Biosynthesis of Rishirilide B

Abstract
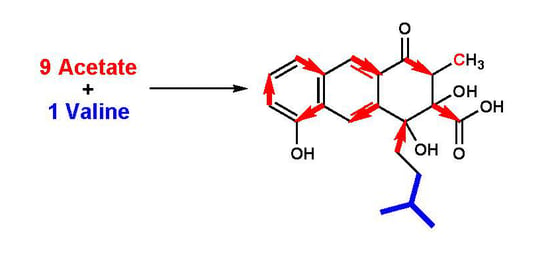
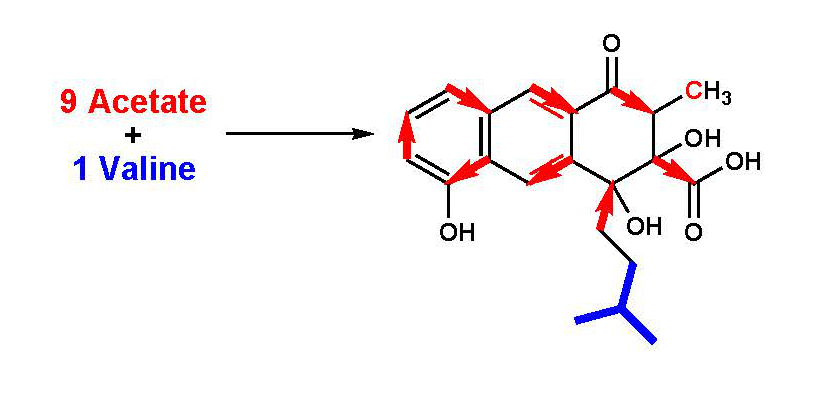
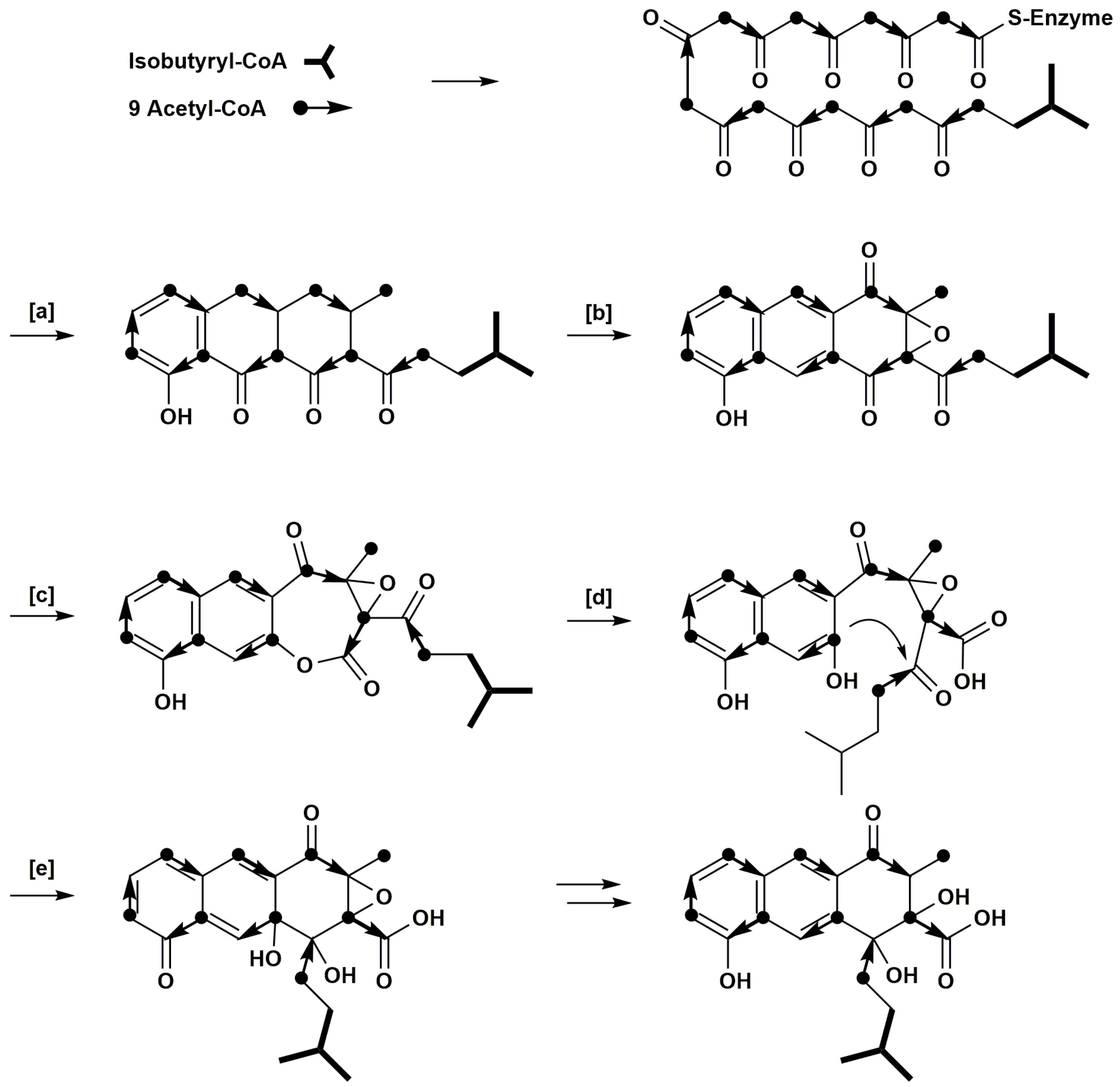
1. Introduction
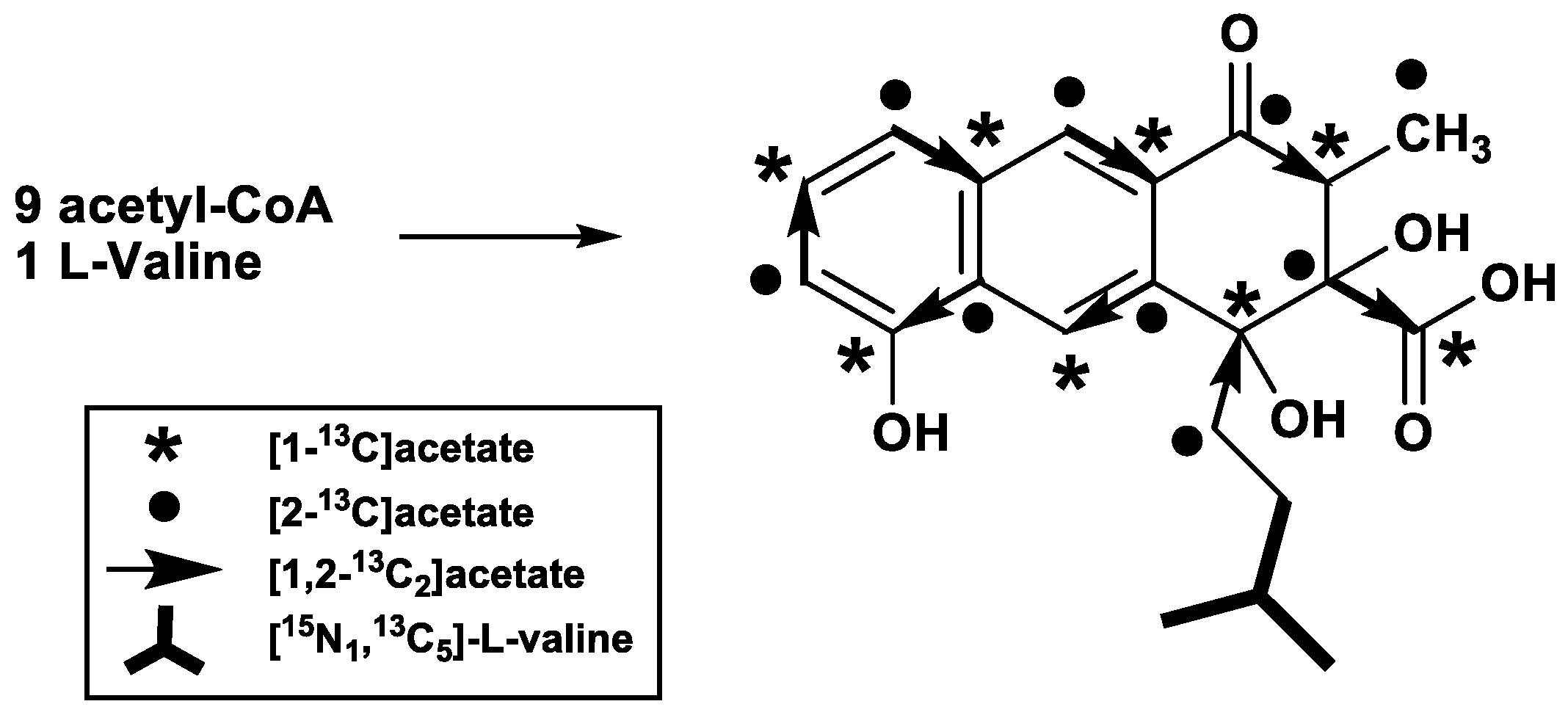
2. Results
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Cultivation
4.2. Feeding Experiments
4.3. Analysis of Rishirilide B by HPLC/MS
4.4. Isolation and Purification of Rishirilide B
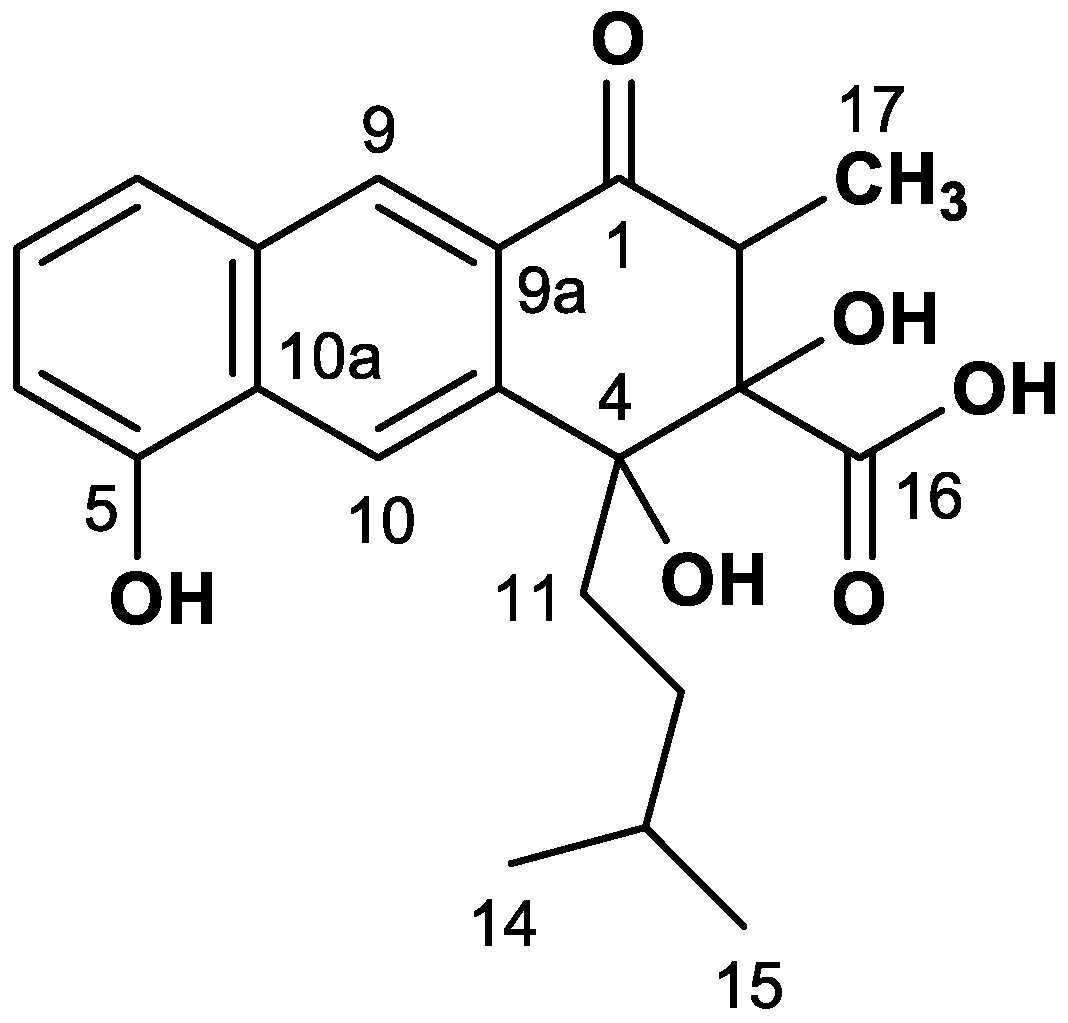
4.5. NMR Analysis of Rishirilide B
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Hagan, D. Biosynthesis of polyketide metabolites. Nat. Prod. Rep. 1992, 9, 447–479. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, H.; Nakayama, Y.; Takahashi, M.; Uetsuki, S.; Kido, M. Structures of rishirilides A and B, α2-macroglobulin inhibitors produced by Streptomyces rishiriensis OFR-1056. J. Antibiot. 1984, XXXVII, 9–11. [Google Scholar]
- Yan, X.; Probst, K.; Linnenbrink, A.; Arnold, M.; Paululat, T.; Zeeck, A.; Bechthold, A. Cloning and heterologous expression of three type II PKS gene clusters from Streptomyces bottropensis. Chembiochem 2012, 13, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.S.; Hertweck, C. Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat. Prod. Rep. 2002, 19, 70–99. [Google Scholar] [PubMed]
- Ubukata, M.; Cheng, X.C.; Uzawa, J.; Isono, K. Biosynthesis of the dialkylmaleic anhydride-containing antibiotics, tautomycin and tautomycetin. J. Chem. Soc. Perkin Trans. 1 1995, 2399–2404. [Google Scholar] [CrossRef]
- Kingston, D.G.; Kolpak, M.X.; LeFevre, J.W.; Borup-Grochtmann, I. Biosynthesis of antibiotics of the virginiamycin family. 3. Biosynthesis of virginiamycin M1. J. Am. Chem. Soc. 1983, 105, 5106–5110. [Google Scholar] [CrossRef]
- Omura, S.; Tsuzuki, K.; Tanaka, Y.; Sakakibara, H.; Aizawa, M.; Lukacs, G. Valine as a precursor of n-butyrate unit in the biosynthesis of macrolide aglycone. J. Antibiot. 1983, 614–616. [Google Scholar] [CrossRef]
- Chan, Y.A.; Podevels, A.M.; Kevany, B.M.; Thomas, M.G. Biosynthesis of polyketide synthase extender units. Nat. Prod. Rep. 2009, 26, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef] [PubMed]
- Schipper, D.; Van Der Baan, J.L.; Bickelhaupt, F. Biosynthesis of malonomicin. Part 1. 13C nuclear magnetic resonance spectrum and feeding experiments with 13C-labelled precursors. J. Chem. Soc. Perkin Trans. 1 1979, 0, 2017–2022. [Google Scholar] [CrossRef]
- Paululat, T.; Gutterer, J.M.; Fiedler, H.-P.; Zeeck, A. Biosynthesis of polyketomycin produced by Streptomyces diastatochromogenes Tü 6028. J. Antibiot. (Tokyo) 1999, 52, 96–101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elson, S.W.; Oliver, R.S. Studies on the biosynthesis of clavulanic acid. Incorporation of 13C-labelled precursors. J. Antibiot. (Tokyo) 1978, 31, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hou, X.-F.; Qi, L.-H.; Yin, Y.; Li, Q.; Pan, H.-X.; Chen, X.-Y.; Tang, G.-L. Biosynthesis of trioxacarcin revealing a different starter unit and complex tailoring steps for type II polyketide synthase. Chem. Sci. 2015, 6, 3440–3447. [Google Scholar] [CrossRef]
- Thomas, R. A biosynthetic classification of fungal and streptomycete fused-ring aromatic polyketides. ChemBioChem 2001, 2, 612–627. [Google Scholar] [CrossRef]
- Fujii, I.; Ebizuka, Y.; Sankawa, U. A novel anthraquinone ring cleavage enzyme from Aspergillus terreus. J. Biochem. 1988, 103, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Beam, M.P.; Bossermann, M.A.; Noinaj, N.; Wehenkel, M.; Rohr, J. Crystal structure of Baeyer-Villiger monooxygenase MtmOIV, the key enzyme of the mithramycin biosynthetic pathway. J. Biochem. 2009, 48, 4476–4487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, G.; Liu, X.; Xu, M.; Wang, Y.; Zhang, F.; Xu, L.; Lv, J.; Long, Q.; Kang, Q.; Ou, H.Y.; Wang, Y.; Rohr, J.; Deng, Z.; Jiang, M.; Lin, S.; Tao, M. Formation of an angular aromatic polyketide from a linear anthrene precursor via oxidative rearrangement. Cell Chem. Biol. 2017, 24, 881–891. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xiang, L.; Kalaitzis, J.A.; Moore, B.S. EncM, a versatile enterocin biosynthetic enzyme involved in Favorskii oxidative rearrangement, aldol condensation, and heterocycle-forming reactions. Proc. Natl. Acad. Sci. USA 2009, 101, 15609–15614. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Miyanaga, A.; Michaudel, Q.; Stull, F.; Louie, G.; Noel, J.P.; Baran, P.S.; Palfey, B.; Moore, B.S. Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature 2013, 503, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Tibrewal, N.; Pahari, P.; Wang, G.; Kharel, M.K.; Morris, C.; Downey, T.; Hou, Y.; Bugni, T.S.; Rohr, J. Baeyer-Villiger C-C bond cleavage reaction in gilvocarcin and jadomycin biosynthesis. J. Am. Chem. Soc. 2012, 134, 18181–18184. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.R.; Yu, X.; Patel, A.B.; Calveras, J.; Barajas, J.F.; Sakaki, E.; Metsä-Ketelä, M.; Liu, H.-W.; Rohr, J.; Tsai, S.-C. Insights into complex oxidation during BE-7585A biosynthesis: Structural determination and analysis of the polyketide monooxygenase BexE. ACS Chem. Biol. 2016, 11, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.I.; Townsend, C.A.; Okada, K.; Kajiwara, M.; Cushley, R.J.; Whitman, P.J. Biosynthesis of corrins. II. Incorporation of 13C-labeled substrates in vitamins B12. J. Am. Chem. Soc. 1974, 96, 8069–8080. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pos. | δC (ppm) | I(b) (%) | II(a) (%) | III JCC (Hz) | III(c) J Partner | IV JCC (Hz) | IV J Partner |
|---|---|---|---|---|---|---|---|
| 1 | 197.3 | −0.4 | 3.2 | 41 | C-2 | (d) | |
| 2 | 47.9 | 3.8 | −0.6 | 41 | C-1 | (d) | |
| 3 | 83.0 | 0.8 | 4.4 | 51 | C-16 | (d) | |
| 4 | 76.6 | 4.9 | −0.3 | 39 | C-11 | (d) | |
| 4a | 140.8 | − | 6.3 | 64 | C-10 | (d) | |
| 5 | 152.9 | 4.6 | −0.5 | 64 | C-10a | (d) | |
| 6 | 109.9 | −0.4 | 2.6 | 55 | C-7 | (d) | |
| 7 | 126.0 | 7.9 | −0.4 | 55 | C-6 | (d) | |
| 8 | 119.6 | −0.3 | 1.3 | 55 | C-8a | (d) | |
| 8a | 132.2 | 5.1 | −0.6 | 55 | C-8 | (d) | |
| 9 | 125.3 | 0.4 | 3.8 | 64 | C-9a | (d) | |
| 9a | 130.3 | 4.2 | 0.4 | 64 | C-9 | (d) | |
| 10 | 119.3 | 6.7 | −0.3 | 64 | C-10a | (d) | |
| 10a | 126.3 | −0.8 | 2.0 | 64 | C-10 | (d) | |
| 11 | 35.0 | −0.3 | 2.4 | 39 | C-4 | (d) | |
| 12 | 31.1 | −0.03 | −0.2 | - | - | 35 | C-13(c) |
| 13 | 27.9 | −0.2 | −0.1 | - | - | 35,35 | C-12(c), C-14(c), C-15(c) |
| 14 | 22.4 | (b) | (b) | - | - | 35 | C-13(c) |
| 15 | 22.6 | −0.1 | −0.4 | - | - | 35 | C-13(c) |
| 16 | 173.8 | 5.0 | −0.1 | 51 | C-3 | (d) | |
| 17 | 10.1 | −0.3 | 1.4 | - | - | (d) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarzer, P.; Wunsch-Palasis, J.; Bechthold, A.; Paululat, T. Biosynthesis of Rishirilide B. Antibiotics 2018, 7, 20. https://doi.org/10.3390/antibiotics7010020
Schwarzer P, Wunsch-Palasis J, Bechthold A, Paululat T. Biosynthesis of Rishirilide B. Antibiotics. 2018; 7(1):20. https://doi.org/10.3390/antibiotics7010020
Chicago/Turabian StyleSchwarzer, Philipp, Julia Wunsch-Palasis, Andreas Bechthold, and Thomas Paululat. 2018. "Biosynthesis of Rishirilide B" Antibiotics 7, no. 1: 20. https://doi.org/10.3390/antibiotics7010020
APA StyleSchwarzer, P., Wunsch-Palasis, J., Bechthold, A., & Paululat, T. (2018). Biosynthesis of Rishirilide B. Antibiotics, 7(1), 20. https://doi.org/10.3390/antibiotics7010020