Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials


Abstract
1. Introduction
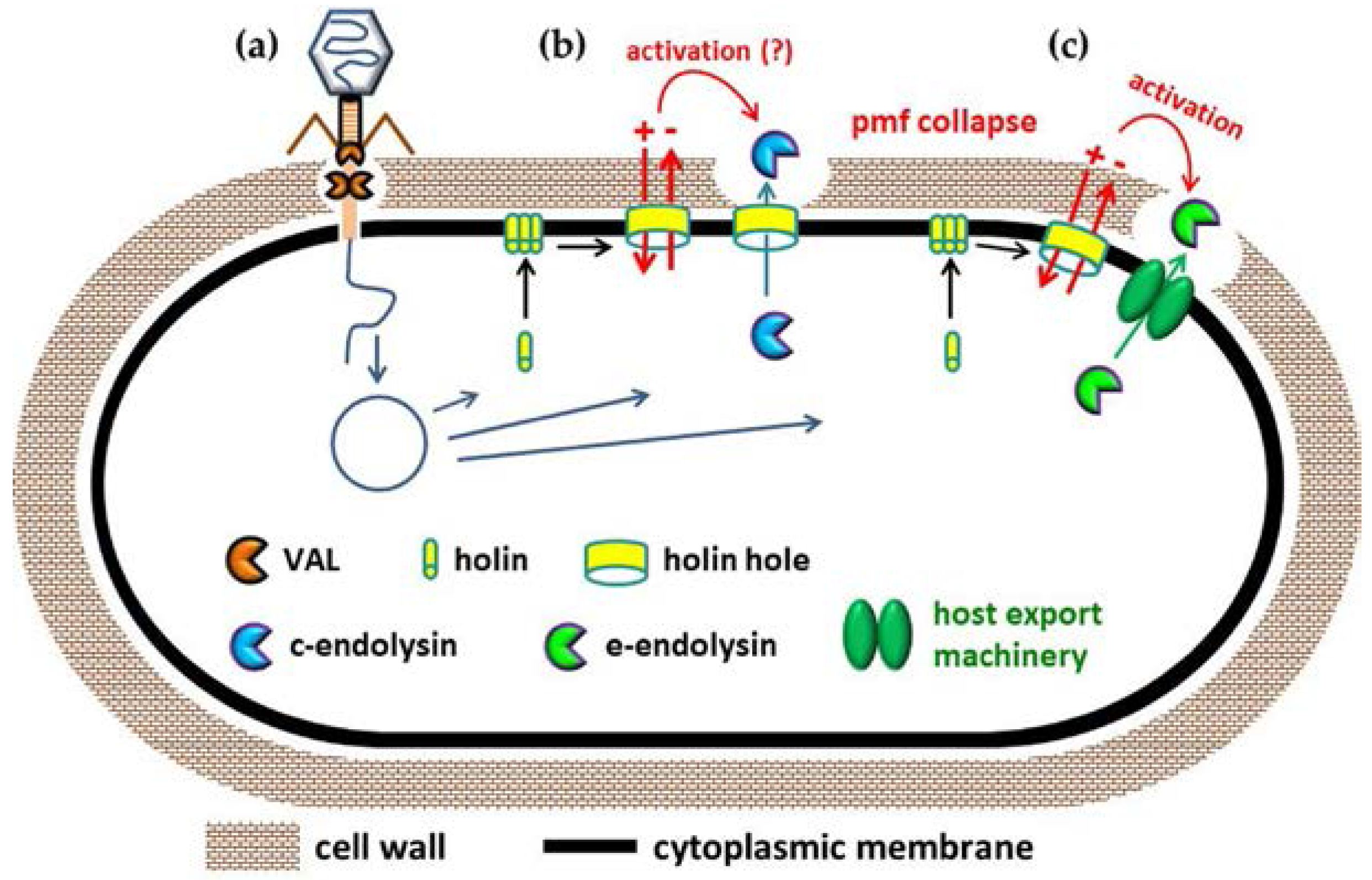
2. Mode of Action of VALs and Endolysins during Phage Infection
3. Enzymatic Activity of PLEs
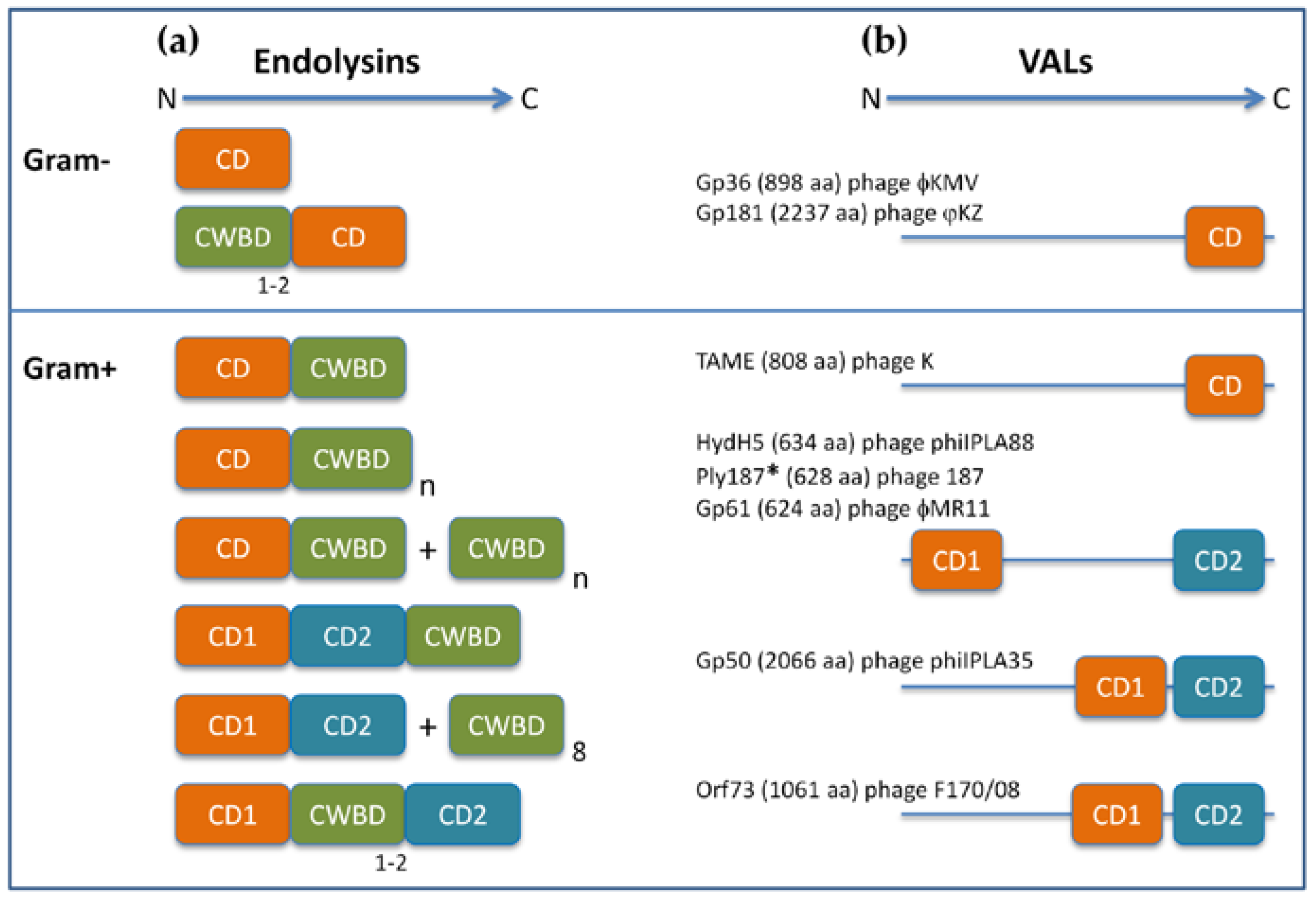
4. Domain Architecture of PLEs
5. Improving the Potential of PLEs as Antibacterials through Protein Engineering
5.1. Generation of Chimeolysins with Increased Lytic Spectrum and Activity
5.2. Other Enginnering Approaches to Expand the Lytic Spectrum and Activity of PLEs
5.3. Improving the Production, Solubility, and Stability of PLEs
5.4. Minimizing Development of Resistance to PLEs
5.5. Enhancing PLEs as Antibacterials towards Gram-Negative Bacteria
5.6. Targeting Intracellular Bacteria with PLEs
6. Conclusions
Conflicts of Interest
References
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- WHO. The World Is Running Out of Antibiotics, WHO Report Confirms. 2017. Available online: http://www.who.int/mediacentre/news/releases/2017/running-out-antibiotics/en/ (accessed on 3 January 2018).
- O’Neill, J. Tackling Drug-Restistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf (accessed on 1 March 2018).
- Adeyi, O.O.; Baris, E.; Jonas, O.B.; Irwin, A.; Berthe, F.C.J.; Le Gall, F.G.; Marquez, P.V.; Nikolic, I.A.; Plante, C.A.; Schneidman, M.; et al. Drug-Resistant Infections: A Threat to Our Economic Future; Final Report; World Bank Group: Washington, DC, USA, 2017. [Google Scholar]
- Kmietowicz, Z. Few novel antibiotics in the pipeline, WHO warns. BMJ 2017, 358, j4339. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Yan, J.; Mao, J.; Xie, J. Bacteriophage polysaccharide depolymerases and biomedical applications. BioDrugs 2014, 28, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and phage-derived proteins—Application approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Oliveira, H.; Melo, L.D.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Moak, M.; Molineux, I.J. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol. Microbiol. 2004, 51, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Catalão, M.J.; Gil, F.; Moniz-Pereira, J.; São-José, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. FEMS Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group a streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.; Donovan, D.M. Endolysins as antimicrobials. Adv. Virus Res. 2012, 83, 299–365. [Google Scholar] [CrossRef] [PubMed]
- Pastagia, M.; Schuch, R.; Fischetti, V.A.; Huang, D.B. Lysins: The arrival of pathogen-directed anti-infectives. J. Med. Microbiol. 2013, 62, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef] [PubMed]
- Haddad Kashani, H.; Schmelcher, M.; Sabzalipoor, H.; Seyed Hosseini, E.; Moniri, R. Recombinant endolysins as potential therapeutics against antibiotic-resistant Staphylococcus aureus: Current status of research and novel delivery strategies. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Martínez, B.; Donovan, D.M.; Rodríguez, A.; García, P. Bacteriophage virion-associated peptidoglycan hydrolases: Potential new enzybiotics. Crit. Rev. Microbiol. 2013, 39, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Azeredo, J.; Lavigne, R.; Kluskens, L.D. Bacteriophage endolysins as a response to emerging foodborne pathogens. Trends Food Sci. Technol. 2012, 28, 103–115. [Google Scholar] [CrossRef]
- Schmelcher, M.; Loessner, M.J. Bacteriophage endolysins: Applications for food safety. Curr. Opin. Biotechnol. 2016, 37, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Gutiérrez, D.; Donovan, D.M.; Martínez, B.; Rodríguez, A.; García, P. Phage lytic proteins: Biotechnological applications beyond clinical antimicrobials. Crit. Rev. Biotechnol. 2016, 36, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnol. Adv. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage t4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Shneider, M.M. Contractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.I.; van Raaij, M.J.; Leiman, P.G. Contractile injection systems of bacteriophages and related systems. Mol. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.R.; Cardarelli, L.; Pell, L.G.; Radford, D.R.; Maxwell, K.L. Long noncontractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 115–142. [Google Scholar] [CrossRef] [PubMed]
- Cumby, N.; Reimer, K.; Mengin-Lecreulx, D.; Davidson, A.R.; Maxwell, K.L. The phage tail tape measure protein, an inner membrane protein and a periplasmic chaperone play connected roles in the genome injection process of E. coli phage HK97. Mol. Microbiol. 2015, 96, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. The bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science 2013, 339, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Morais, M.C.; Cohen, D.N.; Bowman, V.D.; Anderson, D.L.; Rossmann, M.G. Crystal and cryoem structural studies of a cell wall degrading enzyme in the bacteriophage phi29 tail. Proc. Natl. Acad. Sci. USA 2008, 105, 9552–9557. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiang, Y. Membrane penetration by bacterial viruses. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Do we have the hole story yet? Curr. Opin. Microbiol. 2013, 16, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.J.; Melo-Cristino, J.; Ramirez, M. Export of the pneumococcal phage SV1 lysin requires choline-containing teichoic acids and is holin-independent. Mol. Microbiol. 2013, 87, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; São-José, C. More than a hole: The holin lethal function may be required to fully sensitize bacteria to the lytic action of canonical endolysins. Mol. Microbiol. 2016, 102, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Labischinski, H.; Maidhof, H. Bacterial peptidoglycan: Overview and evolving concepts. In Bacterial Cell Wall; Ghuysen, J.M., Hakenbeck, R., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 23–38. [Google Scholar] [CrossRef]
- Oliveira, H.; Melo, L.D.; Santos, S.B.; Nóbrega, F.L.; Ferreira, E.C.; Cerca, N.; Azeredo, J.; Kluskens, L.D. Molecular aspects and comparative genomics of bacteriophage endolysins. J. Virol. 2013, 87, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Ton-That, H.; Faull, K.F.; Schneewind, O. Multiple enzymatic activities of the murein hydrolase from staphylococcal phage φ11. Identification of a d-alanyl-glycine endopeptidase activity. J. Biol. Chem. 1999, 274, 15847–15856. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, D.G.; Dong, S.; Baker, J.R.; Engler, J.A. The bifunctional peptidoglycan lysin of Streptococcus agalactiae bacteriophage B30. Microbiology 2004, 150, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Schuch, R.; Chahales, P.; Zhu, S.; Fischetti, V.A. Plyc: A multimeric bacteriophage lysin. Proc. Natl. Acad. Sci. USA 2006, 103, 10765–10770. [Google Scholar] [CrossRef] [PubMed]
- Linden, S.B.; Zhang, H.; Heselpoth, R.D.; Shen, Y.; Schmelcher, M.; Eichenseher, F.; Nelson, D.C. Biochemical and biophysical characterization of PlyGRCS, a bacteriophage endolysin active against methicillin-resistant Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2015, 99, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Mesnage, S.; Dellarole, M.; Baxter, N.J.; Rouget, J.B.; Dimitrov, J.D.; Wang, N.; Fujimoto, Y.; Hounslow, A.M.; Lacroix-Desmazes, S.; Fukase, K.; et al. Molecular basis for bacterial peptidoglycan recognition by LysM domains. Nat. Commun. 2014, 5, 4269. [Google Scholar] [CrossRef] [PubMed]
- Gründling, A.; Schneewind, O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J. Bacteriol. 2006, 188, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Eugster, M.R.; Haug, M.C.; Huwiler, S.G.; Loessner, M.J. The cell wall binding domain of listeria bacteriophage endolysin PlyP35 recognizes terminal GlcNac residues in cell wall teichoic acid. Mol. Microbiol. 2011, 81, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- López, R.; García, E.; García, P.; García, J.L. The pneumococcal cell wall degrading enzymes: A modular design to create new lysins? Microb. Drug Resist. 1997, 3, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.M.; Hatfull, G.F. Mycobacteriophage endolysins: Diverse and modular enzymes with multiple catalytic activities. PLoS ONE 2012, 7, e34052. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; López, R.; García, J.L. Chimeric phage-bacterial enzymes: A clue to the modular evolution of genes. Proc. Natl. Acad. Sci. USA 1990, 87, 8125–8129. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Gerstmans, H.; Thorpe, S.; Mesnage, S.; Lavigne, R.; Briers, Y. DUF3380 domain from a salmonella phage endolysin shows potent N-Acetylmuramidase activity. Appl. Environ. Microbiol. 2016, 82, 4975–4981. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Lavigne, R. Breaking barriers: Expansion of the use of endolysins as novel antibacterials against Gram-negative bacteria. Future Microbiol. 2015, 10, 377–390. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.; Buckle, A.M.; Mitchell, M.S.; Hoopes, J.T.; Gallagher, D.T.; Heselpoth, R.D.; Shen, Y.; Reboul, C.F.; Law, R.H.; Fischetti, V.A.; et al. X-ray crystal structure of the streptococcal specific phage lysin PlyC. Proc. Natl. Acad. Sci. USA 2012, 109, 12752–12757. [Google Scholar] [CrossRef] [PubMed]
- Proença, D.; Velours, C.; Leandro, C.; Garcia, M.; Pimentel, M.; São-José, C. A two-component, multimeric endolysin encoded by a single gene. Mol. Microbiol. 2015, 95, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Leicht, S.; Krichel, B.; Mertens, H.D.; Thompson, A.; Krijgsveld, J.; Svergun, D.I.; Gómez-Torres, N.; Garde, S.; Uetrecht, C.; et al. Crystal structure of the CTP1L endolysin reveals how its activity is regulated by a secondary translation product. J. Biol. Chem. 2016, 291, 4882–4893. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Briers, Y.; Hertveldt, K.; Robben, J.; Volckaert, G. Identification and characterization of a highly thermostable bacteriophage lysozyme. Cell. Mol. Life Sci. 2004, 61, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Miroshnikov, K.; Chertkov, O.; Nekrasov, A.; Mesyanzhinov, V.; Volckaert, G.; Lavigne, R. The structural peptidoglycan hydrolase gp181 of bacteriophage φkz. Biochem. Biophys. Res. Commun. 2008, 374, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.D.; Rajagopalan, S.S.; Sundarrajan, S.; George, S.E.; Asrani, J.Y.; Pillai, R.; Chikkamadaiah, R.; Durgaiah, M.; Sriram, B.; Padmanabhan, S. A novel bacteriophage tail-associated muralytic enzyme (TAME) from phage K and its development into a potent antistaphylococcal protein. BMC Microbiol. 2011, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.; Martínez, B.; Zhou, Y.; Rodríguez, A.; Donovan, D.M.; García, P. Lytic activity of the virion-associated peptidoglycan hydrolase HydH5 of Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88. BMC Microbiol. 2011, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Rashel, M.; Uchiyama, J.; Takemura, I.; Hoshiba, H.; Ujihara, T.; Takatsuji, H.; Honke, K.; Matsuzaki, S. Tail-associated structural protein gp61 of Staphylococcus aureus phage φMR11 has bifunctional lytic activity. FEMS Microbiol. Lett. 2008, 284, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Gutiérrez, D.; Martínez, B.; Rodríguez, A.; Götz, F.; García, P. The tape measure protein of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA35 has an active muramidase domain. Appl. Environ. Microbiol. 2012, 78, 6369–6371. [Google Scholar] [CrossRef] [PubMed]
- Proença, D.; Leandro, C.; Garcia, M.; Pimentel, M.; São-José, C. EC300: A phage-based, bacteriolysin-like protein with enhanced antibacterial activity against Enterococcus faecalis. Appl. Microbiol. Biotechnol. 2015, 99, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yu, J.; Wei, H. Engineered bacteriophage lysins as novel anti-infectives. Front. Microbiol. 2014, 5, 542. [Google Scholar] [CrossRef] [PubMed]
- Croux, C.; Ronda, C.; López, R.; García, J.L. Interchange of functional domains switches enzyme specificity: Construction of a chimeric pneumococcal-clostridial cell wall lytic enzyme. Mol. Microbiol. 1993, 9, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Tchang, V.S.; Loessner, M.J. Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb. Biotechnol. 2011, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, S.; Coffey, A.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. The recombinant phage lysin LysK has a broad spectrum of lytic activity against clinically relevant staphylococci, including methicillin-resistant Staphylococcus aureus. J. Bacteriol. 2005, 187, 7161–7164. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.C.; Foster-Frey, J.; Stodola, A.J.; Anacker, D.; Donovan, D.M. Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene 2009, 443, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Powell, A.M.; Becker, S.C.; Camp, M.J.; Donovan, D.M. Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl. Environ. Microbiol. 2012, 78, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Linden, S.B.; Wang, J.; Yu, J.; Nelson, D.C.; Wei, H. A chimeolysin with extended-spectrum streptococcal host range found by an induced lysis-based rapid screening method. Sci. Rep. 2015, 5, 17257. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bi, Y.; Shang, X.; Wang, M.; Linden, S.B.; Li, Y.; Nelson, D.C.; Wei, H. Antibiofilm activities of a novel chimeolysin against streptococcus mutans under physiological and cariogenic conditions. Antimicrob. Agents Chemother. 2016, 60, 7436–7443. [Google Scholar] [CrossRef] [PubMed]
- Díez-Martínez, R.; De Paz, H.D.; García-Fernández, E.; Bustamante, N.; Euler, C.W.; Fischetti, V.A.; Menendez, M.; García, P. A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. J. Antimicrob. Chemother. 2015, 70, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, R.; Domenech, M.; Iglesias-Bexiga, M.; Menéndez, M.; García, P. Csl2, a novel chimeric bacteriophage lysin to fight infections caused by Streptococcus suis, an emerging zoonotic pathogen. Sci. Rep. 2017, 7, 16506. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, B.; Fresco-Taboada, A.; Iglesias-Bexiga, M.; Menéndez, M.; García, P. PL3 amidase, a tailor-made lysin constructed by domain shuffling with potent killing activity against pneumococci and related species. Front. Microbiol. 2016, 7, 1156. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Martínez, B.; Rodríguez, A.; Donovan, D.M.; García, P. Enhanced staphylolytic activity of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 HydH5 virion-associated peptidoglycan hydrolase: Fusions, deletions, and synergy with LysH5. Appl. Environ. Microbiol. 2012, 78, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Martínez, B.; Donovan, D.M.; García, P.; Rodríguez, A. Potential of the virion-associated peptidoglycan hydrolase HydH5 and its derivative fusion proteins in milk biopreservation. PLoS ONE 2013, 8, e54828. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; González, S.; Campelo, A.B.; Martínez, B.; Rodríguez, A.; García, P. Downregulation of autolysin-encoding genes by phage-derived lytic proteins inhibits biofilm formation in Staphylococcus aureus. Antimicrob. Agents Chemother. 2017, 61, e02724-16. [Google Scholar] [CrossRef] [PubMed]
- Drilling, A.J.; Cooksley, C.; Chan, C.; Wormald, P.J.; Vreugde, S. Fighting sinus-derived Staphylococcus aureus biofilms in vitro with a bacteriophage-derived muralytic enzyme. Int. Forum Allergy Rhinol. 2016, 6, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.R.; Paul, V.D.; George, S.; Sundarrajan, S.; Kumar, N.; Hebbur, M.; Veena, A.; Maheshwari, U.; Appaiah, C.B.; Chidambaran, M.; et al. Properties and mutation studies of a bacteriophage-derived chimeric recombinant staphylolytic protein P128: Comparison to recombinant lysostaphin. Bacteriophage 2013, 3, e26564. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Schmelcher, M.; Harty, W.J.; Foster-Frey, J.; Donovan, D.M. Chimeric Ply187 endolysin kills Staphylococcus aureus more effectively than the parental enzyme. FEMS Microbiol. Lett. 2013, 342, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Donovan, D.M.; Kumar, A. Intravitreal injection of the chimeric phage endolysin Ply187 protects mice from Staphylococcus aureus endophthalmitis. Antimicrob. Agents Chemother. 2014, 58, 4621–4629. [Google Scholar] [CrossRef] [PubMed]
- Daniel, A.; Euler, C.; Collin, M.; Chahales, P.; Gorelick, K.J.; Fischetti, V.A. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Pastagia, M.; Euler, C.; Chahales, P.; Fuentes-Duculan, J.; Krueger, J.G.; Fischetti, V.A. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob. Agents Chemother. 2011, 55, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; Proença, D.; Cantante, C.; Silva, F.A.; Leandro, C.; Lourenço, S.; Milheiriço, C.; de Lencastre, H.; Cavaco-Silva, P.; Pimentel, M.; et al. Novel chimerical endolysins with broad antimicrobial activity against methicillin-resistant Staphylococcus aureus. Microb. Drug Resist. 2012, 18, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.M.; Seal, B.S.; Garrish, J.K.; Oakley, B.B.; Hiett, K.; Yeh, H.Y.; Woolsey, R.; Schegg, K.M.; Line, J.E.; Donovan, D.M. A thermophilic phage endolysin fusion to a Clostridium perfringens-specific cell wall binding domain creates an anti-Clostridium antimicrobial with improved thermostability. Viruses 2015, 7, 3019–3034. [Google Scholar] [CrossRef] [PubMed]
- Low, L.Y.; Yang, C.; Perego, M.; Osterman, A.; Liddington, R. Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. J. Biol. Chem. 2011, 286, 34391–34403. [Google Scholar] [CrossRef] [PubMed]
- Verbree, C.T.; Dätwyler, S.M.; Meile, S.; Eichenseher, F.; Donovan, D.M.; Loessner, M.J.; Schmelcher, M. Corrected and republished from: Identification of peptidoglycan hydrolase constructs with synergistic staphylolytic activity in cow’s milk. Appl. Environ. Microbiol. 2018, 84, e02134-17. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J.; Gaeng, S.; Scherer, S. Evidence for a holin-like protein gene fully embedded out of frame in the endolysin gene of Staphylococcus aureus bacteriophage 187. J. Bacteriol. 1999, 181, 4452–4460. [Google Scholar] [PubMed]
- Rodríguez-Rubio, L.; Quiles-Puchalt, N.; Martínez, B.; Rodríguez, A.; Penadés, J.R.; García, P. The peptidoglycan hydrolase of Staphylococcus aureus bacteriophage 11 plays a structural role in the viral particle. Appl. Environ. Microbiol. 2013, 79, 6187–6190. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Y.; Yu, J.; Huang, Y.; Zhang, X.E.; Wei, H. Novel chimeric lysin with high-level antimicrobial activity against methicillin-resistant Staphylococcus aureus in vitro and in vivo. Antimicrob. Agents Chemother. 2014, 58, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Y.; Huang, Y.; Yu, J.; Wei, H. Degradation of methicillin-resistant Staphylococcus aureus biofilms using a chimeric lysin. Biofouling 2014, 30, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Wang, J.; Yang, H.; Wei, C.; Yu, J.; Zhang, Y.; Huang, Y.; Zhang, X.E.; Wei, H. Construction of a chimeric lysin Ply187N-V12C with extended lytic activity against staphylococci and streptococci. Microb. Biotechnol. 2015, 8, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, H.; Wang, J.; Yu, J.; Wei, H. A novel chimeric lysin with robust antibacterial activity against planktonic and biofilm methicillin-resistant Staphylococcus aureus. Sci. Rep. 2017, 7, 40182. [Google Scholar] [CrossRef] [PubMed]
- Donovan, D.M.; Dong, S.; Garrett, W.; Rousseau, G.M.; Moineau, S.; Pritchard, D.G. Peptidoglycan hydrolase fusions maintain their parental specificities. Appl. Environ. Microbiol. 2006, 72, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J.; Kramer, K.; Ebel, F.; Scherer, S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol. Microbiol. 2002, 44, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.; Schuch, R.; Pelzek, A.J.; Buckle, A.M.; McGowan, S.; Wilce, M.C.; Rossjohn, J.; Russell, R.; Nelson, D.; Fischetti, V.A.; et al. The 1.6 a crystal structure of the catalytic domain of PlyB, a bacteriophage lysin active against Bacillus anthracis. J. Mol. Biol. 2007, 366, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.C.; Swift, S.; Korobova, O.; Schischkova, N.; Kopylov, P.; Donovan, D.M.; Abaev, I. Lytic activity of the staphylolytic Twort phage endolysin CHAP domain is enhanced by the SH3b cell wall binding domain. FEMS Microbiol. Lett. 2015, 362, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, H.; Yu, J.; Wei, H. Molecular dissection of phage lysin PlySs2: Integrity of the catalytic and cell wall binding domains is essential for its broad lytic activity. Virol. Sin. 2015, 30, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.J.; Gasson, M.J.; Narbad, A. Genomic sequence of bacteriophage ATCC 8074-B1 and activity of its endolysin and engineered variants against Clostridium sporogenes. Appl. Environ. Microbiol. 2012, 78, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Horgan, M.; O’Flynn, G.; Garry, J.; Cooney, J.; Coffey, A.; Fitzgerald, G.F.; Ross, R.P.; McAuliffe, O. Phage lysin LysK can be truncated to its CHAP domain and retain lytic activity against live antibiotic-resistant staphylococci. Appl. Environ. Microbiol. 2009, 75, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Donovan, D.M.; Foster-Frey, J.; Dong, S.; Rousseau, G.M.; Moineau, S.; Pritchard, D.G. The cell lysis activity of the Streptococcus agalactiae bacteriophage B30 endolysin relies on the cysteine, histidine-dependent amidohydrolase/peptidase domain. Appl. Environ. Microbiol. 2006, 72, 5108–5112. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.; Keary, R.; McAuliffe, O.; Ross, R.P.; O’Mahony, J.; Coffey, A. Bacteriophage-derived peptidase CHAPK eliminates and prevents staphylococcal biofilms. Int. J. Microbiol. 2013, 2013, 625341. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.; Casey, P.G.; Hill, C.; Gahan, C.G.M.; Ross, R.P.; McAuliffe, O.; O’Mahony, J.; Maher, F.; Coffey, A. The truncated phage lysin CHAPK eliminates Staphylococcus aureus in the nares of mice. Bioeng. Bugs 2010, 1, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.C.; Dong, S.; Baker, J.R.; Foster-Frey, J.; Pritchard, D.G.; Donovan, D.M. LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol. Lett. 2009, 294, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Sass, P.; Bierbaum, G. Lytic activity of recombinant bacteriophage φ11 and φ12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl. Environ. Microbiol. 2007, 73, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Benešík, M.; Nováček, J.; Janda, L.; Dopitová, R.; Pernisová, M.; Melková, K.; Tišáková, L.; Doškař, J.; Žídek, L.; Hejátko, J.; et al. Role of SH3b binding domain in a natural deletion mutant of Kayvirus endolysin LysF1 with a broad range of lytic activity. Virus Genes 2017. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; Bao, H.; Wang, X.; Wang, R. The lytic activity of recombinant phage lysin LysKΔamidase against staphylococcal strains associated with bovine and human infections in the Jiangsu province of China. Res. Vet. Sci. 2017, 111, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Low, L.Y.; Yang, C.; Perego, M.; Osterman, A.; Liddington, R.C. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J. Biol. Chem. 2005, 280, 35433–35439. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.J.; Garefalaki, V.; Spoerl, R.; Narbad, A.; Meijers, R. Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J. Bacteriol. 2011, 193, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Fischetti, V.A. Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl. Microbiol. Biotechnol. 2007, 74, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Donovan, D.M.; Foster-Frey, J. LambdaSa2 prophage endolysin requires Cpl-7-binding domains and amidase-5 domain for antimicrobial lysis of streptococci. FEMS Microbiol. Lett. 2008, 287, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Heselpoth, R.D.; Nelson, D.C. A new screening method for the directed evolution of thermostable bacteriolytic enzymes. J. Vis. Exp. 2012, 4216. [Google Scholar] [CrossRef] [PubMed]
- Díez-Martínez, R.; de Paz, H.D.; de Paz, H.; Bustamante, N.; García, E.; Menéndez, M.; García, P. Improving the lethal effect of Cpl-7, a pneumococcal phage lysozyme with broad bactericidal activity, by inverting the net charge of its cell wall-binding module. Antimicrob. Agents Chemother. 2013, 57, 5355–5365. [Google Scholar] [CrossRef] [PubMed]
- Heselpoth, R.D.; Yin, Y.; Moult, J.; Nelson, D.C. Increasing the stability of the bacteriophage endolysin plyc using rationale-based foldx computational modeling. Protein Eng. Des. Sel. 2015, 28, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Resch, G.; Moreillon, P.; Fischetti, V.A. A stable phage lysin (Cpl-1) dimer with increased antipneumococcal activity and decreased plasma clearance. Int. J. Antimicrob. Agents 2011, 38, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.C.; Roach, D.R.; Chauhan, V.S.; Shen, Y.; Foster-Frey, J.; Powell, A.M.; Bauchan, G.; Lease, R.A.; Mohammadi, H.; Harty, W.J.; et al. Triple-acting lytic enzyme treatment of drug-resistant and intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 25063. [Google Scholar] [CrossRef] [PubMed]
- Haddad Kashani, H.; Fahimi, H.; Dasteh Goli, Y.; Moniri, R. A novel chimeric endolysin with antibacterial activity against methicillin-resistant Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2017, 7, 290. [Google Scholar] [CrossRef] [PubMed]
- Pohane, A.A.; Joshi, H.; Jain, V. Molecular dissection of phage endolysin: An interdomain interaction confers host specificity in Lysin A of Mycobacterium phage D29. J. Biol. Chem. 2014, 289, 12085–12095. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Euler, C.W.; Delaune, A.; Fischetti, V.A. Using a novel lysin to help control Clostridium difficile infections. Antimicrob. Agents Chemother. 2015, 59, 7447–7457. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.M.; Sánchez-Pérez, A.; Calo-Mata, P.; Villa, T.G. Antimicrobial peptides (AMPS): Ancient compounds that represent novel weapons in the fight against bacteria. Biochem. Pharmacol. 2017, 133, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Wittekind, M.; Schuch, R. Cell wall hydrolases and antibiotics: Exploiting synergy to create efficacious new antimicrobial treatments. Curr. Opin. Microbiol. 2016, 33, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Shah, A.; Mond, J. Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob. Agents Chemother. 2003, 47, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Filatova, L.Y.; Donovan, D.M.; Becker, S.C.; Lebedev, D.N.; Priyma, A.D.; Koudriachova, H.V.; Kabanov, A.V.; Klyachko, N.L. Physicochemical characterization of the staphylolytic LysK enzyme in complexes with polycationic polymers as a potent antimicrobial. Biochimie 2013, 95, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Resch, G.; Moreillon, P.; Fischetti, V.A. PEGylating a bacteriophage endolysin inhibits its bactericidal activity. AMB Express 2011, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Bacteriophage lytic enzymes: Novel anti-infectives. Trends Microbiol. 2005, 13, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Martínez, B.; Rodríguez, A.; Donovan, D.M.; Götz, F.; García, P. The phage lytic proteins from the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 display multiple active catalytic domains and do not trigger staphylococcal resistance. PLoS ONE 2013, 8, e64671. [Google Scholar] [CrossRef] [PubMed]
- Thandar, M.; Lood, R.; Winer, B.Y.; Deutsch, D.R.; Euler, C.W.; Fischetti, V.A. Novel engineered peptides of a phage lysin as effective antimicrobials against multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2016, 60, 2671–2679. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.Y.; You, R.I.; Lai, M.J.; Lin, N.T.; Chen, L.K.; Chang, K.C. Highly potent antimicrobial modified peptides derived from the Acinetobacter baumannii phage endolysin LysAB2. Sci. Rep. 2017, 7, 11477. [Google Scholar] [CrossRef] [PubMed]
- Lukacik, P.; Barnard, T.J.; Keller, P.W.; Chaturvedi, K.S.; Seddiki, N.; Fairman, J.W.; Noinaj, N.; Kirby, T.L.; Henderson, J.P.; Steven, A.C.; et al. Structural engineering of a phage lysin that targets Gram-negative pathogens. Proc. Natl. Acad. Sci. USA 2012, 109, 9857–9862. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Liu, J.; Ma, Q.; Zhu, R.; Guo, Z.; Gao, C.; Wang, S.; Yu, L.; Gu, J.; Hu, D.; et al. The N-terminal and central domain of colicin A enables phage lysin to lyse Escherichia coli extracellularly. Antonie Van Leeuwenhoek 2017, 110, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Walmagh, M.; Van Puyenbroeck, V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Oliveira, H.; Azeredo, J.; Verween, G.; Pirnay, J.P.; et al. Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. mBio 2014, 5, e01379-14. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Walmagh, M.; Grymonprez, B.; Biebl, M.; Pirnay, J.P.; Defraine, V.; Michiels, J.; Cenens, W.; Aertsen, A.; Miller, S.; et al. Art-175 is a highly efficient antibacterial against multidrug-resistant strains and persisters of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 3774–3784. [Google Scholar] [CrossRef] [PubMed]
- Defraine, V.; Schuermans, J.; Grymonprez, B.; Govers, S.K.; Aertsen, A.; Fauvart, M.; Michiels, J.; Lavigne, R.; Briers, Y. Efficacy of artilysin art-175 against resistant and persistent Acinetobacter baumannii. Antimicrob. Agents Chemother. 2016, 60, 3480–3488. [Google Scholar] [CrossRef] [PubMed]
- Schirmeier, E.; Zimmermann, P.; Hofmann, V.; Biebl, M.; Gerstmans, H.; Maervoet, V.E.; Briers, Y. Inhibitory and bactericidal effect of Artilysin® Art-175 against colistin-resistant mcr-1-positive Escherichia coli isolates. Int. J. Antimicrob. Agents 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gu, J.; Lv, M.; Guo, Z.; Yan, G.; Yu, L.; Du, C.; Feng, X.; Han, W.; Sun, C.; et al. The antibacterial activity of E. coli bacteriophage lysin lysep3 is enhanced by fusing the Bacillus amyloliquefaciens bacteriophage endolysin binding domain D8 to the C-terminal region. J. Microbiol. 2017, 55, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Guo, Z.; Gao, C.; Zhu, R.; Wang, S.; Yu, L.; Qin, W.; Xia, X.; Gu, J.; Yan, G.; et al. Enhancement of the direct antimicrobial activity of Lysep3 against Escherichia coli by inserting cationic peptides into its C-terminus. Antonie Van Leeuwenhoek 2017, 110, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Wu, S.J.; Chang, T.W.; Wang, C.F.; Suen, C.S.; Hwang, M.J.; Chang, M.D.; Chen, Y.T.; Liao, Y.D. Outer membrane protein I of Pseudomonas aeruginosa is a target of cationic antimicrobial peptide/protein. J. Biol. Chem. 2010, 285, 8985–8994. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, M.; Yu, J.; Wei, H. Antibacterial activity of a novel peptide-modified lysin against Acinetobacter baumannii and Pseudomonas aeruginosa. Front. Microbiol. 2015, 6, 1471. [Google Scholar] [CrossRef] [PubMed]
- Orito, Y.; Morita, M.; Hori, K.; Unno, H.; Tanji, Y. Bacillus amyloliquefaciens phage endolysin can enhance permeability of Pseudomonas aeruginosa outer membrane and induce cell lysis. Appl. Microbiol. Biotechnol. 2004, 65, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Chang, W.L.; Gutiérrez, D.; Lavigne, R.; Martínez, B.; Rodríguez, A.; Govers, S.K.; Aertsen, A.; Hirl, C.; Biebl, M.; et al. ‘Artilysation’ of endolysin λSa2lys strongly improves its enzymatic and antibacterial activity against streptococci. Sci. Rep. 2016, 6, 35382. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T. Classical labeling of bacterial pathogens according to their lifestyle in the host: Inconsistencies and alternatives. Front. Microbiol. 2012, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Borysowski, J.; Górski, A. Fusion to cell-penetrating peptides will enable lytic enzymes to kill intracellular bacteria. Med. Hypotheses 2010, 74, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Dietz, G.P. Cell-penetrating peptide technology to deliver chaperones and associated factors in diseases and basic research. Curr. Pharm. Biotechnol. 2010, 11, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Barros, M.; Vennemann, T.; Gallagher, D.T.; Yin, Y.; Linden, S.B.; Heselpoth, R.D.; Spencer, D.J.; Donovan, D.M.; Moult, J.; et al. A bacteriophage endolysin that eliminates intracellular streptococci. eLife 2016, 5, e13152. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Chimeolysin | CD Source | CWBD Source | Susceptible Bacteria | In Vivo Assay(s) | Outcome | Reference |
|---|---|---|---|---|---|---|
| EAD118_III_CBDPSA | Endopeptidase CD of Ply118 (endolysin L. monocytogenes phage A118) | PlyPSA (endolysin L. monocytogenes phage PSA) | L. monocytogenes | Not reported | 3-fold higher activity compared with parental PlyPSA | [65] |
| λSA2-E-Lyso-SH3b and λSA2-E-LysK-SH3b | Endopeptidase CD of λSA2 (endolysin S. agalactiae prophage λSA2) | SH3b-like CWBD of Lysostaphin or of LysK (endolysin of S. aureus phage K) | Staphylococci, streptococci | Mouse model of mastitis | Efficient activity extended to S. aureus while retaining significant streptolytic activity | [67,68] |
| ClyR | Amidase CD (CHAP) of PlyC (endolysin streptococcal phage C1) | PlySs2 (endolysin S. suis prophage) | Several streptococcal species (including S. pneumoniae), E. faecalis, S. aureus | Murine models of S. agalactiae systemic infection and of S. mutans dental colonization | Higher activity and broader lytic spectrum than the parental and other streptococcal endolysins. Stable under storage | [69,70] |
| Cpl-711 | Muramidase CD of Cpl-7S (improved variant of pneumococcal endolysin Cpl-7, see below) | Cpl-1 (endolysin pneumococcal phage Cpl-1) | S. pneumoniae, S. mitis | Murine bacteraemia model | Greater killing and antibiofilm activity than parental endolysins in vitro. Superior protection compared with Cpl-1 in a mouse model of bacteraemia | [71] |
| Csl2 | Muramidase CD of Cpl-7 (endolysin pneumococcal phage Cp-7) | LySMP (endolysin S. suis phage SMP) | S. suis, S. pseudopneumonia, S. mitis, S. oralis | Adult zebrafish model of infection | Superior bactericidal and antibiofilm activity than parental LysSMP | [72] |
| PL3 | Amidase CD of Pal (endolysin pneumococcal phage Dp-1) | First two choline-binding repeats of Pal and the last four of LytA (major pneumococcal autolysin) | S. pneumonia and other choline-containing streptococci | Zebrafish embryo infection model | Superior bactericidal activity than parental enzymes and high stability | [73] |
| CHAPSH3b | Endopeptidase CD (CHAP) of HydH5 (VAL S. aureus phage phiIPLA88) | SH3b-like CWBD of lysostaphin (bacteriolysin S. simulans) | S. aureus, S. epidermidis | Not reported | Thermostability. Much higher activity than the parental HydH5 | [59,74,75,76] |
| P128 | Putative endopeptidase (CHAP) of Orf56 (VAL S. aureus phage K) | SH3b-like CWBD of lysostaphin (bacteriolysin S. simulans) | S. aureus, S. epidermidis, S. carnosus, S. simulans | Rat nasal colonization model (S. aureus USA 300) | P128 has much higher killing activity than the isolated CHAP CD of Orf56. Effective antibiofilm activity. Better thermostability than lysostaphin | [58,77,78] |
| Ply187AN-KSH3b | Putative endopeptidase CD of Ply187 (PLE from S. aureus phage 187) | SH3b-like CWBD of LysK (endolysin of S. aureus phage K) | S. aureus and other staphylococcal species | Mouse model of S. aureus Endophthalmitis | More active than native Ply187 and Ply187AN truncated Enzyme. Effective antibiofilm activity | [79,80] |
| EC300 | Putative endopeptidase CD (M23) of Orf73 (putative VAL E. faecalis phage F170/08) | Oligomerization-prone CWBD of Lys170 (endolysin E. faecalis phage F170/08) | Multidrug-resistant E. faecalis, including VRE | Not reported | In contrast to the parental endolysin, EC300 lysis E. faecalis actively growing in rich medium | [62] |
| ClyS | Endopeptidase CD of PlyTW (endolysin S. aureus phage Twort) | Endolysin S. aureus phage phiNM3 (highly soluble CWBD not related to the very common SH3b) | S. aureus, S. sciuri, S. simulans, S. epidermidis | Different murine colonization/infection models (nasal, skin and systemic) | Broad-spectrum activity and high solubility when compared to most staphylococcal endolysins | [81,82] |
| Lys168-87 | Putative endopeptidase CD of Lys168 (endolysin E. faecalis phage F168/08) | Putative CWBD of Lys87 (endolysin S. aureus phage F87s/06) | Staphylococci, E. faecalis, E. faecium, S. pyogenes | Not reported | High solubility compared to most native PLEs targeting S. aureus. Expanded spectrum of activity | [83] |
| PlyGVE2CpCWB | Amidase CD of PlyGVE2 (endolysin Geobacillus phage ФGVE2) | PlyCP26F (endolysin C. perfringens phage ФCP26F) | C. perfringens | Not reported | Better thermostability than parental PlyCP26F | [84] |
| Engineering Approach | Example(s) 1 | Engineering Details 1 | Susceptible Bacteria | In Vivo Assay(s) | Outcome | Reference |
|---|---|---|---|---|---|---|
| Fusion to lytic enzymes | B30-443-Lyso B30-182-Lyso | Fusion of S. agalactiae phage B30 endolysin (or of its endopeptidase CD) to S. simulans lysostaphin | Several streptococcal species, including pathogens and dairy bacteria. S. aureus | Not reported | Lytic spectrum extended to S. aureus and increased activity (B30-182-Lyso) | [94] |
| Domain deletion | CHAPK | CHAPK corresponds to the endopeptidase (CHAP) CD of LysK (first 165 aa of de endolysin of S. aureus phage K) | S. aureus | S aureus elimination in the nares of mice | Higher lytic activity than LysK | [100,102,103] |
| PlyLCAT (amidase) PlyBa04CAT (muramidase) | Deletion of the C-ter CWBDs of PlyL and PlyBa04, the endolysins of B. anthracis λBa02 Prophage and B. anthracis phage Ba04, respectively | B. cereus, B. megaterium, B. anthracis, B. subtilis | Not reported | Extended lytic spectrum. Enhanced lytic activity (especially against B. subtilis in the case of PlyLCAT) | [85,108] | |
| CD27L1-179 (N-ter amidase CD) | Deletion of the C-ter CWBD of the clostridial endolysin CD27L | Clostridium spp. (including C. difficile), Bacillus spp., Listeria spp. | Not reported | Increased lytic activity and spectrum extended to two additional Listeria sp. | [109] | |
| PlyGBS94 | PlyGBS94 corresponds to the first 146 aa of native PlyGBS (endolysin S. agalactiae phage NCTC 11261), carrying only a endopeptidase CD | Group B streptococci (Streptococcus agalactiae) | Not reported | ~25-fold increase of specific activity | [110] | |
| λSa2-ECC | Deletion of C-ter glycosidase CD of λSA2 endolysin (S. agalactiae prophage λSA2) | Several streptococcal species and few S. aureus strains | Not reported | Increased activity towards certain streptococcal strains and few S. aureus strains | [111] | |
| Domain addition | HydH5SH3b | Addition of lysostaphin CWBD (SH3b) to VAL HydH5 of S. aureus phage phiIPLA88 | S. aureus, S. epidermidis | Not reported | Higher activity than the parental HydH5 | [74] |
| Domain duplication | EAD_CBD500-500 | Extra copy of CWBD added to Ply500 (endolysin L. monocytogenes phage A500) | Essentially Listeria spp. | Not reported | Much higher affinity improves endolysin activity at high salt concentrations | [65] |
| Random mutagenesis | PlyGBS90-1 | Frameshift mutation truncates PlyGBS at aa 141 and adds 13 aa | Group B streptococci (Streptococcus agalactiae) | Decolonization in a mouse vaginal model | ~28-fold increase of specific activity, although less stable than native PlyGBS in certain conditions. Improved killing activity in vivo | [110] |
| 29C3 mutant of PlyC | Mutation-prone PCR of PlyCA subunit of PlyC (endolysin streptococcal phage C1) | S. pyogenes | Not reported | The 29C3 mutant exhibits higher thermostability than PlyC, which should translate into extended shelf life | [112] | |
| Site-directed mutagenesis | Cpl-7S | 15 aa substitutions added positive charges to the CWBD of pneumococcal endolysin Cpl-7 (from −14.93 to +3.0 at neutral pH) | S. pneumoniae, E. faecalis, S. mitis, S. pyogenes, and, to a lesser extent, S. dysgalactiae and S. iniae. E. coli and P. putida in presence of carvacrol | Zebrafish embryo infection model (S. pneumoniae and S. pyogenes) | Improved killing activity compared to the native Cpl-7 endolysin | [113] |
| (PlyC)T406R | T406R substitution in PlyCA subunit of PlyC (endolysin streptococcal phage C1) | S. pyogenes | Not reported | Thermostabilization of PlyC (16-fold increase of half-life at 45 °C), although with moderate loss of lytic activity in vitro | [114] | |
| Multimerization | Cpl-1 dimer | Cpl-1C45S,D324C. Introduction of Cys residues at aa position 324 allowed intermolecular disulphide bonding. The C45S substitution avoided unwanted interactions with this Cys residue | S. pneumoniae | Not reported | 2-fold increase of antipneumococcal activity and ~10-fold decrease in plasma clearance (mice) compared to native Cpl-1 | [115] |
| Mixed approaches | L98WCD27L1-179 | Deletion of CD27L C-ter CWBD and L98W mutation in CD27L CD | Clostridium spp. (including C. difficile), Bacillus spp., Listeria spp. | Not reported | The L98W mutation further increased lytic activity of CD27L1-179 against L. monocytogenes | [109] |
| K-L K-L-PTD L-K L-K-PTD (triple-CD PLEs, i.e., 3 distinct CDs) | LysK/Lysostaphin chimeras added or not of protein transduction domains (PTD). K-L: CHAP-Amidase CDs of LysK fused to lysostaphin. L-K: LysK CDs inserted between the CD (M23) and CWBD (SH3b) of lysostaphin | S. aureus and coagulase negative staphylococci | Decolonization in rat nasal model. Murine model of mastitis | The presence of 3 distinct CDs in the chimeras reduces emergence of resistant strains. Superior killing activity of L-K in rat nasal model | [116] | |
| CHAP-Amidase | Codon-optimized CHAP and amidase CDs of LysK (endolysin S. aureus phage K) connected by the linker GSH6GS. No CWBD | S. aureus, S. epidermidis, E. faecium, and E. faecalis | Not reported | Enhanced production, stability, and solubility by improving codon-usage and the properties of primary, secondary, and tertiary structures | [117] |
| Engineering Approach | Example(s) | Engineering Details 1 | Susceptible Bacteria | In Vivo Assay(s) | Outcome | Reference |
|---|---|---|---|---|---|---|
| Fusion to domains targeting OM receptor/transport systems | Pesticin-T4 lysozyme hybrid | Pesticin (bacteriocin) domain targeting FyuA (OMP) fused to the N-ter of E. coli phage T4 endolysin | FyuA-expressing pathogenic bacteria (Y. pestis, Y. pseudotuberculosis, uropathogenic E. coli) | - | The hybrid protein crosses the OM through FyuA-mediated transport | [131] |
| Fusion to domains or peptides that destabilize the OM | LoGT-001 LoGT-008 | LoGT-001: PCNP (polycationic nonapeptide) connected to the N-ter of OBPgp279 (endolysin P. fluorescens phage OBP) LoGT-008: PCNP connected to the N-ter of PVP-SE1gp146 (endolysin S. enterica phage PVP-SE1) | P. aeruginosa. Other Artilysins of the LoGT series also killed effectively A. baumannii and E. coli (≥1 Log reduction). Killing of S. Typhimurium required EDTA | C. elegans infection assay (LoGT-008) | The PCNP tag increased the intrinsic antibacterial of two modular endolysins (OBPgp279 and PVPSE1gp146) by facilitation OM crossing | [133] |
| Art-175 | Antimicrobial peptide SMAP-29 fused to the N-ter of mutated KZ144 (endolysin P. aeruginosa phage φKZ) | P. aeruginosa (and few other Pseudomonas spp.), K. pneumoniae, A. baumannii, colistin-resistant E. coli | - | In contrast to KZ144, Art-175 crosses the outer membrane and efficiently kills target cells. Capacity to eliminate P. aeruginosa and A. baumannii persister cells. Art-175 outcompetes conventional antibiotics in bactericidal activity against A. baumannii | [134,135,136] | |
| Lysep3-D8 | Lysep3 (endolysin E. coli phage vB_EcoM-ep3) fused to region D8 of the endolysin of B. amyloliquefaciens phage Morita2001 | E. coli, P. aeruginosa (3 strains), A. baumannii (1 strain), Streptococcus sp. (1 strain) | - | In contrast to isolated Lyse3 and D8, Lysep3-D8 has bactericidal activity | [137] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
São-José, C. Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials. Antibiotics 2018, 7, 29. https://doi.org/10.3390/antibiotics7020029
São-José C. Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials. Antibiotics. 2018; 7(2):29. https://doi.org/10.3390/antibiotics7020029
Chicago/Turabian StyleSão-José, Carlos. 2018. "Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials" Antibiotics 7, no. 2: 29. https://doi.org/10.3390/antibiotics7020029
APA StyleSão-José, C. (2018). Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials. Antibiotics, 7(2), 29. https://doi.org/10.3390/antibiotics7020029