Acyltransferases as Tools for Polyketide Synthase Engineering



Abstract
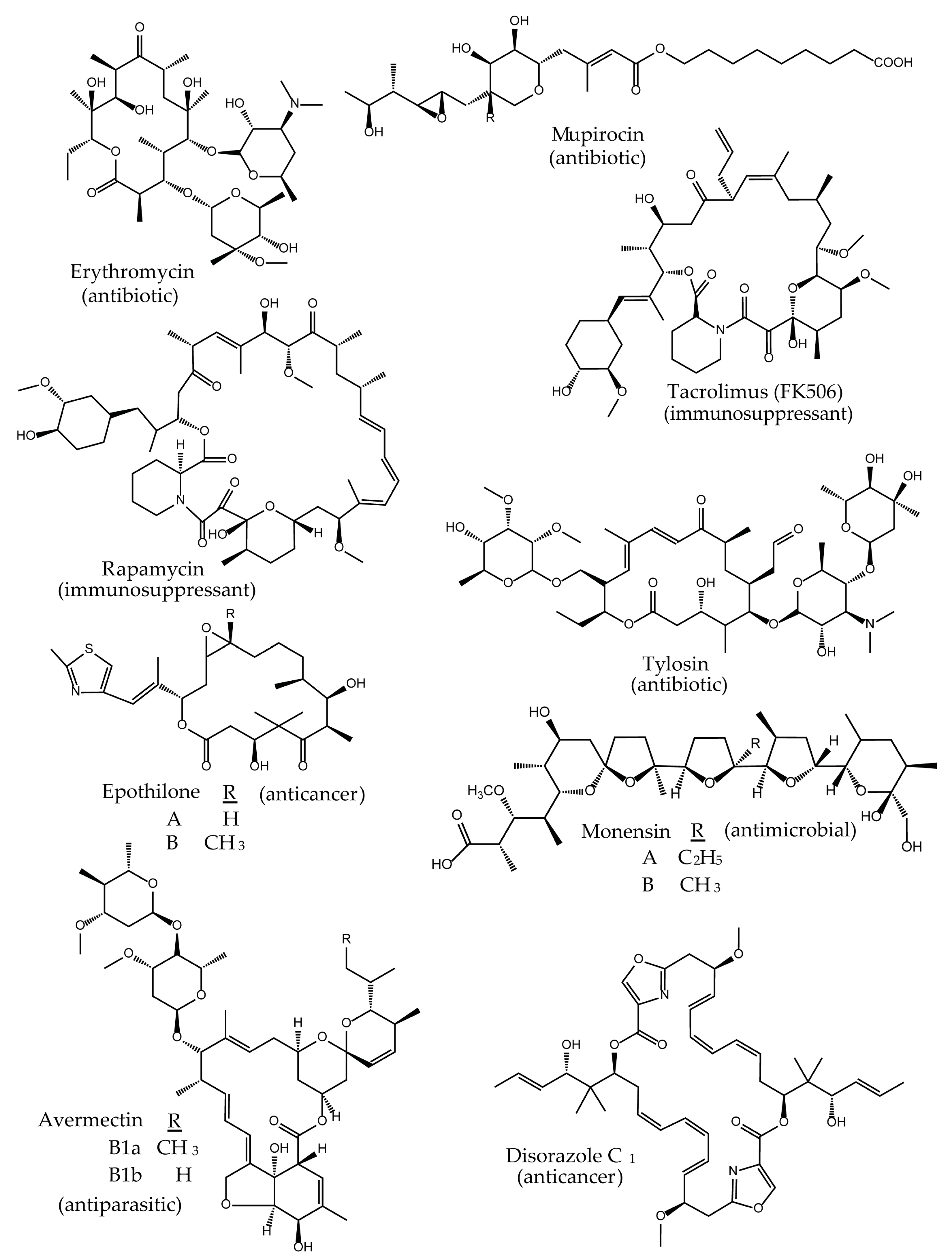
1. Introduction
2. Polyketide Synthases and the Essential Acyltransferase Domains
2.1. Acyltransferase Substrates and Loading of the Acyl Carrier Protein
2.1.1. Provision of Malonyl-CoA and Methylmalonyl-CoA Precursors
2.1.2. Provision of Ethylmalonyl-CoA and Exotic Alkylmalonyl-CoA Precursors
2.2. Mechanistic and Structural Insights into Acyltransferases and Polyketide Synthase Modules
2.2.1. Substrate Recognition and Acyltransferase-Acyl Carrier Protein Interactions
2.2.2. Interactions between the Acyltransferase, Ketosynthase and the Acyl Carrier Protein
2.2.3. Docking Domains: Intersubunit Communication in Polyketide Assembly Lines
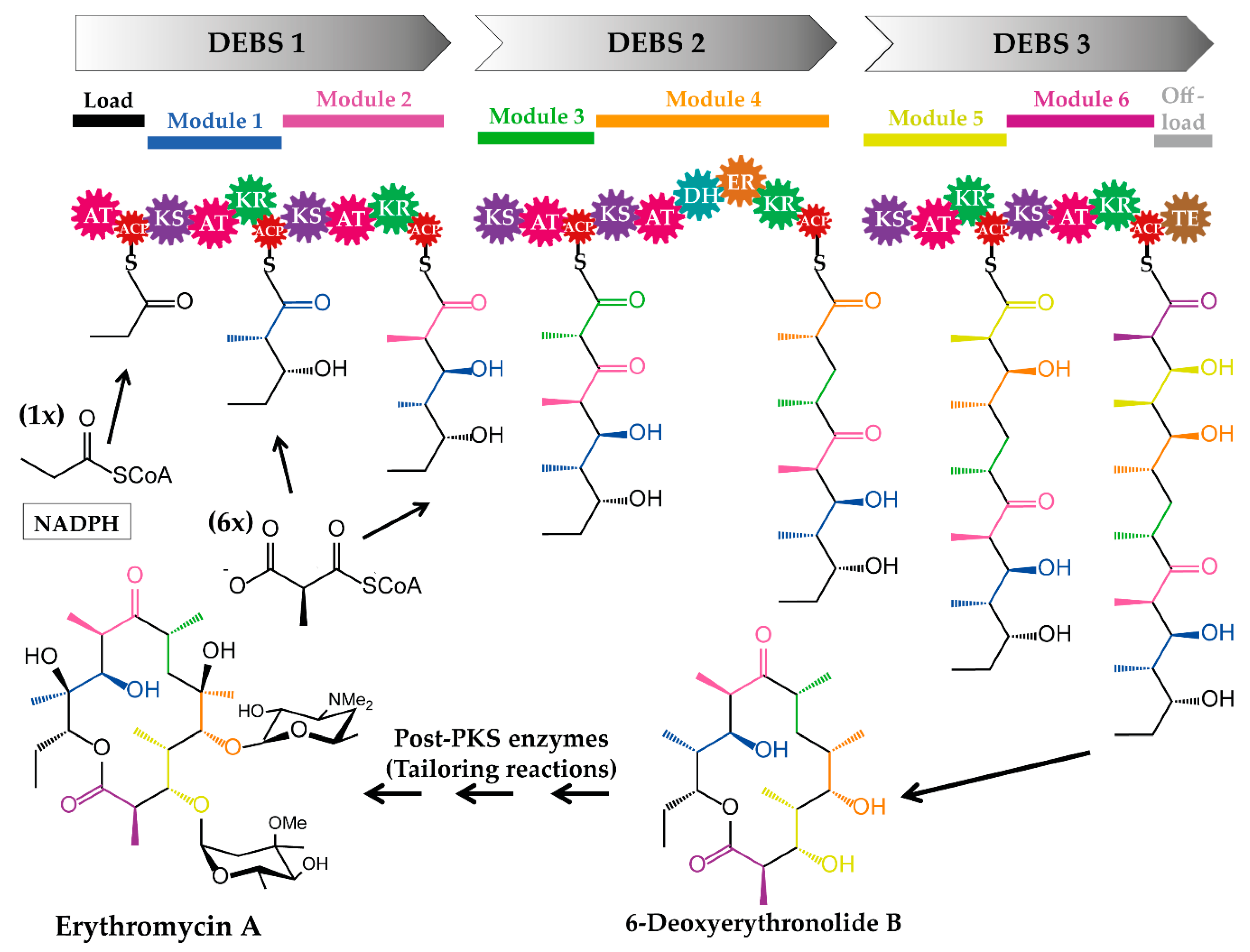
3. Strategies of Acyltransferase-Based Polyketide Engineering
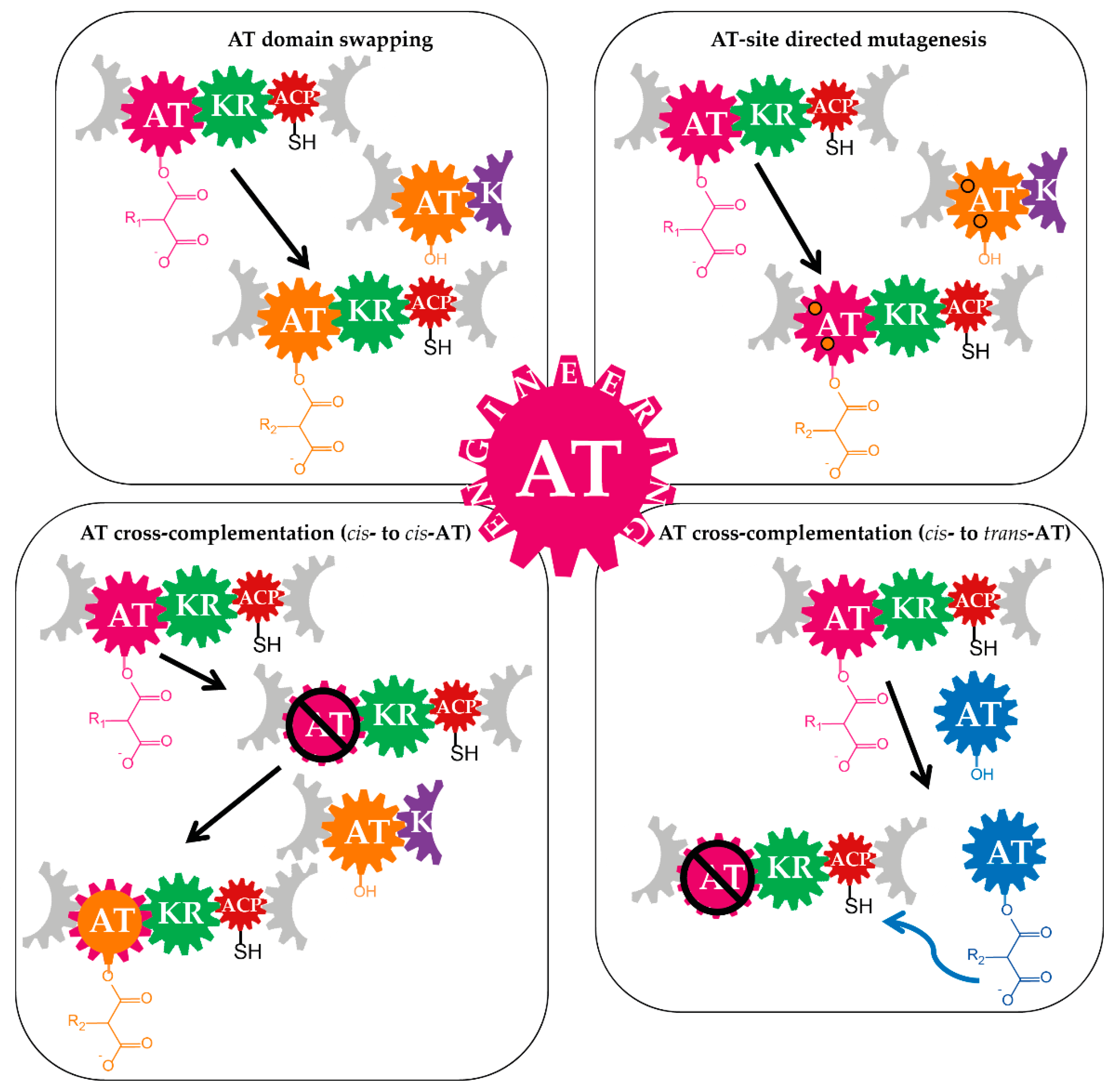
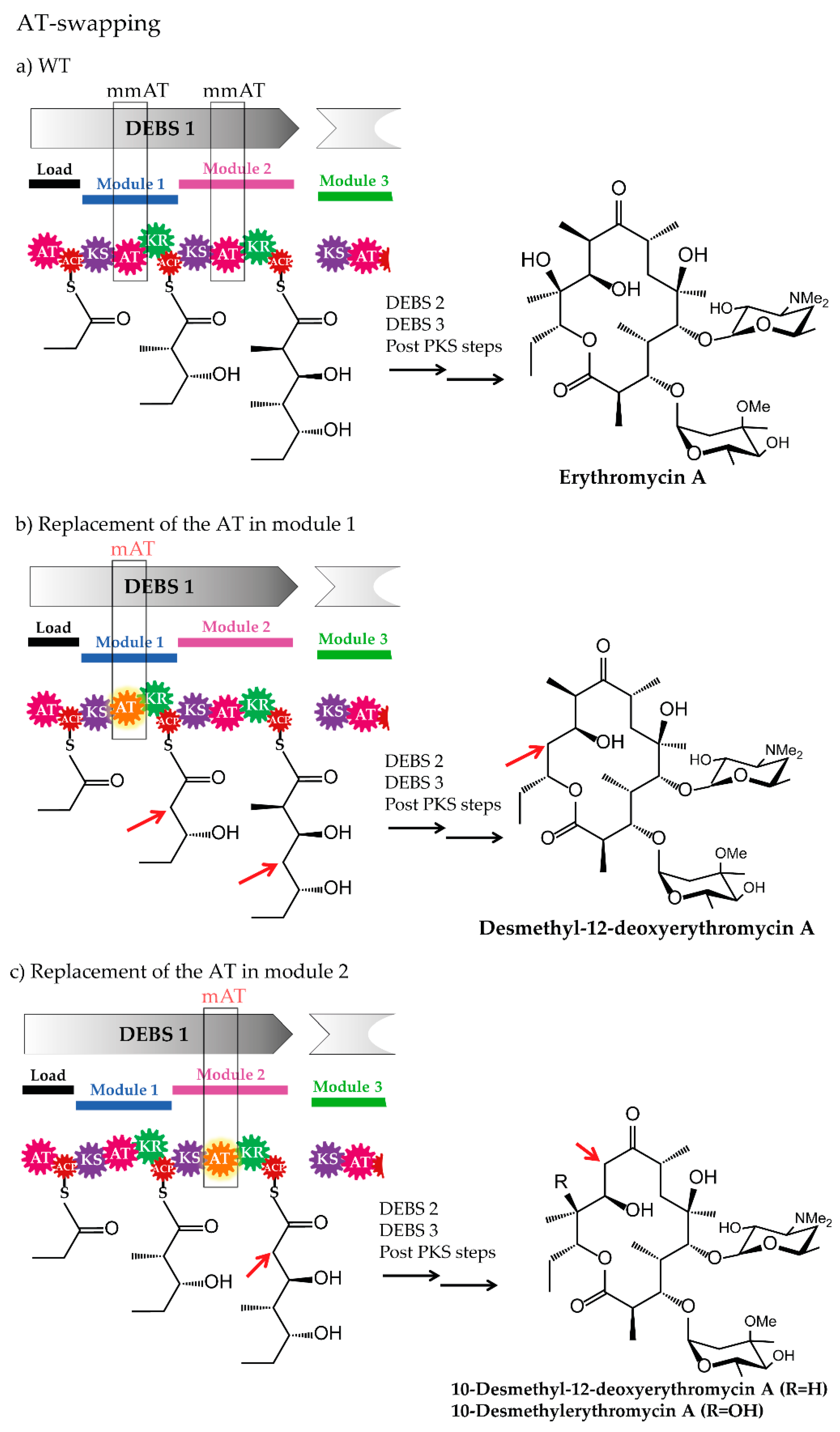
3.1. Domain-Swapping
3.1.1. Acyltransferase Domain Substitution and the Provision of the Required Non-Native Precursors
3.1.2. Examples of Acyltransferase Domain Substitution by Exchanging the Entire Module and the Supply of Non-Native Precursors
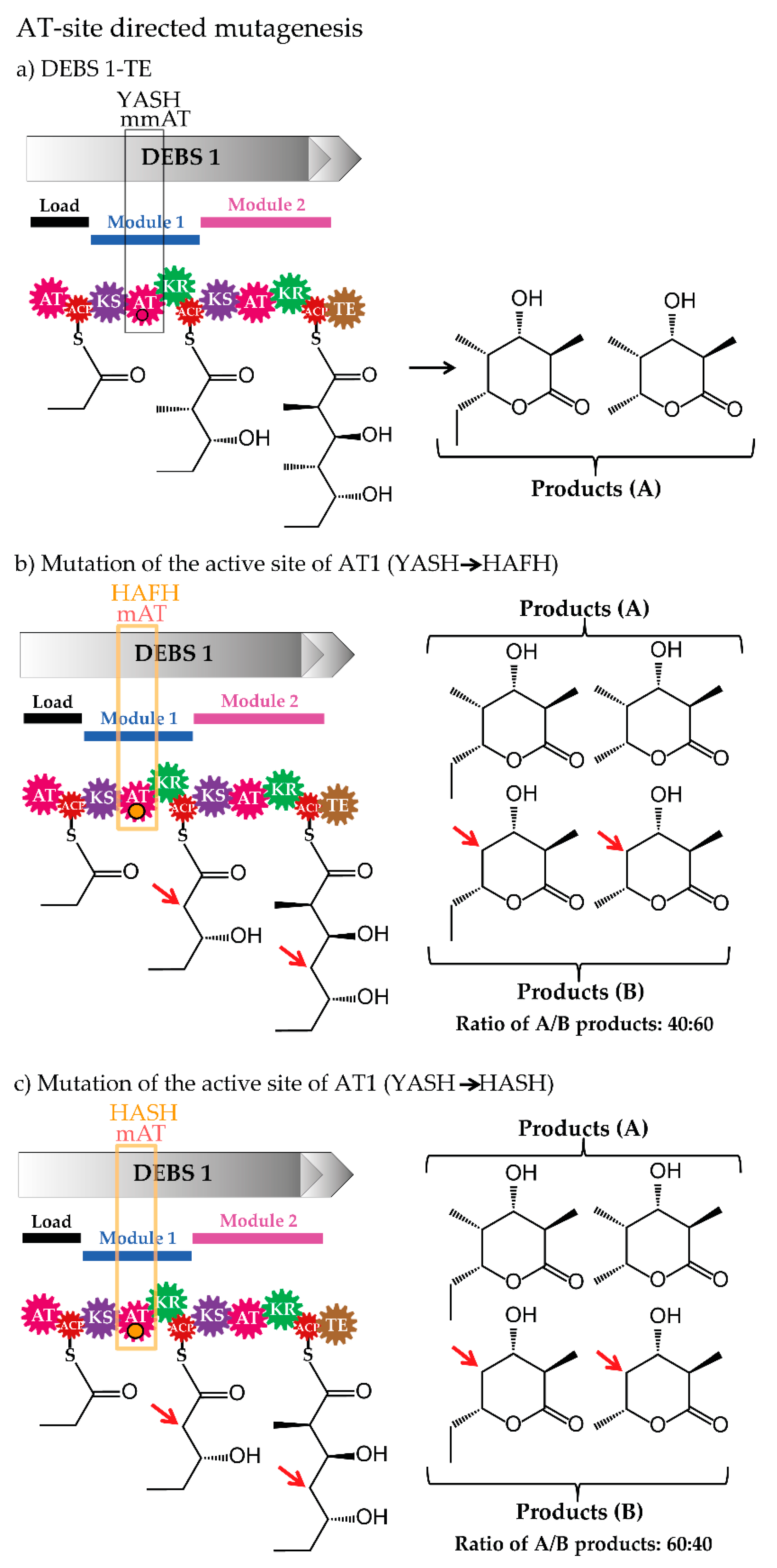
3.2. Site-Specific Mutagenesis of Acyltransferases
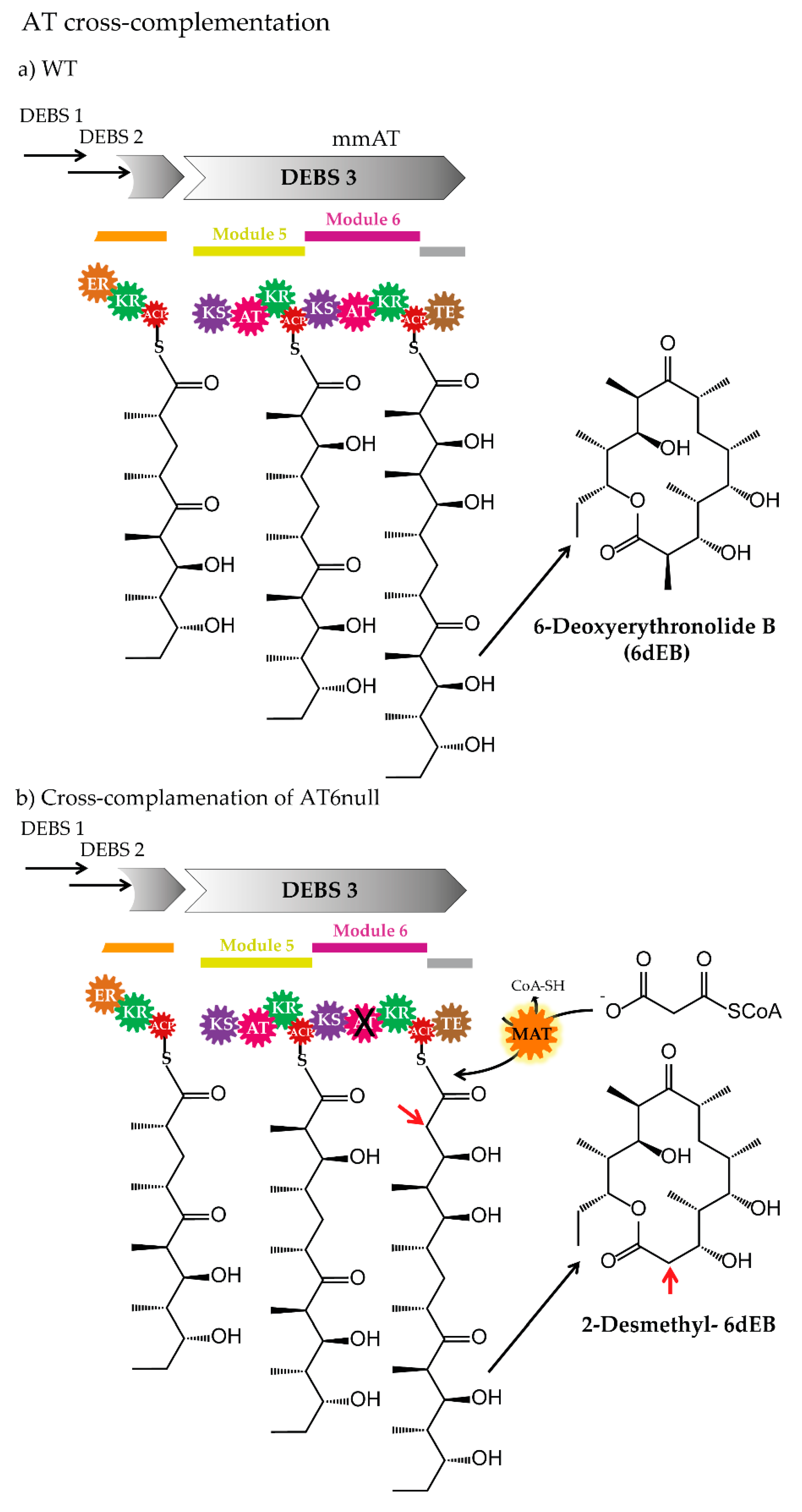
3.3. Cross-Acyltransferase Complementation
4. Advances in Natural Science and Future Perspectives of AT-Based PKS Engineering
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| ACP | acyl carrier protein |
| AT | acyltransferase |
| CCR | crotonyl-CoA carboxylase/reductase |
| CCRC | crotonyl-CoA reductase/carboxylase |
| CoA | coenzyme A |
| CRISPR-Cas9 | clustered regularly interspersed palindromic repeats; CRISPR associated (Cas) protein (Cas9) |
| Cryo-EM | cryo-electron microscopy |
| KS | ketosynthase |
| DD | docking domains |
| DEBS | 6-deoxyerythromycin B synthase |
| DH | dehydrogenase |
| ECR | enoyl-CoA carboxylase/reductase |
| ER | enoylreductase |
| KR | ketoreductase |
| NRPS | nonribosomal peptide synthetase |
| PCC | propionyl-CoA carboxylase |
| PKS | polyketide synthase |
| PPTase | 4-phosphopantetheinyl transferase |
| Red/ET | homologous recombination system based on the Red operon of lambda phage or RecE/RecT from Rac phage |
| SNAC | N-acetylcysteamine |
| TAR | transformation-associated recombination |
| TE | thioesterase |
| USER | uracil-excision based cloning |
| YCC | acyl-CoA carboxylase |
References
- Cane, D.E.; Walsh, C.T.; Khosla, C. Harnessing the biosynthetic code: Combinations, permutations, and mutations. Science 1998, 282, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. Engl. 2009, 48, 4688–4716. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, G.B.; Balachandran, L. Antibacterial agents from actinomycetes—A review. Front. Biosci. 2012, 4, 240–253. [Google Scholar] [CrossRef]
- Vannucchi, V. Clinical study of a new antibiotic; erythromycin. Riv. Crit. Clin. Med. 1952, 52, 128–136. [Google Scholar] [PubMed]
- Haight, T.H.; Finland, M. Laboratory and clinical studies on erythromycin. N. Engl. J. Med. 1952, 247, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wuite, J.; Davies, B.I.; Go, M.; Lambers, J.; Jackson, D.; Mellows, G. Pseudomonic acid: A new topical antimicrobial agent. Lancet 1983, 2, 394. [Google Scholar] [CrossRef]
- Casewell, M.W.; Hill, R.L. Mupirocin (‘pseudomonic acid’)—A promising new topical antimicrobial agent. J. Antimicrob. Chemother. 1987, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. (Tokyo) 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hatanaka, H.; Hashimoto, M.; Nishiyama, M.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; Imanaka, H. FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. (Tokyo) 1987, 40, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hatanaka, H.; Miyata, S.; Inamura, N.; Nishiyama, M.; Yajima, T.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J. Antibiot. (Tokyo) 1987, 40, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar] [PubMed]
- Gerth, K.; Bedorf, N.; Hofle, G.; Irschik, H.; Reichenbach, H. Epothilons A and B: Antifungal and cytotoxic compounds from Sorangium cellulosum (myxobacteria). Production, physico-chemical and biological properties. J. Antibiot. (Tokyo) 1996, 49, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Lam, N.Y.S. Challenges and discoveries in the total synthesis of complex polyketide natural products. J. Antibiot. (Tokyo) 2018, 71, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Mapperson, R.R.; Kotiw, M.; Davis, R.A.; Dearnaley, J.D. The diversity and antimicrobial activity of Preussia sp. endophytes isolated from Australian dry rainforests. Curr. Microbiol. 2014, 68, 30–37. [Google Scholar] [CrossRef] [PubMed]
- El-Hawary, S.S.; Mohammed, R.; AbouZid, S.F.; Bakeer, W.; Ebel, R.; Sayed, A.M.; Rateb, M.E. Solamargine production by a fungal endophyte of Solanum nigrum. J. Appl. Microbiol. 2016, 120, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Iftime, D.; Kulik, A.; Hartner, T.; Rohrer, S.; Niedermeyer, T.H.; Stegmann, E.; Weber, T.; Wohlleben, W. Identification and activation of novel biosynthetic gene clusters by genome mining in the kirromycin producer Streptomyces collinus Tü 365. J. Ind. Microbiol. Biotechnol. 2016, 43, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Teanpaisan, R.; Kawsud, P.; Pahumunto, N.; Puripattanavong, J. Screening for antibacterial and antibiofilm activity in Thai medicinal plant extracts against oral microorganisms. J. Tradit. Complement Med. 2017, 7, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.J.; Yao, S.; Guo, X.; Yue, B.S.; Ma, X.Y.; Li, J. Bioactivity-guided screening of wound-healing active constituents from American cockroach (Periplaneta americana). Molecules 2018, 23, 101. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Haydock, S.F.; Roberts, G.A.; Bevitt, D.J.; Leadlay, P.F. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 1990, 348, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Donadio, S.; Staver, M.J.; McAlpine, J.B.; Swanson, S.J.; Katz, L. Modular organization of genes required for complex polyketide biosynthesis. Science 1991, 252, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef]
- Müller, R. Don’t classify polyketide synthases. Chem. Biol. 2004, 11, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Stuken, A.; Orr, R.J.; Svendsen, H.M.; Jakobsen, K.S. Biosynthesis and molecular genetics of polyketides in marine dinoflagellates. Mar. Drugs 2010, 8, 1011–1048. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Du, L. Iterative polyketide biosynthesis by modular polyketide synthases in bacteria. Appl. Microbiol. Biotechnol. 2016, 100, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Ogata, H.; Goto, S. Type III polyketide synthases: Functional classification and phylogenomics. ChemBioChem 2017, 18, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Ogata, H.; Goto, S. Corrigendum: Type III polyketide synthases: Functional classification and phylogenomics. ChemBioChem 2017, 18, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hertweck, C. Functional analysis of the aureothin iterative type I polyketide synthase. ChemBioChem 2005, 6, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.J.; Pang, L.M.; Ding, Y.; Cheang, Q.W.; Le Mai Hoang, K.; Thi Tran, H.; Li, J.; Liu, X.W.; Kanagasundaram, Y.; Yang, L.; et al. Identification of a biosynthetic gene cluster for the polyene macrolactam sceliphrolactam in a Streptomyces strain isolated from mangrove sediment. Sci. Rep. 2018, 8, 1594. [Google Scholar] [CrossRef] [PubMed]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef] [PubMed]
- Musiol, E.M.; Weber, T. Discrete acyltransferases involved in polyketide biosynthesis. Med. Chem. Comm. 2012, 3, 871–886. [Google Scholar] [CrossRef]
- Helfrich, E.J.; Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2016, 33, 231–316. [Google Scholar] [CrossRef] [PubMed]
- Hajra, A.K. Dihydroxyacetone phosphate acyltransferase. Biochim. Biophys. Acta 1997, 1348, 27–34. [Google Scholar] [CrossRef]
- Barney, B.M.; Wahlen, B.D.; Garner, E.; Wei, J.; Seefeldt, L.C. Differences in substrate specificities of five bacterial wax ester synthases. Appl. Environ. Microbiol. 2012, 78, 5734–5745. [Google Scholar] [CrossRef] [PubMed]
- Wenning, L.; Yu, T.; David, F.; Nielsen, J.; Siewers, V. Establishing very long-chain fatty alcohol and wax ester biosynthesis in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2017, 114, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, P. Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem 2003, 4, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Minowa, Y.; Araki, M.; Kanehisa, M. Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J. Mol. Biol. 2007, 368, 1500–1517. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Ishida, K.; Traitcheva, N.; Busch, B.; Dahse, H.M.; Hertweck, C. Freedom and constraint in engineered noncolinear polyketide assembly lines. Chem. Biol. 2015, 22, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Reid, R.; Piagentini, M.; Rodriguez, E.; Ashley, G.; Viswanathan, N.; Carney, J.; Santi, D.V.; Hutchinson, C.R.; McDaniel, R. A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry 2003, 42, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Mohanty, D. Modeling holo-ACP:DH and holo-ACP:KR complexes of modular polyketide synthases: A docking and molecular dynamics study. BMC Struct. Biol. 2012, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.; Anand, S.; Mohanty, D. Prediction of inter domain interactions in modular polyketide synthases by docking and correlated mutation analysis. J. Biomol. Struct. Dyn. 2013, 31, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hashimoto, T.; Qin, B.; Hashimoto, J.; Kozone, I.; Kawahara, T.; Okada, M.; Awakawa, T.; Ito, T.; Asakawa, Y.; et al. Characterization of giant modular PKSs provides insight into genetic mechanism for structural diversification of aminopolyol polyketides. Angew. Chem. Int. Ed. Engl. 2017, 56, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Keatinge-Clay, A.T. Polyketide synthase modules redefined. Angew. Chem. Int. Ed. Engl. 2017, 56, 4658–4660. [Google Scholar] [CrossRef] [PubMed]
- Vander Wood, D.A.; Keatinge-Clay, A.T. The modules of trans-acyltransferase assembly lines redefined with a central acyl carrier protein. Proteins 2018, 86, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Cimermancic, P.; Sali, A.; Takano, E.; Fischbach, M.A. A systematic computational analysis of biosynthetic gene cluster evolution: Lessons for engineering biosynthesis. PLoS Comput. Biol. 2014, 10, e1004016. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.; Freeman, M.F.; Piel, J. The catalytic diversity of multimodular polyketide synthases: Natural product biosynthesis beyond textbook assembly rules. Top. Curr. Chem. 2011. [Google Scholar] [CrossRef]
- Khosla, C.; Herschlag, D.; Cane, D.E.; Walsh, C.T. Assembly line polyketide synthases: Mechanistic insights and unsolved problems. Biochemistry 2014, 53, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.J.; Khosla, C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J. R. Soc. Interface 2013, 10, 20130297. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.L.; Yadav, V.G. Towards precision engineering of canonical polyketide synthase domains: Recent advances and future prospects. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Pereda, A.; Stassi, D.L.; Zeidner, D.; Summers, R.G.; Jackson, M.; Shivakumar, A.; Kakavas, S.; Staver, M.J.; Donadio, S.; et al. Acyltransferase domain substitutions in erythromycin polyketide synthase yield novel erythromycin derivatives. J. Bacteriol. 1997, 179, 6416–6425. [Google Scholar] [CrossRef] [PubMed]
- Oliynyk, M.; Brown, M.J.; Cortes, J.; Staunton, J.; Leadlay, P.F. A hybrid modular polyketide synthase obtained by domain swapping. Chem. Biol. 1996, 3, 833–839. [Google Scholar] [CrossRef]
- Lau, J.; Fu, H.; Cane, D.E.; Khosla, C. Dissecting the role of acyltransferase domains of modular polyketide synthases in the choice and stereochemical fate of extender units. Biochemistry 1999, 38, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.; Timoney, M.; Bycroft, M.; Cortes, J.; Thomas, I.P.; Wilkinson, B.; Kellenberger, L.; Hanefeld, U.; Galloway, I.S.; Staunton, J.; et al. Knowledge-based design of bimodular and trimodular polyketide synthases based on domain and module swaps: A route to simple statin analogues. Chem. Biol. 1999, 6, 731–741. [Google Scholar] [CrossRef]
- Liu, L.; Thamchaipenet, A.; Fu, H.; Betlach, M.; Ashley, G. Biosynthesis of 2-nor-6-deoxyerythronolide B by rationally designed domain substitution. J. Am. Chem. Soc. 1997, 119, 10553–10554. [Google Scholar] [CrossRef]
- Petkovic, H.; Sandmann, A.; Challis, I.R.; Hecht, H.J.; Silakowski, B.; Low, L.; Beeston, N.; Kuscer, E.; Garcia-Bernardo, J.; Leadlay, P.F.; et al. Substrate specificity of the acyl transferase domains of EpoC from the epothilone polyketide synthase. Org. Biomol. Chem. 2008, 6, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Stassi, D.L.; Kakavas, S.J.; Reynolds, K.A.; Gunawardana, G.; Swanson, S.; Zeidner, D.; Jackson, M.; Liu, H.; Buko, A.; Katz, L. Ethyl-substituted erythromycin derivatives produced by directed metabolic engineering. Proc. Natl. Acad. Sci. USA 1998, 95, 7305–7309. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Bai, L.; Xue, Q.; Revill, W.P.; Yu, T.W.; Floss, H.G. Functional expression of genes involved in the biosynthesis of the novel polyketide chain extension unit, methoxymalonyl-acyl carrier protein, and engineered biosynthesis of 2-desmethyl-2-methoxy-6-deoxyerythronolide B. J. Am. Chem. Soc. 2002, 124, 5268–5269. [Google Scholar] [CrossRef] [PubMed]
- Reeves, C.D.; Chung, L.M.; Liu, Y.; Xue, Q.; Carney, J.R.; Revill, W.P.; Katz, L. A new substrate specificity for acyl transferase domains of the ascomycin polyketide synthase in Streptomyces hygroscopicus. J. Biol. Chem. 2002, 277, 9155–9159. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Piagentini, M.; Rascher, A.; Tian, Z.Q.; Buchanan, G.O.; Regentin, R.; Hu, Z.; Hutchinson, C.R.; McDaniel, R. Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem. Biol. 2004, 11, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, F.; Petkovic, H.; Kendrew, S.G.; Low, L.; Wilkinson, B.; Lill, R.; Cortes, J.; Rudd, B.A.; Staunton, J.; Leadlay, P.F. Active-site residue, domain and module swaps in modular polyketide synthases. J. Ind. Microbiol. Biotechnol. 2003, 30, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Sundermann, U.; Bravo-Rodriguez, K.; Klopries, S.; Kushnir, S.; Gomez, H.; Sanchez-Garcia, E.; Schulz, F. Enzyme-directed mutasynthesis: A combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem. Biol. 2013, 8, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Reeves, C.D.; Murli, S.; Ashley, G.W.; Piagentini, M.; Hutchinson, C.R.; McDaniel, R. Alteration of the substrate specificity of a modular polyketide synthase acyltransferase domain through site-specific mutations. Biochemistry 2001, 40, 15464–15470. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Rodriguez, K.; Klopries, S.; Koopmans, K.R.M.; Sundermann, U.; Yahiaoui, S.; Arens, J.; Kushnir, S.; Schulz, F.; Sanchez-Garcia, E. Substrate flexibility of a mutated acyltransferase domain and implications for polyketide biosynthesis. Chem. Biol. 2015, 22, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Klopries, S.; Bravo-Rodriguez, K.; Koopmans, K.R.; Sundermann, U.; Yahiaoui, S.; Arens, J.; Kushnir, S.; Sanchez-Garcia, E.; Schulz, F. Data in support of substrate flexibility of a mutated acyltransferase domain and implications for polyketide biosynthesis. Data Brief. 2015, 5, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, I.; Kasey, C.; McArthur, J.B.; Lowell, A.N.; Chemler, J.A.; Li, S.; Hansen, D.A.; Sherman, D.H.; Williams, G.J. Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering. ACS Chem. Biol. 2017, 12, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Koppisch, A.T.; Cane, D.E.; Khosla, C. Enhancing the modularity of the modular polyketide synthases: Transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J. Am. Chem. Soc. 2003, 125, 14307–14312. [Google Scholar] [CrossRef] [PubMed]
- Lopanik, N.B.; Shields, J.A.; Buchholz, T.J.; Rath, C.M.; Hothersall, J.; Haygood, M.G.; Hakansson, K.; Thomas, C.M.; Sherman, D.H. In vivo and in vitro trans-acylation by BryP, the putative bryostatin pathway acyltransferase derived from an uncultured marine symbiont. Chem. Biol. 2008, 15, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.K.; Cronan, J.E., Jr. Activation of long chain fatty acids with acyl carrier protein: Demonstration of a new enzyme, acyl-acyl carrier protein synthetase, in Escherichia coli. Proc. Natl. Acad. Sci. USA 1976, 73, 4374–4378. [Google Scholar] [CrossRef] [PubMed]
- Quadri, L.E.; Weinreb, P.H.; Lei, M.; Nakano, M.M.; Zuber, P.; Walsh, C.T. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 1998, 37, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Pavlidou, M.; Pross, E.K.; Musiol, E.M.; Kulik, A.; Wohlleben, W.; Weber, T. The phosphopantetheinyl transferase KirP activates the ACP and PCP domains of the kirromycin NRPS/PKS of Streptomyces collinus Tü 365. FEMS Microbiol. Lett. 2011, 319, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Coughlin, J.M.; Lim, S.K.; Shen, B. Type I polyketide synthases that require discrete acyltransferases. Methods Enzymol. 2009, 459, 165–186. [Google Scholar] [PubMed]
- Oliynyk, M.; Stark, C.B.; Bhatt, A.; Jones, M.A.; Hughes-Thomas, Z.A.; Wilkinson, C.; Oliynyk, Z.; Demydchuk, Y.; Staunton, J.; Leadlay, P.F. Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin in Streptomyces cinnamonensis and evidence for the role of monB and monC genes in oxidative cyclization. Mol. Microbiol. 2003, 49, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.; Stark, C.B.; Harvey, B.M.; Gallimore, A.R.; Demydchuk, Y.A.; Spencer, J.B.; Staunton, J.; Leadlay, P.F. Accumulation of an E,E,E-triene by the monensin-producing polyketide synthase when oxidative cyclization is blocked. Angew. Chem. Int. Ed. Engl. 2005, 44, 7075–7078. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Rodriguez, K.; Ismail-Ali, A.F.; Klopries, S.; Kushnir, S.; Ismail, S.; Fansa, E.K.; Wittinghofer, A.; Schulz, F.; Sanchez-Garcia, E. Predicted incorporation of non-native substrates by a polyketide synthase yields bioactive natural product derivatives. ChemBioChem 2014, 15, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Guchhait, R.B.; Polakis, S.E.; Dimroth, P.; Stoll, E.; Moss, J.; Lane, M.D. Acetyl coenzyme A carboxylase system of Escherichia coli. Purification and properties of the biotin carboxylase, carboxyltransferase, and carboxyl carrier protein components. J. Biol. Chem. 1974, 249, 6633–6645. [Google Scholar] [PubMed]
- Bramwell, H.; Hunter, I.S.; Coggins, J.R.; Nimmo, H.G. Propionyl-CoA carboxylase from Streptomyces coelicolor A3(2): Cloning of the gene encoding the biotin-containing subunit. Microbiology 1996, 142 Pt 3, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Gago, G.; Kurth, D.; Diacovich, L.; Tsai, S.C.; Gramajo, H. Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J. Bacteriol. 2006, 188, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Diacovich, L.; Peiru, S.; Kurth, D.; Rodriguez, E.; Podesta, F.; Khosla, C.; Gramajo, H. Kinetic and structural analysis of a new group of acyl-CoA carboxylases found in Streptomyces coelicolor A3(2). J. Biol. Chem. 2002, 277, 31228–31236. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Keatinge-Clay, A. Enzymatic extender unit generation for in vitro polyketide synthase reactions: Structural and functional showcasing of Streptomyces coelicolor MatB. Chem. Biol. 2011, 18, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pohl, N.L.; Hans, M.; Lee, H.Y.; Kim, Y.S.; Cane, D.E.; Khosla, C. Remarkably broad substrate tolerance of malonyl-CoA synthetase, an enzyme capable of intracellular synthesis of polyketide precursors. J. Am. Chem. Soc. 2001, 123, 5822–5823. [Google Scholar] [CrossRef] [PubMed]
- An, J.H.; Kim, Y.S. A gene cluster encoding malonyl-CoA decarboxylase (MatA), malonyl-CoA synthetase (MatB) and a putative dicarboxylate carrier protein (MatC) in Rhizobium trifolii--cloning, sequencing, and expression of the enzymes in Escherichia coli. Eur. J. Biochem. 1998, 257, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Chae, H.Z. Purification and properties of malonyl-CoA synthetase from Rhizobium japonicum. Biochem. J. 1991, 273 Pt 3, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, S.; Park, J.W.; Yoon, Y.J.; Lee, H.C.; Sohng, J.K. Metabolic engineering of Streptomyces venezuelae for malonyl-CoA biosynthesis to enhance heterologous production of polyketides. Biotechnol. Lett. 2010, 32, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, I.; Williams, G.J. Mutant malonyl-CoA synthetases with altered specificity for polyketide synthase extender unit generation. ChemBioChem 2011, 12, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Crosby, H.A.; Rank, K.C.; Rayment, I.; Escalante-Semerena, J.C. Structure-guided expansion of the substrate range of methylmalonyl coenzyme A synthetase (MatB) of Rhodopseudomonas palustris. Appl. Environ. Microbiol. 2012, 78, 6619–6629. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Watanabe, K.; Wojcicki, W.A.; Wang, C.C.; Tang, Y. Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem. Biol. 2006, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Kim, D.H.; Lee, J.H.; Park, J.W.; Basnet, D.B.; Ban, Y.H.; Yoo, Y.J.; Chen, S.W.; Park, S.R.; Choi, E.A.; et al. Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J. Am. Chem. Soc. 2011, 133, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Klopries, S.; Sundermann, U.; Schulz, F. Quantification of N-acetylcysteamine activated methylmalonate incorporation into polyketide biosynthesis. Beilstein. J. Org. Chem. 2013, 9, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.T.; Pohl, N.L.; Kealey, J.T.; Khosla, C. Tolerance and specificity of recombinant 6-methylsalicyclic acid synthase. Metab. Eng. 1999, 1, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Musiol-Kroll, E.M.; Zubeil, F.; Schafhauser, T.; Hartner, T.; Kulik, A.; McArthur, J.; Koryakina, I.; Wohlleben, W.; Grond, S.; Williams, G.J.; et al. Polyketide bioderivatization using the promiscuous acyltransferase KirCII. ACS Synth. Biol. 2017, 6, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Berg, I.A.; Brecht, V.; Muller, M.; Fuchs, G.; Alber, B.E. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 10631–10636. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Brecht, V.; Fuchs, G.; Muller, M.; Alber, B.E. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl-thioester reductase. Proc. Natl. Acad. Sci. USA 2009, 106, 8871–8876. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Podevels, A.M.; Kevany, B.M.; Thomas, M.G. Biosynthesis of polyketide synthase extender units. Nat. Prod. Rep. 2009, 26, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.; Moore, B.S. Recent advances in the biosynthesis of unusual polyketide synthase substrates. Nat. Prod. Rep. 2016, 33, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Riveros-Rosas, H.; Julian-Sanchez, A.; Villalobos-Molina, R.; Pardo, J.P.; Pina, E. Diversity, taxonomy and evolution of medium-chain dehydrogenase/reductase superfamily. Eur. J. Biochem. 2003, 270, 3309–3334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mori, T.; Zheng, Q.; Awakawa, T.; Yan, Y.; Liu, W.; Abe, I. Rational control of polyketide extender units by structure-based engineering of a crotonyl-CoA carboxylase/reductase in antimycin biosynthesis. Angew. Chem. Int. Ed. Engl. 2015, 54, 13462–13465. [Google Scholar] [CrossRef] [PubMed]
- Kundert, J.; Gulder, T.A. Extending polyketide structural diversity by using engineered carboxylase/reductase enzymes. Angew. Chem. Int. Ed. Engl. 2016, 55, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.; Valentic, T.R.; Miyazawa, T.; Withall, D.M.; Song, L.; Milligan, J.C.; Osada, H.; Takahashi, S.; Tsai, S.C.; Challis, G.L. A crotonyl-CoA reductase-carboxylase independent pathway for assembly of unusual alkylmalonyl-CoA polyketide synthase extender units. Nat. Commun. 2016, 7, 13609. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Schulz, F. The stereochemistry of complex polyketide biosynthesis by modular polyketide synthases. Molecules 2011, 16, 6092–6115. [Google Scholar] [CrossRef] [PubMed]
- Marsden, A.F.; Caffrey, P.; Aparicio, J.F.; Loughran, M.S.; Staunton, J.; Leadlay, P.F. Stereospecific acyl transfers on the erythromycin-producing polyketide synthase. Science 1994, 263, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.; Martin, C.J.; Wilkinson, C.J.; Staunton, J.; Leadlay, P.F. Skipping in a hybrid polyketide synthase. Evidence for ACP-to-ACP chain transfer. Chem. Biol. 2002, 9, 781–787. [Google Scholar] [CrossRef]
- Moss, S.J.; Martin, C.J.; Wilkinson, B. Loss of co-linearity by modular polyketide synthases: a mechanism for the evolution of chemical diversity. Nat. Prod. Rep. 2004, 21, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Kalkreuter, E.; Williams, G.J. Engineering enzymatic assembly lines for the production of new antimicrobials. Curr. Opin. Microbiol. 2018, 45, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Khosla, C.; Tang, Y.; Chen, A.Y.; Schnarr, N.A.; Cane, D.E. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 2007, 76, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.C.; Wakil, S.J. Studies on the mechanism of fatty acid synthesis. XXVI. Purification and properties of malonyl-coenzyme A--acyl carrier protein transacylase of Escherichia coli. Arch. Biochem. Biophys. 1971, 143, 493–505. [Google Scholar] [CrossRef]
- Wong, F.T.; Jin, X.; Mathews, I.I.; Cane, D.E.; Khosla, C. Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry 2011, 50, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.T.; Chen, A.Y.; Cane, D.E.; Khosla, C. Protein-protein recognition between acyltransferases and acyl carrier proteins in multimodular polyketide synthases. Biochemistry 2010, 49, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.J.; Cane, D.E.; Khosla, C. Mechanism and specificity of an acyltransferase domain from a modular polyketide synthase. Biochemistry 2013, 52, 1839–1841. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Thomas, M.G. Recognition of (2S)-aminomalonyl-acyl carrier protein (ACP) and (2R)-hydroxymalonyl-ACP by acyltransferases in zwittermicin A biosynthesis. Biochemistry 2010, 49, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kevany, B.M.; Dyer, D.H.; Thomas, M.G.; Forest, K.T. A polyketide synthase acyltransferase domain structure suggests a recognition mechanism for its hydroxymalonyl-acyl carrier protein substrate. PLoS ONE 2014, 9, e110965. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K.; Kamishohara, M.; Odagawa, A.; Matsuoka, M.; Kawai, H. Vicenistatin, a novel 20-membered macrocyclic lactam antitumor antibiotic. J. Antibiot. (Tokyo) 1993, 46, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Cieslak, J.; Shinohara, Y.; Kudo, F.; Eguchi, T. The crystal structure of the adenylation enzyme VinN reveals a unique beta-amino acid recognition mechanism. J. Biol. Chem. 2014, 289, 31448–31457. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Iwasawa, S.; Shinohara, Y.; Kudo, F.; Eguchi, T. Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis. Proc. Natl. Acad. Sci. USA 2016, 113, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Tsuji, S.Y.; Cane, D.E.; Khosla, C. Assessing the balance between protein-protein interactions and enzyme-substrate interactions in the channeling of intermediates between polyketide synthase modules. J. Am. Chem. Soc. 2001, 123, 6465–6474. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Cane, D.E.; Khosla, C. Quantitative analysis of the relative contributions of donor acyl carrier proteins, acceptor ketosynthases, and linker regions to intermodular transfer of intermediates in hybrid polyketide synthases. Biochemistry 2002, 41, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Schnarr, N.A.; Kim, C.Y.; Cane, D.E.; Khosla, C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 2006, 128, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J.; Muller, R. Protein-protein interactions in multienzyme megasynthetases. ChemBioChem 2008, 9, 826–848. [Google Scholar] [CrossRef] [PubMed]
- Charkoudian, L.K.; Liu, C.W.; Capone, S.; Kapur, S.; Cane, D.E.; Togni, A.; Seebach, D.; Khosla, C. Probing the interactions of an acyl carrier protein domain from the 6-deoxyerythronolide B synthase. Protein Sci. 2011, 20, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Chen, A.Y.; Cane, D.E.; Khosla, C. Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. USA 2010, 107, 22066–22071. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Whicher, J.R.; Hansen, D.A.; Hale, W.A.; Chemler, J.A.; Congdon, G.R.; Narayan, A.R.; Hakansson, K.; Sherman, D.H.; Smith, J.L.; et al. Structure of a modular polyketide synthase. Nature 2014, 510, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Whicher, J.R.; Dutta, S.; Hansen, D.A.; Hale, W.A.; Chemler, J.A.; Dosey, A.M.; Narayan, A.R.; Hakansson, K.; Sherman, D.H.; Smith, J.L.; et al. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature 2014, 510, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Lowry, B.; Li, X.; Robbins, T.; Cane, D.E.; Khosla, C. A turnstile mechanism for the controlled growth of biosynthetic intermediates on assembly line polyketide synthases. ACS Cent. Sci. 2016, 2, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Khosla, C.; Kapur, S.; Cane, D.E. Revisiting the modularity of modular polyketide synthases. Curr. Opin. Chem. Biol. 2009, 13, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, R.W.; Nietlispach, D.; Wheatcroft, M.P.; Leadlay, P.F.; Weissman, K.J. The structure of docking domains in modular polyketide synthases. Chem. Biol. 2003, 10, 723–731. [Google Scholar] [CrossRef]
- Dorival, J.; Annaval, T.; Risser, F.; Collin, S.; Roblin, P.; Jacob, C.; Gruez, A.; Chagot, B.; Weissman, K.J. Characterization of intersubunit communication in the virginiamycin trans-acyl transferase polyketide synthase. J. Am. Chem. Soc. 2016, 138, 4155–4167. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wagner, D.T.; Zhang, Z.; Moretto, L.; Addison, J.D.; Keatinge-Clay, A.T. Portability and structure of the four-helix bundle docking domains of trans-acyltransferase modular polyketide synthases. ACS Chem. Biol. 2016, 11, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Gay, D.C.; Gay, G.; Axelrod, A.J.; Jenner, M.; Kohlhaas, C.; Kampa, A.; Oldham, N.J.; Piel, J.; Keatinge-Clay, A.T. A close look at a ketosynthase from a trans-acyltransferase modular polyketide synthase. Structure 2014, 22, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.L.; Cheng, Y.Q.; Shen, B. Leinamycin biosynthesis revealing unprecedented architectural complexity for a hybrid polyketide synthase and nonribosomal peptide synthetase. Chem. Biol. 2004, 11, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Lohman, J.R.; Ma, M.; Osipiuk, J.; Nocek, B.; Kim, Y.; Chang, C.; Cuff, M.; Mack, J.; Bigelow, L.; Li, H.; et al. Structural and evolutionary relationships of “AT-less” type I polyketide synthase ketosynthases. Proc. Natl. Acad. Sci. USA 2015, 112, 12693–12698. [Google Scholar] [CrossRef] [PubMed]
- Jenner, M.; Kosol, S.; Griffiths, D.; Prasongpholchai, P.; Manzi, L.; Barrow, A.S.; Moses, J.E.; Oldham, N.J.; Lewandowski, J.R.; Challis, G.L. Mechanism of intersubunit ketosynthase-dehydratase interaction in polyketide synthases. Nat. Chem. Biol. 2018, 14, 270–275. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, J.P. Evidence for a sex factor in Streptomyces erythreus. J. Bacteriol. 1985, 164, 969–971. [Google Scholar] [PubMed]
- Kao, C.M.; Katz, L.; Khosla, C. Engineered biosynthesis of a complete macrolactone in a heterologous host. Science 1994, 265, 509–512. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, R.; Ebert-Khosla, S.; Hopwood, D.A.; Khosla, C. Engineered biosynthesis of novel polyketides. Science 1993, 262, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Ziermann, R.; Betlach, M.C. Recombinant polyketide synthesis in Streptomyces: Engineering of improved host strains. Biotechniques 1999, 26, 106–110. [Google Scholar] [PubMed]
- McDaniel, R.; Thamchaipenet, A.; Gustafsson, C.; Fu, H.; Betlach, M.; Ashley, G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc. Natl. Acad. Sci. USA 1999, 96, 1846–1851. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.M.; Luo, G.; Katz, L.; Cane, D.E.; Khosla, C. Engineered biosynthesis of a triketide lactone from an incomplete modular polyketide synthase. J. Am. Chem. Soc. 1994, 116, 11612–11613. [Google Scholar] [CrossRef]
- Cortes, J.; Wiesmann, K.E.; Roberts, G.A.; Brown, M.J.; Staunton, J.; Leadlay, P.F. Repositioning of a domain in a modular polyketide synthase to promote specific chain cleavage. Science 1995, 268, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. Genetic engineering of modular PKSs: From combinatorial biosynthesis to synthetic biology. Nat. Prod. Rep. 2016, 33, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Pieper, R.; Rosa, A.; Khosla, C.; Cane, D.E. Erythromycin biosynthesis: Exploiting the catalytic versatility of the modular polyketide synthase. Bioorg. Med. Chem. 1996, 4, 995–999. [Google Scholar] [CrossRef]
- Rowe, C.J.; Cortes, J.; Gaisser, S.; Staunton, J.; Leadlay, P.F. Construction of new vectors for high-level expression in actinomycetes. Gene 1998, 216, 215–223. [Google Scholar] [CrossRef]
- Robertsen, H.L.; Musiol-Kroll, E.M.; Ding, L.; Laiple, K.J.; Hofeditz, T.; Wohlleben, W.; Lee, S.Y.; Grond, S.; Weber, T. Filling the gaps in the kirromycin biosynthesis: Deciphering the role of genes involved in ethylmalonyl-CoA supply and tailoring reactions. Sci. Rep. 2018, 8, 3230. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J.; Leadlay, P.F. Combinatorial biosynthesis of reduced polyketides. Nat. Rev. Microbiol. 2005, 3, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Maervoet, V.E.T.; Briers, Y. Synthetic biology of modular proteins. Bioengineered 2017, 8, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Backman, T.W.H.; Keasling, J.D.; Katz, L. Synthetic biology of polyketide synthases. J. Ind. Microbiol. Biotechnol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Donadio, S.; Sosio, M. Strategies for combinatorial biosynthesis with modular polyketide synthases. Comb. Chem. High Throughput Screen 2003, 6, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, Z.; Zhao, H.; Ang, E.L. Recent advances in combinatorial biosynthesis for drug discovery. Drug Des. Devel. Ther. 2015, 9, 823–833. [Google Scholar] [PubMed]
- Smanski, M.J.; Zhou, H.; Claesen, J.; Shen, B.; Fischbach, M.A.; Voigt, C.A. Synthetic biology to access and expand nature's chemical diversity. Nat. Rev. Microbiol. 2016, 14, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Deng, K.; Wang, G.; Baidoo, E.E.; Northen, T.R.; Adams, P.D.; Katz, L.; Keasling, J.D. Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth. Biol. 2017, 6, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Marsden, A.F.; Wilkinson, B.; Cortes, J.; Dunster, N.J.; Staunton, J.; Leadlay, P.F. Engineering broader specificity into an antibiotic-producing polyketide synthase. Science 1998, 279, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, R.S.; Tsuji, S.Y.; Cane, D.E.; Khosla, C. Dissecting and exploiting intermodular communication in polyketide synthases. Science 1999, 284, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, L.S.; Lill, R.E.; Wilkinson, B.; Sheridan, R.M.; Vousden, W.A.; Kaja, A.L.; Crouse, G.D.; Gifford, J.; Graupner, P.R.; Karr, L.; et al. Engineering of the spinosyn PKS: Directing starter unit incorporation. J. Nat. Prod. 2006, 69, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Dutton, C.J.; Gibson, S.P.; Goudie, A.C.; Holdom, K.S.; Pacey, M.S.; Ruddock, J.C.; Bu'Lock, J.D.; Richards, M.K. Novel avermectins produced by mutational biosynthesis. J. Antibiot. (Tokyo) 1991, 44, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Goudie, A.C.; Evans, N.A.; Gration, K.A.; Bishop, B.F.; Gibson, S.P.; Holdom, K.S.; Kaye, B.; Wicks, S.R.; Lewis, D.; Weatherley, A.J.; et al. Doramectin—A potent novel endectocide. Vet. Parasitol. 1993, 49, 5–15. [Google Scholar] [CrossRef]
- Palaniappan, N.; Kim, B.S.; Sekiyama, Y.; Osada, H.; Reynolds, K.A. Enhancement and selective production of phoslactomycin B, a protein phosphatase IIa inhibitor, through identification and engineering of the corresponding biosynthetic gene cluster. J. Biol. Chem. 2003, 278, 35552–35557. [Google Scholar] [CrossRef] [PubMed]
- Ghatge, M.; Palaniappan, N.; Das Choudhuri, S.; Reynolds, K. Genetic manipulation of the biosynthetic process leading to phoslactomycins, potent protein phosphatase 2A inhibitors. J. Ind. Microbiol. Biotechnol. 2006, 33, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escribano, J.P.; Alt, S.; Bibb, M.J. Next generation sequencing of actinobacteria for the discovery of novel natural products. Mar. Drugs 2016, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; de Los Santos, E.L.C.; Kim, H.U.; Nave, M.; et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic. Acids. Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.; Gokhale, R.S.; Mohanty, D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J. Mol. Biol. 2003, 328, 335–363. [Google Scholar] [CrossRef]
- Khayatt, B.I.; Overmars, L.; Siezen, R.J.; Francke, C. Classification of the adenylation and acyl-transferase activity of NRPS and PKS systems using ensembles of substrate specific hidden Markov models. PLoS ONE 2013, 8, e62136. [Google Scholar] [CrossRef] [PubMed]
- Haydock, S.F.; Aparicio, J.F.; Molnar, I.; Schwecke, T.; Khaw, L.E.; Konig, A.; Marsden, A.F.; Galloway, I.S.; Staunton, J.; Leadlay, P.F. Divergent sequence motifs correlated with the substrate specificity of (methyl)malonyl-CoA:acyl carrier protein transacylase domains in modular polyketide synthases. FEBS Lett. 1995, 374, 246–248. [Google Scholar] [CrossRef]
- Mathews, I.I.; Allison, K.; Robbins, T.; Lyubimov, A.Y.; Uervirojnangkoorn, M.; Brunger, A.T.; Khosla, C.; DeMirci, H.; McPhillips, S.E.; Hollenbeck, M.; et al. The conformational flexibility of the acyltransferase from the disorazole polyketide synthase is revealed by an X-ray free-electron laser using a room-temperature sample delivery method for serial crystallography. Biochemistry 2017, 56, 4751–4756. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.T.; Zhang, Z.; Meoded, R.A.; Cepeda, A.J.; Piel, J.; Keatinge-Clay, A.T. Structural and functional studies of a pyran synthase domain from a trans-acyltransferase assembly line. ACS Chem. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Skiba, M.A.; Maloney, F.P.; Dan, Q.; Fraley, A.E.; Aldrich, C.C.; Smith, J.L.; Brown, W.C. PKS-NRPS enzymology and structural biology: Considerations in protein production. Methods Enzymol. 2018, 604, 45–88. [Google Scholar] [PubMed]
- Gay, D.C.; Wagner, D.T.; Meinke, J.L.; Zogzas, C.E.; Gay, G.R.; Keatinge-Clay, A.T. The LINKS motif zippers trans-acyltransferase polyketide synthase assembly lines into a biosynthetic megacomplex. J. Struct. Biol. 2016, 193, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Ji, J.; Zhou, Z.; Yu, J.; Zhu, H.; Su, Z.; Zhang, L.; Zheng, J. Structural and functional analysis of the loading acyltransferase from avermectin modular polyketide synthase. ACS Chem. Biol. 2015, 10, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Bai, L.F.; Ran, X.X.; Jiang, X.H.; Wu, H.; Zhang, W.; Jin, M.Y.; Li, Y.Q.; Jiang, H. Biochemical characterization of a malonyl-specific acyltransferase domain of FK506 biosynthetic polyketide synthase. Protein Pept. Lett. 2015, 22, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, J. Recent advances in understanding and engineering polyketide synthesis. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zhang, W. Engineering modular polyketide synthases for production of biofuels and industrial chemicals. Curr. Opin. Biotechnol. 2018, 50, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Herbst, D.A.; Huitt-Roehl, C.R.; Jakob, R.P.; Kravetz, J.M.; Storm, P.A.; Alley, J.R.; Townsend, C.A.; Maier, T. The structural organization of substrate loading in iterative polyketide synthases. Nat. Chem. Biol. 2018, 14, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Puglisi, J.D.; Pande, V.S.; Cane, D.E.; Khosla, C. Precursor directed biosynthesis of an orthogonally functional erythromycin analogue: Selectivity in the ribosome macrolide binding pocket. J. Am. Chem. Soc. 2012, 134, 12259–12265. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Zhang, H.; Tian, W.; Wu, L.; Wang, S.; Zheng, M.; Zhang, J.; Sun, C.; Deng, Z.; et al. Structural basis of a broadly selective acyltransferase from the polyketide synthase of splenocin. Angew Chem. Int. Ed. Engl. 2018, 57, 5823–5827. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.J.; Watts, K.R.; Robbins, T.; Cane, D.E.; Khosla, C. Comparative analysis of the substrate specificity of trans- versus cis-acyltransferases of assembly line polyketide synthases. Biochemistry 2014, 53, 3796–3806. [Google Scholar] [CrossRef] [PubMed]
- Musiol, E.M.; Hartner, T.; Kulik, A.; Moldenhauer, J.; Piel, J.; Wohlleben, W.; Weber, T. Supramolecular templating in kirromycin biosynthesis: The acyltransferase KirCII loads ethylmalonyl-CoA extender onto a specific ACP of the trans-AT PKS. Chem. Biol. 2011, 18, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Laiple, K.J.; Pross, E.K.; Textor, A.; Grond, S.; Welzel, K.; Pelzer, S.; Vente, A.; Wohlleben, W. Molecular analysis of the kirromycin biosynthetic gene cluster revealed beta-alanine as precursor of the pyridone moiety. Chem. Biol. 2008, 15, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Musiol, E.M.; Weber, T.; Williams, G.J. Reprogramming acyl carrier protein interactions of an Acyl-CoA promiscuous trans-acyltransferase. Chem. Biol. 2014, 21, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, I.; McArthur, J.; Randall, S.; Draelos, M.M.; Musiol, E.M.; Muddiman, D.C.; Weber, T.; Williams, G.J. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem. Biol. 2013, 8, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Hans, M.; Hornung, A.; Dziarnowski, A.; Cane, D.E.; Khosla, C. Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J. Am. Chem. Soc. 2003, 125, 5366–5374. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, D.; Hong, S.S.; Na, Z.; Shin, J.C.; Roh, S.H.; Wu, C.Z.; Choi, O.; Lee, K.; Shen, Y.M.; et al. Rational biosynthetic engineering for optimization of geldanamycin analogues. ChemBioChem 2009, 10, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Katz, L. The DEBS paradigm for type I modular polyketide synthases and beyond. Methods Enzymol. 2009, 459, 113–142. [Google Scholar] [PubMed]
- Weber, T.; Charusanti, P.; Musiol-Kroll, E.M.; Jiang, X.; Tong, Y.; Kim, H.U.; Lee, S.Y. Metabolic engineering of antibiotic factories: New tools for antibiotic production in actinomycetes. Trends Biotechnol. 2015, 33, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.; Liu, Y.C.; Cane, D.E.; Khosla, C. Structure and mechanism of assembly line polyketide synthases. Curr. Opin. Struct. Biol. 2016, 41, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.F.; Blake-Hedges, J.M.; Bailey, C.B.; Curran, S.; Keasling, J.D. Engineered polyketides: Synergy between protein and host level engineering. Synth. Syst. Biotechnol. 2017, 2, 147–166. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Jenner, M.; Masschelein, J.; Jones, C.; Bull, M.J.; Harris, S.R.; Hartkoorn, R.C.; Vocat, A.; Romero-Canelon, I.; Coupland, P.; et al. Discovery and biosynthesis of gladiolin: A Burkholderia gladioli antibiotic with promising activity against Mycobacterium tuberculosis. J. Am. Chem. Soc. 2017, 139, 7974–7981. [Google Scholar] [CrossRef] [PubMed]
- Cuskin, F.; Solovyova, A.S.; Lewis, R.J.; Race, P.R. Crystallization and preliminary X-ray analysis of the bacillaene synthase trans-acting acyltransferase PksC. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Pistorius, D.; Muller, R. Discovery of the rhizopodin biosynthetic gene cluster in Stigmatella aurantiaca Sg a15 by genome mining. ChemBioChem 2012, 13, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Musiol, E.M.; Greule, A.; Hartner, T.; Kulik, A.; Wohlleben, W.; Weber, T. The AT(2) domain of KirCI loads malonyl extender units to the ACPs of the kirromycin PKS. ChemBioChem 2013, 14, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Coughlin, J.M.; Ju, J.; Zhu, D.; Wendt-Pienkowski, E.; Zhou, X.; Wang, Z.; Shen, B.; Deng, Z. Oxazolomycin biosynthesis in Streptomyces albus JA3453 featuring an “acyltransferase-less” type I polyketide synthase that incorporates two distinct extender units. J. Biol. Chem. 2010, 285, 20097–20108. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.K.; Hothersall, J.; Cooper, S.M.; Stephens, E.; Simpson, T.J.; Thomas, C.M. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem. Biol. 2003, 10, 419–430. [Google Scholar] [CrossRef]
- El-Sayed, A.K.; Hothersall, J.; Thomas, C.M. Quorum-sensing-dependent regulation of biosynthesis of the polyketide antibiotic mupirocin in Pseudomonas fluorescens NCIMB 10586. Microbiology 2001, 147, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.; Song, K.; Wen, J. Metabolic network model guided engineering ethylmalonyl-CoA pathway to improve ascomycin production in Streptomyces hygroscopicus var. ascomyceticus. Microb. Cell Fact. 2017, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic. Acids. Res. 2012, 40, D13–D25. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic. Acids. Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed]
- Boddy, C.N. Bioinformatics tools for genome mining of polyketide and non-ribosomal peptides. J. Ind. Microbiol. Biotechnol. 2014, 41, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Jenke-Kodama, H.; Dittmann, E. Bioinformatic perspectives on NRPS/PKS megasynthases: Advances and challenges. Nat. Prod. Rep. 2009, 26, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef] [PubMed]
- Sandler, I.; Abu-Qarn, M.; Aharoni, A. Protein co-evolution: How do we combine bioinformatics and experimental approaches? Mol. Biosyst. 2013, 9, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Suplatov, D.; Sharapova, Y.; Timonina, D.; Kopylov, K.; Svedas, V. The visualCMAT: A web-server to select and interpret correlated mutations/co-evolving residues in protein families. J. Bioinform. Comput. Biol. 2017, 16, 1840005. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.H.; Shi, J.; Deane, C.M. Comparing co-evolution methods and their application to template-free protein structure prediction. Bioinformatics 2017, 33, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Milano, T.; Paiardini, A.; Grgurina, I.; Pascarella, S. Type I pyridoxal 5′-phosphate dependent enzymatic domains embedded within multimodular nonribosomal peptide synthetase and polyketide synthase assembly lines. BMC Struct. Biol. 2013, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Sagar, M.; Pandey, N.; Qamar, N.; Singh, B.; Shukla, A. Domain analysis of 3 Keto Acyl-CoA synthase for structural variations in Vitis vinifera and Oryza brachyantha using comparative modelling. Interdiscip. Sci. 2015, 7, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Backman, T.W.H.; Bailey, C.B.; Magnan, C.; Garcia Martin, H.; Katz, L.; Baldi, P.; Keasling, J.D. ClusterCAD: A computational platform for type I modular polyketide synthase design. Nucleic Acids Res. 2018, 46, D509–D515. [Google Scholar] [CrossRef] [PubMed]
- Annaval, T.; Rudolf, J.D.; Chang, C.Y.; Lohman, J.R.; Kim, Y.; Bigelow, L.; Jedrzejczak, R.; Babnigg, G.; Joachimiak, A.; Phillips, G.N., Jr.; et al. Crystal structure of thioesterase SgcE10 supporting common polyene intermediates in 9- and 10-membered enediyne core biosynthesis. ACS Omega 2017, 2, 5159–5169. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. Uncovering the structures of modular polyketide synthases. Nat. Prod. Rep. 2015, 32, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. Correction: Uncovering the structures of modular polyketide synthases. Nat. Prod. Rep. 2017, 34, 1035. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.L.; Matsui, T.; Weiss, T.M.; Khosla, C. Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide B synthase. J. Mol. Biol. 2014, 426, 2229–2245. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Skiniotis, G.; Sherman, D.H. Architecture of the polyketide synthase module: Surprises from electron cryo-microscopy. Curr. Opin. Struct. Biol. 2015, 31, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.N.; Bergendahl, L.T.; Marsh, J.A. Computational modelling of protein complex structure and assembly. Methods Mol. Biol. 2018, 1764, 347–356. [Google Scholar] [PubMed]
- Mertens, H.D.; Svergun, D.I. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 2010, 172, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Khater, S.; Gupta, M.; Agrawal, P.; Sain, N.; Prava, J.; Gupta, P.; Grover, M.; Kumar, N.; Mohanty, D. SBSPKSv2: Structure-based sequence analysis of polyketide synthases and non-ribosomal peptide synthetases. Nucleic Acids Res. 2017, 45, W72–W79. [Google Scholar] [CrossRef] [PubMed]
- Diminic, J.; Zucko, J.; Ruzic, I.T.; Gacesa, R.; Hranueli, D.; Long, P.F.; Cullum, J.; Starcevic, A. Databases of the thiotemplate modular systems (CSDB) and their in silico recombinants (r-CSDB). J. Ind. Microbiol. Biotechnol. 2013, 40, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.; Dorival, J.; Rabeharindranto, H.; Mazon, H.; Chagot, B.; Gruez, A.; Weissman, K.J. Insights into the function of trans-acyl transferase polyketide synthases from the SAXS structure of a complete module. Chem. Sci. 2014, 5, 3081–3095. [Google Scholar]
- Porter, J.L.; Rusli, R.A.; Ollis, D.L. Directed evolution of enzymes for industrial biocatalysis. ChemBioChem 2016, 17, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, J.B.; Reetz, M.T. Biocatalysts for the pharmaceutical industry created by structure-guided directed evolution of stereoselective enzymes. Bioorg Med. Chem. 2017, 26, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Barley, M.H.; Turner, N.J.; Goodacre, R. Recommendations on the implementation of genetic algorithms for the directed evolution of enzymes for industrial purposes. ChemBioChem 2017, 18, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wikmark, Y.; Backvall, J.E.; Reetz, M.T. New concepts for increasing the efficiency in directed evolution of stereoselective enzymes. Chemistry 2016, 22, 5046–5054. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, M.; Miyakawa, T.; Shimizu, S.; Tanokura, M. Enzymes useful for chiral compound synthesis: Structural biology, directed evolution, and protein engineering for industrial use. Appl. Microbiol. Biotechnol. 2016, 100, 5747–5757. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Tibby, M.R.; Wagner, D.T.; Brantley, J.N.; Keatinge-Clay, A.T. Investigating the reactivities of a polyketide synthase module through fluorescent click chemistry. Chem. Commun. (Camb) 2014, 50, 5276–5278. [Google Scholar] [CrossRef] [PubMed]
- Dorrestein, P.C.; Bumpus, S.B.; Calderone, C.T.; Garneau-Tsodikova, S.; Aron, Z.D.; Straight, P.D.; Kolter, R.; Walsh, C.T.; Kelleher, N.L. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry 2006, 45, 12756–12766. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xie, X.; Pashkov, I.; Sawaya, M.R.; Laidman, J.; Zhang, W.; Cacho, R.; Yeates, T.O.; Tang, Y. Directed evolution and structural characterization of a simvastatin synthase. Chem. Biol. 2009, 16, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Rui, Z.; Zhang, W. Engineering biosynthesis of non-ribosomal peptides and polyketides by directed evolution. Curr. Top. Med. Chem. 2016, 16, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhao, X.Q.; Wang, H.N.; Qiu, R.G.; Tang, L. Seamless stitching of biosynthetic gene cluster containing type I polyketide synthases using Red/ET mediated recombination for construction of stably co-existing plasmids. Gene 2015, 554, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Shen, Q.; Bian, X.; Chen, H.; Fu, J.; Wang, H.; Lei, P.; Guo, Z.; Chen, W.; Li, D.; et al. Simple and rapid direct cloning and heterologous expression of natural product biosynthetic gene cluster in Bacillus subtilis via Red/ET recombineering. Sci. Rep. 2016, 6, 34623. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Huang, F.; Stewart, F.A.; Xia, L.; Zhang, Y.; Muller, R. Direct cloning, genetic engineering, and heterologous expression of the syringolin biosynthetic gene cluster in E. coli through Red/ET recombineering. ChemBioChem 2012, 13, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Huang, H.; Min, T.; Hu, H. TAR cloning and integrated overexpression of 6-demethylchlortetracycline biosynthetic gene cluster in Streptomyces aureofaciens. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Larionov, V. Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 2016, 125, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.G.; Salomonsen, B.; Nielsen, M.T.; Nielsen, J.B.; Hansen, N.B.; Nielsen, K.F.; Regueira, T.B.; Nielsen, J.; Patil, K.R.; Mortensen, U.H. Versatile enzyme expression and characterization system for Aspergillus nidulans, with the Penicillium brevicompactum polyketide synthase gene from the mycophenolic acid gene cluster as a test case. Appl. Environ. Microbiol. 2011, 77, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Geu-Flores, F.; Nour-Eldin, H.H.; Nielsen, M.T.; Halkier, B.A. USER fusion: A rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res. 2007, 35, e55. [Google Scholar] [CrossRef] [PubMed]
- Hansen, N.B.; Lubeck, M.; Lubeck, P.S. Advancing USER cloning into simpleUSER and nicking cloning. J. Microbiol. Methods 2014, 96, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Larionov, V.; Kouprina, N. Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res. 2015, 43, e55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Wong, F.T.; Wang, Y.; Luo, S.; Lim, Y.H.; Heng, E.; Yeo, W.L.; Cobb, R.E.; Enghiad, B.; Ang, E.L.; et al. CRISPR-Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat. Chem. Biol. 2017, 13, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zheng, G.; Jiang, W.; Hu, H.; Lu, Y. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wen, S.; Xu, W.; He, Z.; Zhai, G.; Liu, Y.; Deng, Z.; Sun, Y. Highly efficient editing of the actinorhodin polyketide chain length factor gene in Streptomyces coelicolor M145 using CRISPR/Cas9-CodA(sm) combined system. Appl. Microbiol. Biotechnol. 2015, 99, 10575–10585. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Robertsen, H.L.; Blin, K.; Weber, T.; Lee, S.Y. CRISPR-Cas9 toolkit for actinomycete genome editing. Methods Mol. Biol. 2018, 1671, 163–184. [Google Scholar] [PubMed]
- Siegl, T.; Tokovenko, B.; Myronovskyi, M.; Luzhetskyy, A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab. Eng. 2013, 19, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, L.; Barton, K.W.; Zhao, H. Systematic identification of a panel of strong constitutive promoters from Streptomyces albus. ACS Synth. Biol. 2015, 4, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Phelan, R.M.; Sachs, D.; Petkiewicz, S.J.; Barajas, J.F.; Blake-Hedges, J.M.; Thompson, M.G.; Reider Apel, A.; Rasor, B.J.; Katz, L.; Keasling, J.D. Development of next generation synthetic biology tools for use in Streptomyces venezuelae. ACS Synth. Biol. 2017, 6, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Guell, M.; Church, G.M.; Pfeifer, B.A. Heterologous erythromycin production across strain and plasmid construction. Biotechnol. Prog. 2018, 34, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, Z.; Li, M.H.; Wang, J.D.; Bai, H.; Zhou, J.; Zheng, Y.G. High level of spinosad production in the heterologous host Saccharopolyspora erythraea. Appl. Environ. Microbiol. 2016, 82, 5603–5611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, C.; Bai, L. Conversion of the high-yield salinomycin producer Streptomyces albus BK3-25 into a surrogate host for polyketide production. Sci. China Life Sci. 2017, 60, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, J.; Lu, C.; Shen, Y. Heterologous expression of galbonolide biosynthetic genes in Streptomyces coelicolor. Antonie Van Leeuwenhoek 2015, 107, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H. Streptomyces and Saccharopolyspora hosts for heterologous expression of secondary metabolite gene clusters. J. Ind. Microbiol. Biotechnol. 2010, 37, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.S.; Kim, E.; Yoo, Y.J.; Ban, Y.H.; Kim, E.J.; Yoon, Y.J. Characterization and engineering of the ethylmalonyl-CoA pathway towards the improved heterologous production of polyketides in Streptomyces venezuelae. Appl. Microbiol. Biotechnol. 2014, 98, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, A.R.; Dhakal, D.; Jha, A.K.; Sohng, J.K. Herboxidiene biosynthesis, production, and structural modifications: Prospect for hybrids with related polyketide. Appl. Microbiol. Biotechnol. 2015, 99, 8351–8362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, L.; Osburne, M.S.; Pfeifer, B.A. The continuing development of E. coli as a heterologous host for complex natural product biosynthesis. Methods Mol. Biol. 2016, 1401, 121–134. [Google Scholar] [PubMed]
- Sun, L.; Zeng, J.; Zhang, S.; Gladwin, T.; Zhan, J. Effects of exogenous nutrients on polyketide biosynthesis in Escherichia coli. Appl. Microbiol. Biotechnol. 2015, 99, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Kim, W.; Katz, L.; Keasling, J.D. Heterologous production of polyketides by modular type I polyketide synthases in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Kumpfmuller, J.; Methling, K.; Fang, L.; Pfeifer, B.A.; Lalk, M.; Schweder, T. Production of the polyketide 6-deoxyerythronolide B in the heterologous host Bacillus subtilis. Appl. Microbiol. Biotechnol. 2016, 100, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.; Tang, Y.; Li, L. Saccharomyces cerevisiae as a tool for mining, studying and engineering fungal polyketide synthases. Fungal. Genet. Biol. 2016, 89, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Vickery, C.R.; Cardenas, J.; Bowman, M.E.; Burkart, M.D.; Da Silva, N.A.; Noel, J.P. A coupled in vitro/in vivo approach for engineering a heterologous type III PKS to enhance polyketide biosynthesis in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2018, 15, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Li, W.; Zhang, P.; Lyu, H.; Hu, Y.; Yin, W.B. Rational design for heterologous production of aurovertin-type compounds in Aspergillus nidulans. Appl. Microbiol. Biotechnol. 2018, 102, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Gressler, M.; Hortschansky, P.; Geib, E.; Brock, M. A new high-performance heterologous fungal expression system based on regulatory elements from the Aspergillus terreus terrein gene cluster. Front. Microbiol. 2015, 6, 184. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Engineering Strategy | Examples | |||
|---|---|---|---|---|
| Acceptor PKS (Target Assembly Line for the Engineering) | Tool (e.g., Donor AT or Motif) and Origin (Pathway) | Substrate Specificities Native AT (Acceptor PKS)/Used Tool (AT or Motif) | References | |
| AT-swapping | AT from module 1, and 2/erythromycin | “Hyg” AT2 (module 2)/“Hyg” PKS gene, AT (module 14)/rapamycin, Ven AT/unknown PKS-like gene cluster | MM-CoA/M-CoA | [50] |
| AT from module 1, and 2/erythromycin | AT (module 2)/rapamycin | MM-CoA/M-CoA | [51,52] | |
| AT from module 1/erythromycin | AT (module 3)/erythromycin | MM-CoA/MM-CoA | [53] | |
| AT from module 6/erythromycin | AT (module 6)/rapamycin | MM-CoA/M-CoA | [54] | |
| AT from module 1/erythromycin | AT (module 2)/epothilone | MM-CoA/M-CoA | [55] | |
| AT from module 1/erythromycin | AT (module 3)/epothilone | MM-CoA/promiscuous MM, M-CoA | [55] | |
| AT from module 4/erythromycin | AT (module 5)/niddamycin | MM-CoA/EM-CoA | [56] | |
| AT from module 6/erythromycin | hydroxymalonate-specifying fkbA-AT8/ascomycin (FK520) | MM-CoA/Methoxymalonyl-ACP | [57,58] | |
| AT from module 1-5 and 7/geldanamycin | AT (module 2 and 14)/rapamycin | MM-CoA, MeO-ACP/M-CoA | [59] | |
| AT-site directed mutagenesis | AT from module 1 and 4/erythromycin | consensus motifs (YASH/HAFH motif) | MM-CoA/M-CoA | [60,61,62] |
| AT from module 6 of/erythromycin | structurally identified relevant residue | MM-CoA/SNAC-derivatives | [63,64] | |
| AT from module 6/erythromycin | saturation mutagenesis of relevant motifs | MM-CoA/non-natural alkynyl-modified unit | [65] | |
| AT-cross complementation | AT6null (inactivated AT in module 6)/erythromycin | S. coelicolor malonyl-CoA: ACP transacylase | MM-CoA/M-CoA | [66] |
| AT6null (inactivated AT in module 6)/erythromycin | AT (BryP-AT1)/bryostatin | MM-CoA/M-CoA | [67] | |
| AT6null (inactivated AT in module 6)/erythromycin | AT (BryP-AT2)/bryostatin | MM-CoA/MM-CoA | [67] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musiol-Kroll, E.M.; Wohlleben, W. Acyltransferases as Tools for Polyketide Synthase Engineering. Antibiotics 2018, 7, 62. https://doi.org/10.3390/antibiotics7030062
Musiol-Kroll EM, Wohlleben W. Acyltransferases as Tools for Polyketide Synthase Engineering. Antibiotics. 2018; 7(3):62. https://doi.org/10.3390/antibiotics7030062
Chicago/Turabian StyleMusiol-Kroll, Ewa Maria, and Wolfgang Wohlleben. 2018. "Acyltransferases as Tools for Polyketide Synthase Engineering" Antibiotics 7, no. 3: 62. https://doi.org/10.3390/antibiotics7030062
APA StyleMusiol-Kroll, E. M., & Wohlleben, W. (2018). Acyltransferases as Tools for Polyketide Synthase Engineering. Antibiotics, 7(3), 62. https://doi.org/10.3390/antibiotics7030062