, Y.; Gevaert, K.; Tsiotis, G. Proteome studies of bacterial antibiotic resistance mechanisms. J. Proteom. 2014, 97, 88–99. [Google Scholar] [CrossRef]
- Freiberg, C.; Brötz-Oesterhelt, H.; Labischinski, H. The impact of transcriptome and proteome analyses on antibiotic drug discovery. Curr. Opin. Microbiol. 2004, 7, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Bandow, J.E. Proteomic signatures in antibiotic research. Proteomics 2011, 11, 3256–3268. [Google Scholar] [CrossRef]
- Carneiro, D.G.; Clarke, T.; Davies, C.C.; Bailey, D. Identifying novel protein interactions: Proteomic methods, optimisation approaches and data analysis pipelines. Methods 2016, 95, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, S.E.; Lamont, R.J. Tyrosine phosphorylation and bacterial virulence. Int. J. Oral Sci. 2012, 4, 1. [Google Scholar] [CrossRef][Green Version]
- Kobir, A.; Shi, L.; Boskovic, A.; Grangeasse, C.; Franjevic, D.; Mijakovic, I. Protein phosphorylation in bacterial signal transduction. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 989–994. [Google Scholar] [CrossRef]
- Morris, M.K.; Chi, A.; Melas, I.N.; Alexopoulos, L.G. Phosphoproteomics in drug discovery. Drug Discov. Today 2014, 19, 425–432. [Google Scholar] [CrossRef]
- Vihervaara, T.; Suoniemi, M.; Laaksonen, R. Lipidomics in drug discovery. Drug Discov. Today 2014, 19, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Sandra, K.; Sandra, P. Lipidomics from an analytical perspective. Curr. Opin. Chem. Biol. 2013, 17, 847–853. [Google Scholar] [CrossRef]
- Yao, J.; Rock, C.O. Bacterial fatty acid metabolism in modern antibiotic discovery. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1300–1309. [Google Scholar] [CrossRef]
- Lam, S.M.; Shui, G. Lipidomics as a principal tool for advancing biomedical research. J. Genet. Genom. 2013, 40, 375–390. [Google Scholar] [CrossRef]
- Lindon, J.C.; Nicholson, J.K. Analytical technologies for metabonomics and metabolomics, and multi-omic information recovery. TrAC Trends Anal. Chem. 2008, 27, 194–204. [Google Scholar] [CrossRef]
- Gao, P.; Xu, G. Mass-spectrometry-based microbial metabolomics: Recent developments and applications. Anal. Bioanal. Chem. 2015, 407, 669–680. [Google Scholar] [CrossRef]
- Nagana Gowda, G.A.; Raftery, D. Can NMR solve some significant challenges in metabolomics? J. Magn. Reson. 2015, 260, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Hoerr, V.; Duggan, G.E.; Zbytnuik, L.; Poon, K.K.H.; Große, C.; Neugebauer, U.; Methling, K.; Löffler, B.; Vogel, H.J. Characterization and prediction of the mechanism of action of antibiotics through NMR metabolomics. BMC Microbiol. 2016, 16, 1–14. [Google Scholar] [CrossRef]
- Mitsuwan, W.; Olaya-Abril, A.; Calderón-Santiago, M.; Jiménez-Munguía, I.; González-Reyes, J.A.; Priego-Capote, F.; Voravuthikunchai, S.P.; Rodríguez-Ortega, M.J. Integrated proteomic and metabolomic analysis reveals that rhodomyrtone reduces the capsule in Streptococcus pneumoniae. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Chavali, A.K.; D’Auria, K.M.; Hewlett, E.L.; Pearson, R.D.; Papin, J.A. A metabolic network approach for the identification and prioritization of antimicrobial drug targets. Trends Microbiol. 2012, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Milshteyn, A.; Schneider, J.S.; Brady, S.F. Mining the metabiome: identifying novel natural products from microbial communities. Chem. Biol. 2014, 21, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A. Antibiotics from microbes: Converging to kill. Curr. Opin. Microbiol. 2009, 12, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Opportunities for natural products in 21st century antibiotic discovery. Nat. Prod. Rep. 2017, 34, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Barrett, J.F. Empirical antibacterial drug discovery - Foundation in natural products. Biochem. Pharmacol. 2006, 71, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Kealey, C.; Creaven, C.A.; Murphy, C.D.; Brady, C.B. New approaches to antibiotic discovery. Biotechnol. Lett. 2017, 39, 805–817. [Google Scholar] [CrossRef]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Kolter, R.; van Wezel, G.P. Goodbye to brute force in antibiotic discovery? Nat. Microbiol. 2016, 1, 15020. [Google Scholar] [CrossRef]
- Kolmeder, C.A.; de Vos, W.M. Metaproteomics of our microbiome—Developing insight in function and activity in man and model systems. J. Proteom. 2014, 97, 3–16. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Donia, M.S.; Cimermancic, P.; Schulze, C.J.; Wieland Brown, L.C.; Martin, J.; Mitreva, M.; Clardy, J.; Linington, R.G.; Fischbach, M.A. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 2014, 158, 1402–1414. [Google Scholar] [CrossRef]
- Dopazo, J. Genomics and transcriptomics in drug discovery. Drug Discov. Today 2014, 19, 126–132. [Google Scholar] [CrossRef]
- Swinney, D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef]
- Fernandes, P.; Martens, E. Antibiotics in late clinical development. Biochem. Pharmacol. 2017, 133, 152–163. [Google Scholar] [CrossRef]
- Farrell, L.J.; Lo, R.; Wanford, J.J.; Jenkins, A.; Maxwell, A.; Piddock, L.J.V. Revitalizing the drug pipeline: AntibioticDB, an open access database to aid antibacterial research and development. J. Antimicrob. Chemother. 2018, 73, 2284–2297. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclasses | Examples of Marketed Antibiotics |
|---|---|
| Penicillins | Penicillin G, Penicillin V, Ampicillin, Amoxicillin, Bacampicillin, Cloxacillinm, Floxacillin, Mezlocillin, Nafcillin, Oxacillin, Methicillin a, Dicloxacillin a, Carbenicillin b, Idanyl b, Piperacillin b, Ticarcillin b |
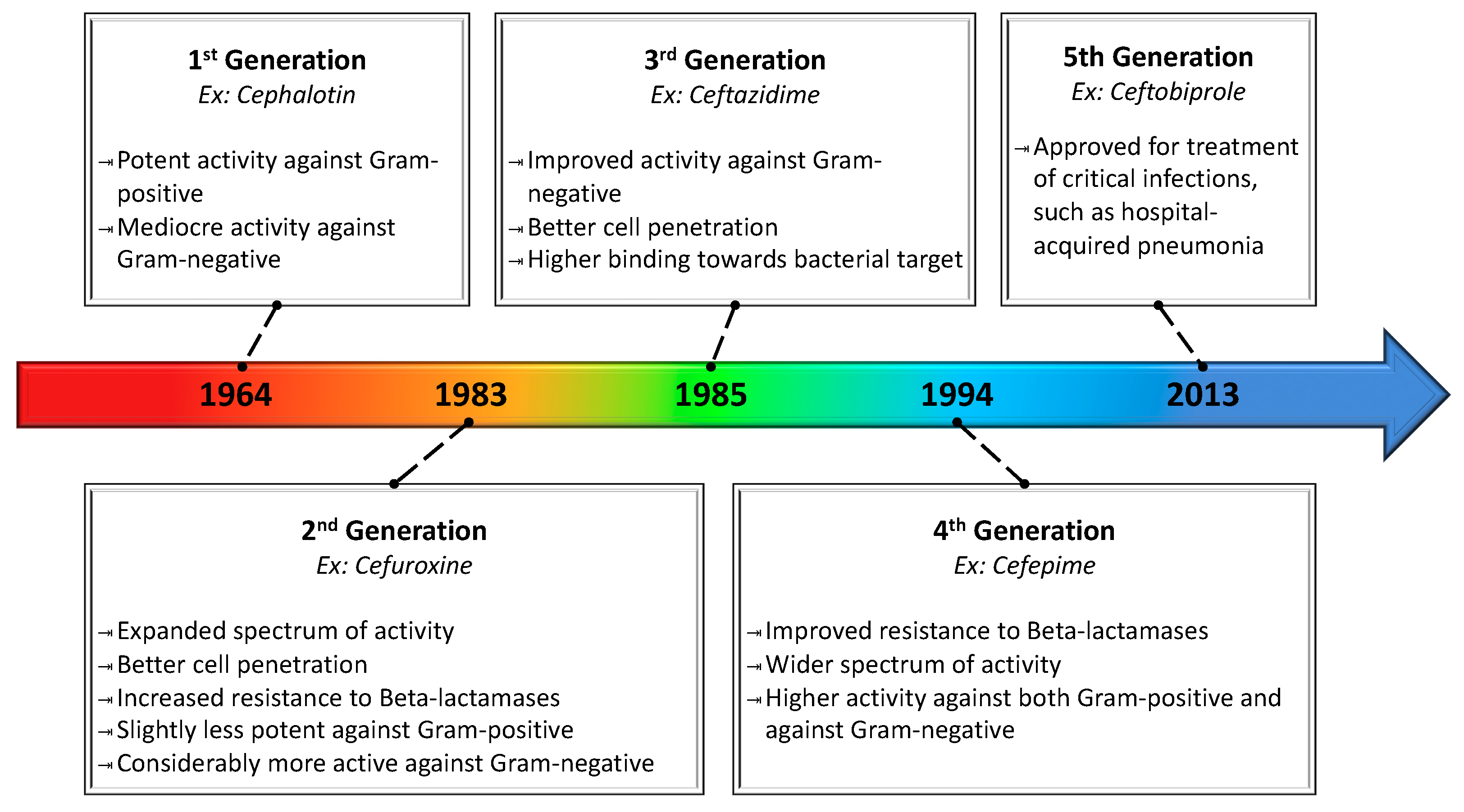
| Cephalosporins | Cefalothin c, Cephradinea c, Cefadroxyl c, Cefazolin c, Cephalexin c, Cefuroxine d, Cefaclor d, Cefotetam d, Cefmetazole d, Cefonicid d, Cefixime e, Ceftibuten e, Cefizoxime e, Ceftriaxone e, Cefamandol e, Cefoperazone e, Cefotaxime e, Proxetil e, Cefprozil e, Ceftazidime e, Cefuroxime Axetil e, Cefpodexime e, Cefepime f, Ceftobiprole g |
| Other Minor Subclasses | Flomoxef h, Latamoxef h, Cefoxitin i, Loracarbef j, Imipenem j, Meropenem j, Panipenem j, Aztreonam k, Carumonam k |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro da Cunha, B.; Fonseca, L.P.; Calado, C.R.C. Antibiotic Discovery: Where Have We Come from, Where Do We Go? Antibiotics 2019, 8, 45. https://doi.org/10.3390/antibiotics8020045
Ribeiro da Cunha B, Fonseca LP, Calado CRC. Antibiotic Discovery: Where Have We Come from, Where Do We Go? Antibiotics. 2019; 8(2):45. https://doi.org/10.3390/antibiotics8020045
Chicago/Turabian StyleRibeiro da Cunha, Bernardo, Luís P. Fonseca, and Cecília R. C. Calado. 2019. "Antibiotic Discovery: Where Have We Come from, Where Do We Go?" Antibiotics 8, no. 2: 45. https://doi.org/10.3390/antibiotics8020045
APA StyleRibeiro da Cunha, B., Fonseca, L. P., & Calado, C. R. C. (2019). Antibiotic Discovery: Where Have We Come from, Where Do We Go? Antibiotics, 8(2), 45. https://doi.org/10.3390/antibiotics8020045