Diversification of Secondary Metabolite Biosynthetic Gene Clusters Coincides with Lineage Divergence in Streptomyces


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
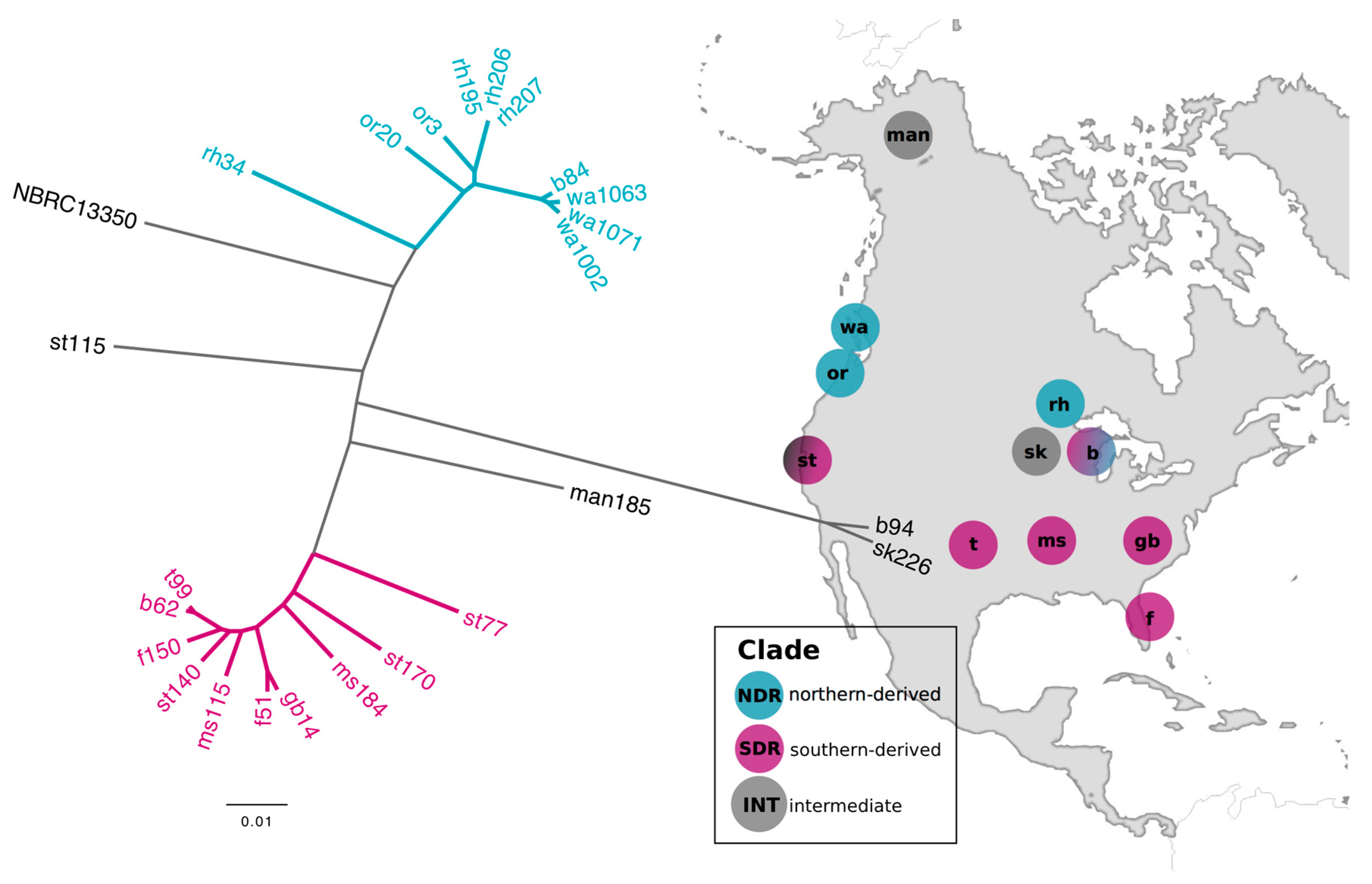
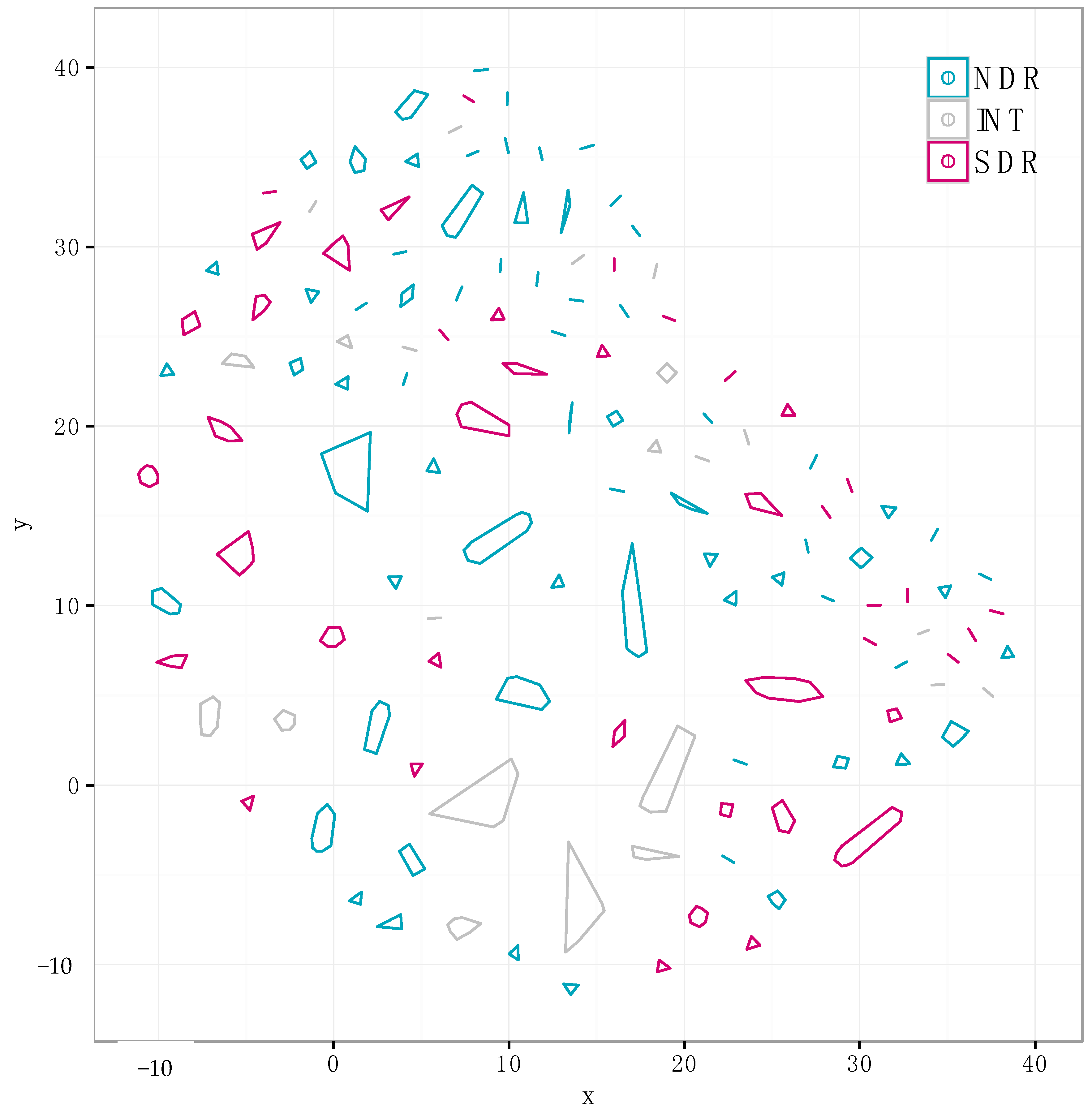
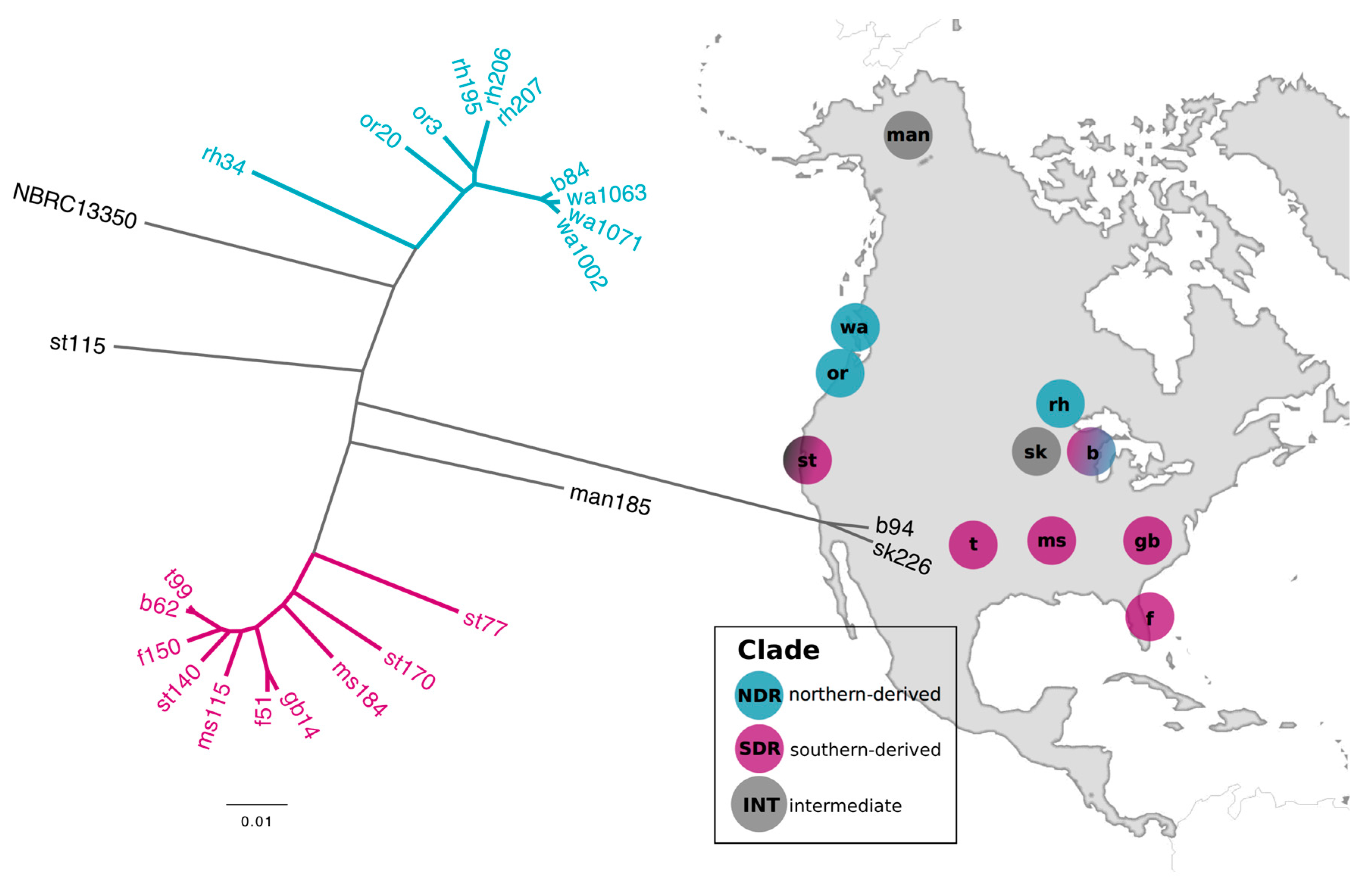
2.1. Genomic Divergence between Streptomyces Sister-Taxa
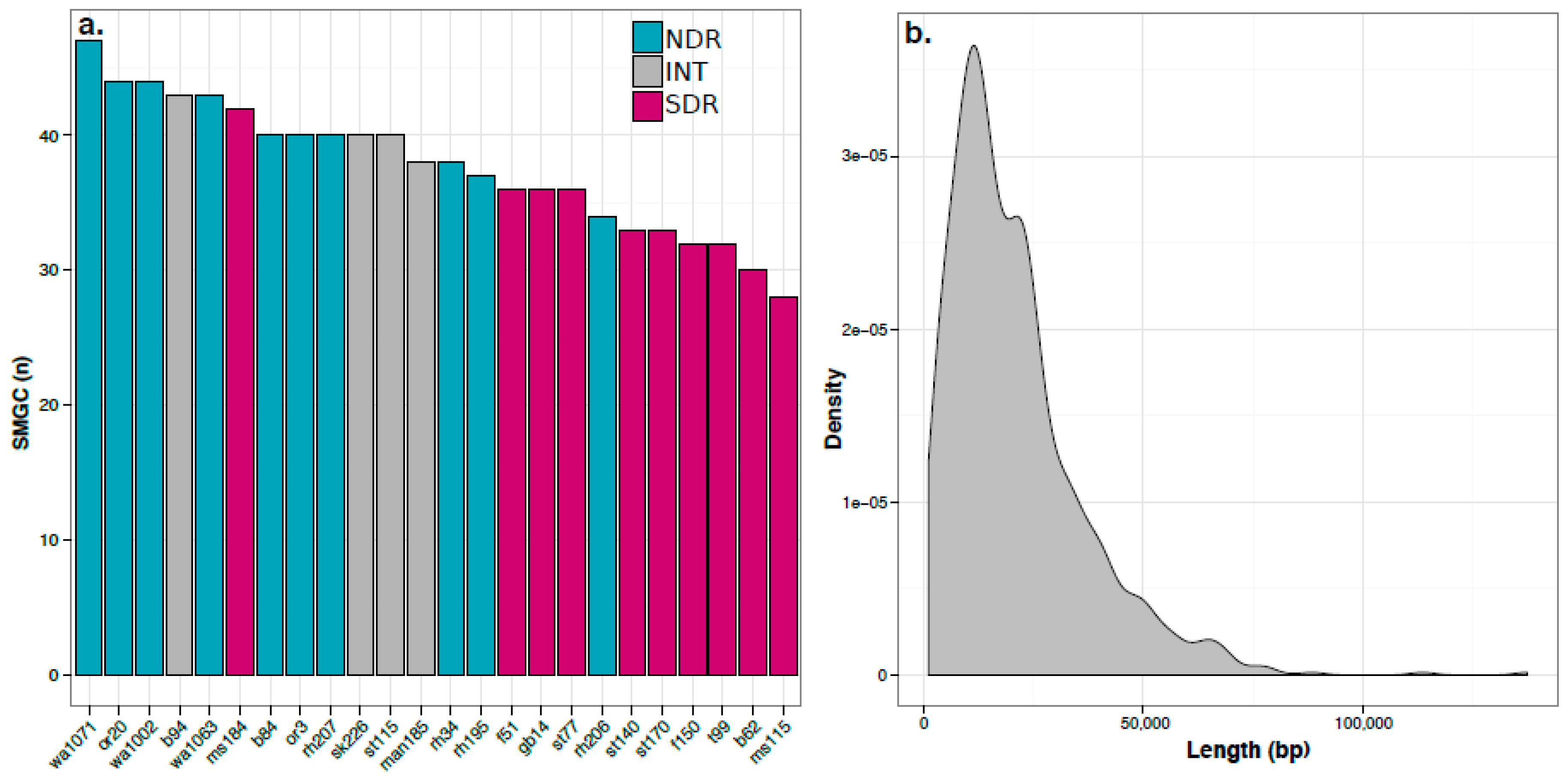
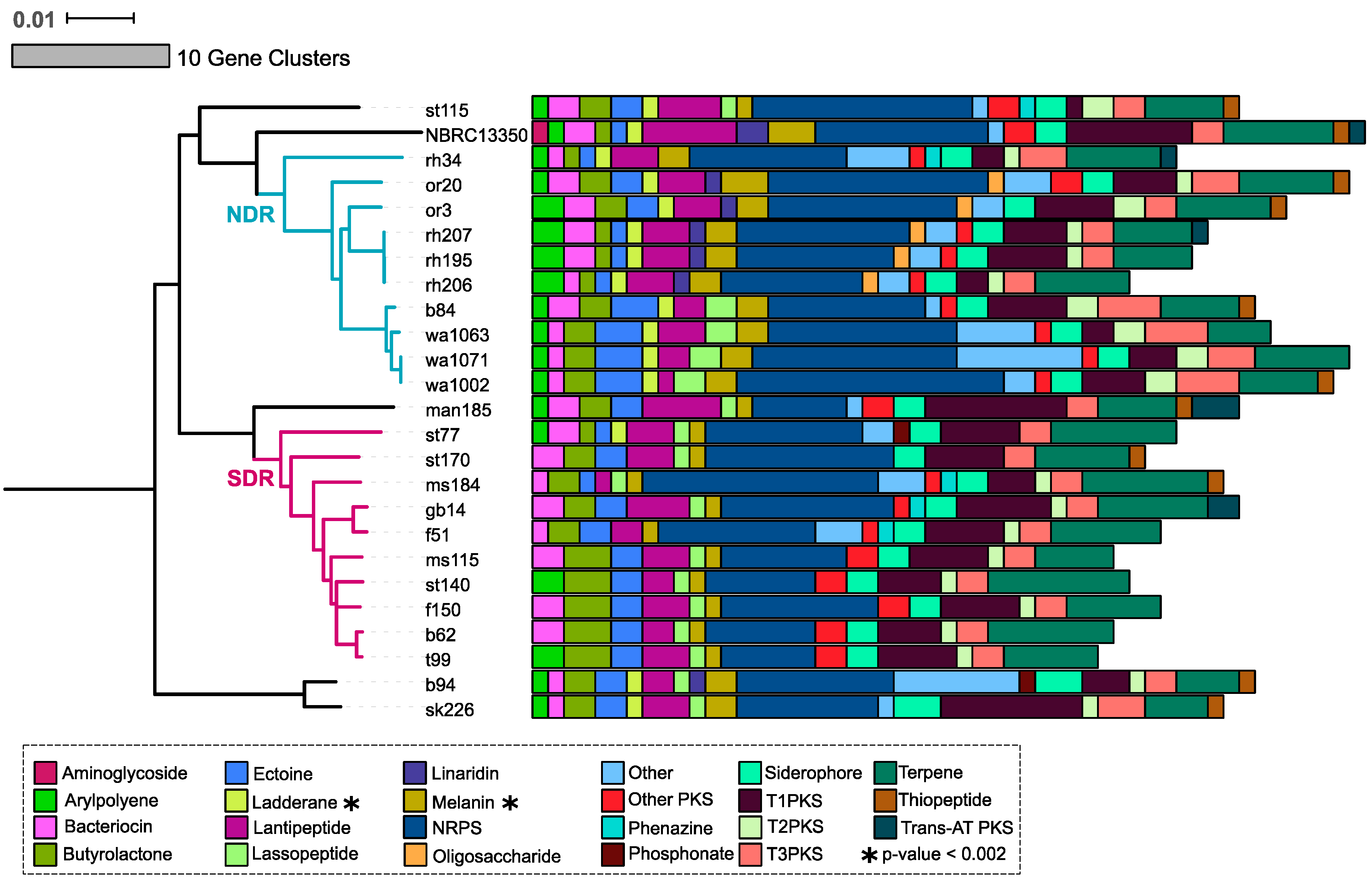
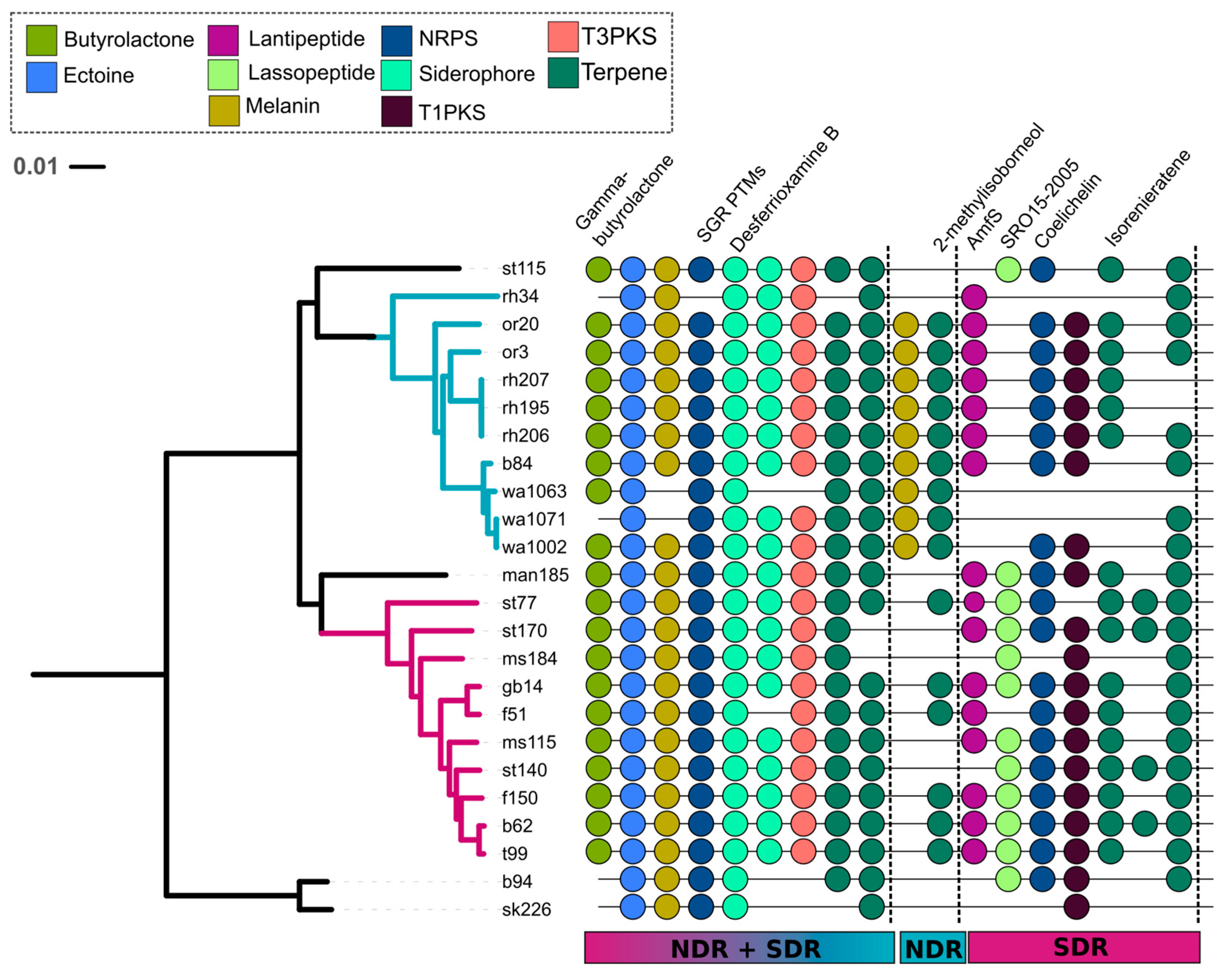
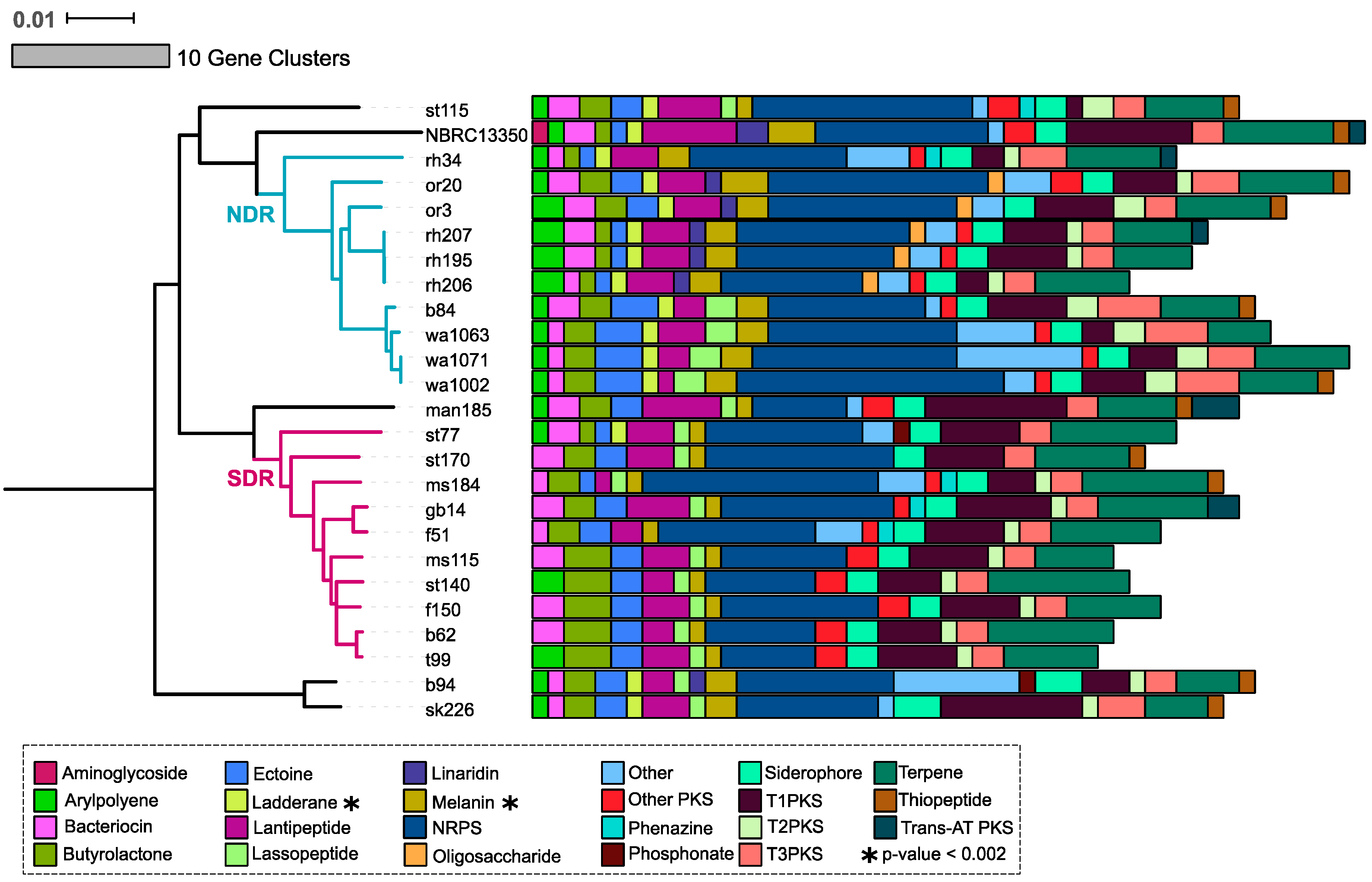
2.2. Secondary Metabolite Biosynthetic Gene Cluster (SMGC) Identification and Classification
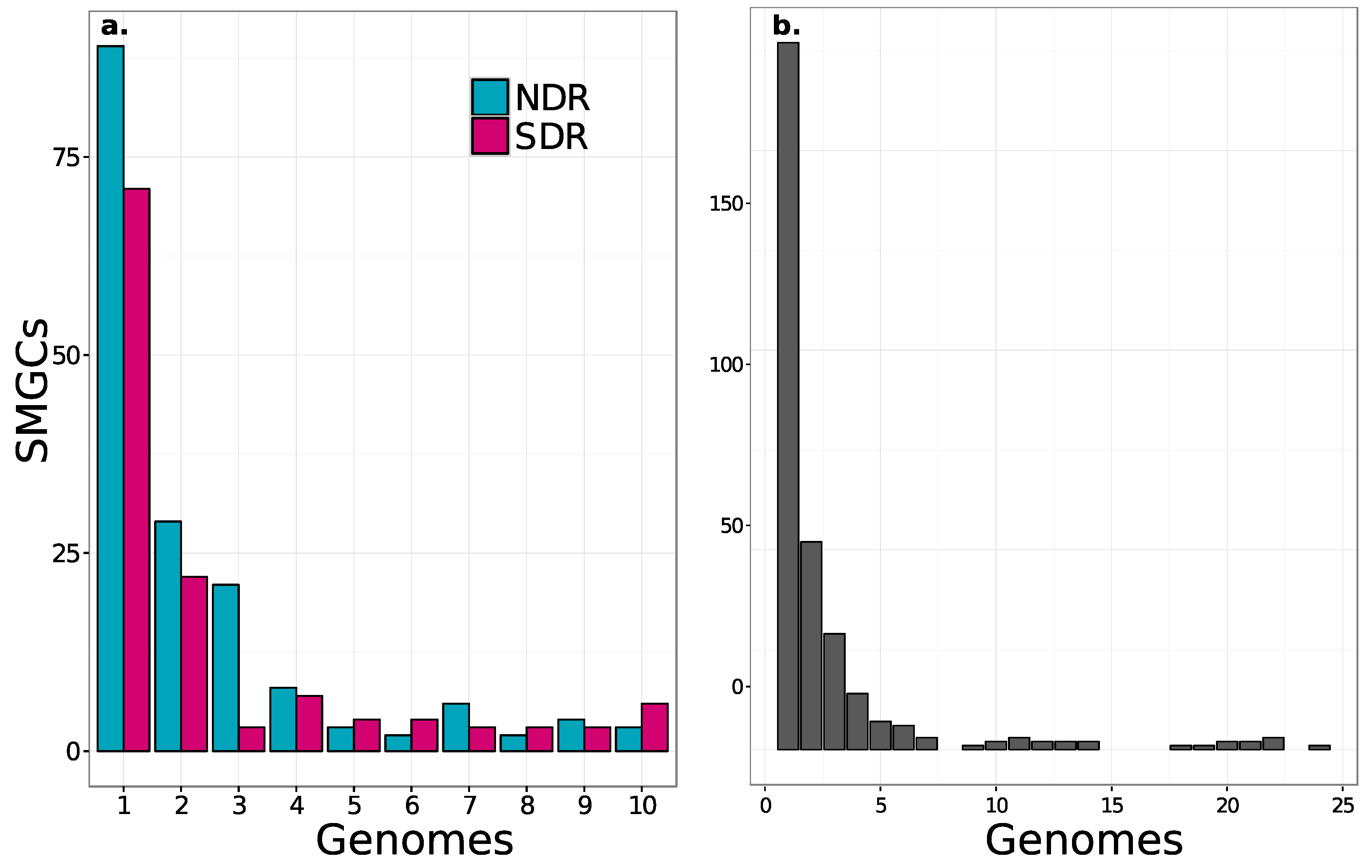
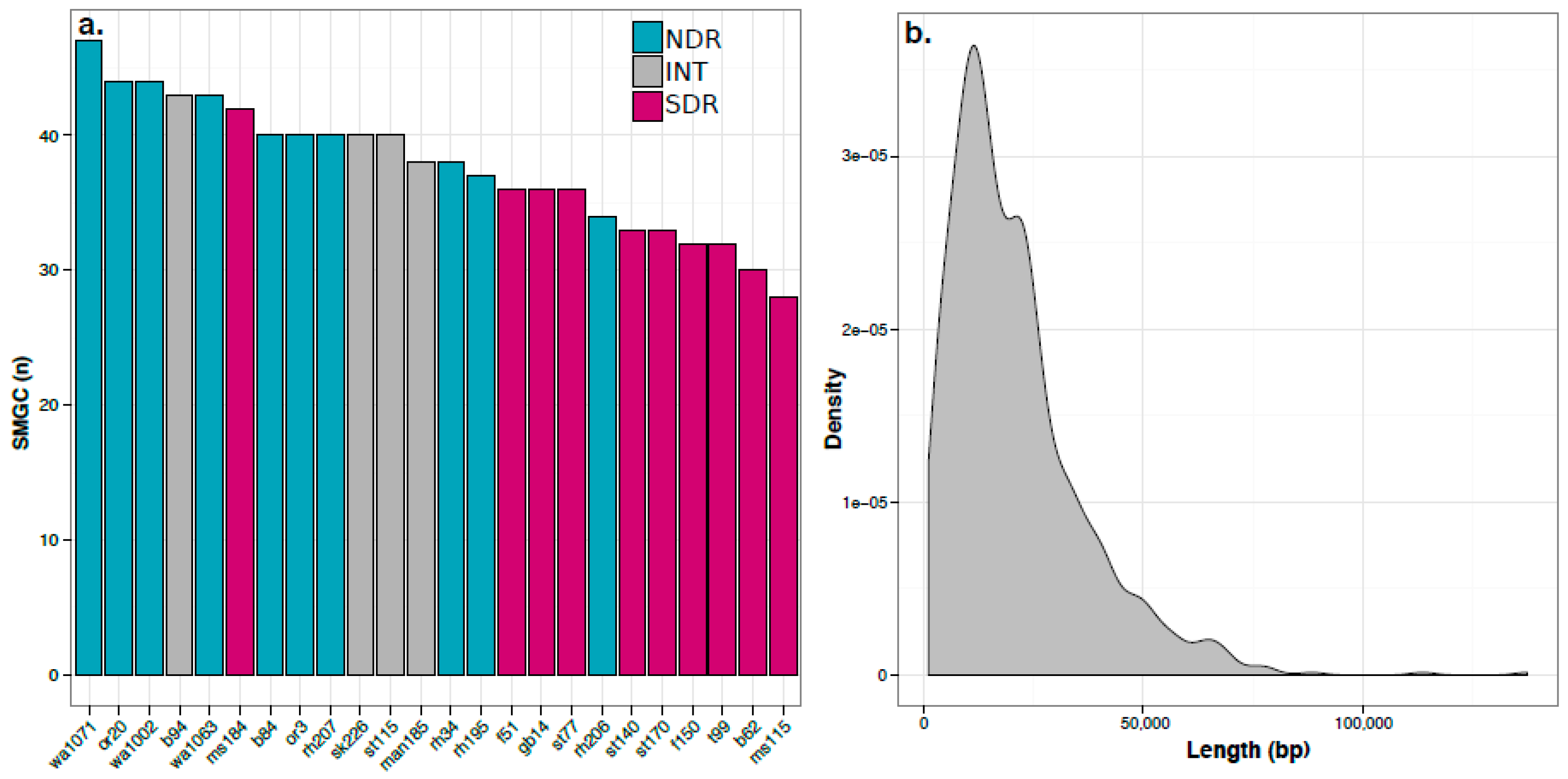
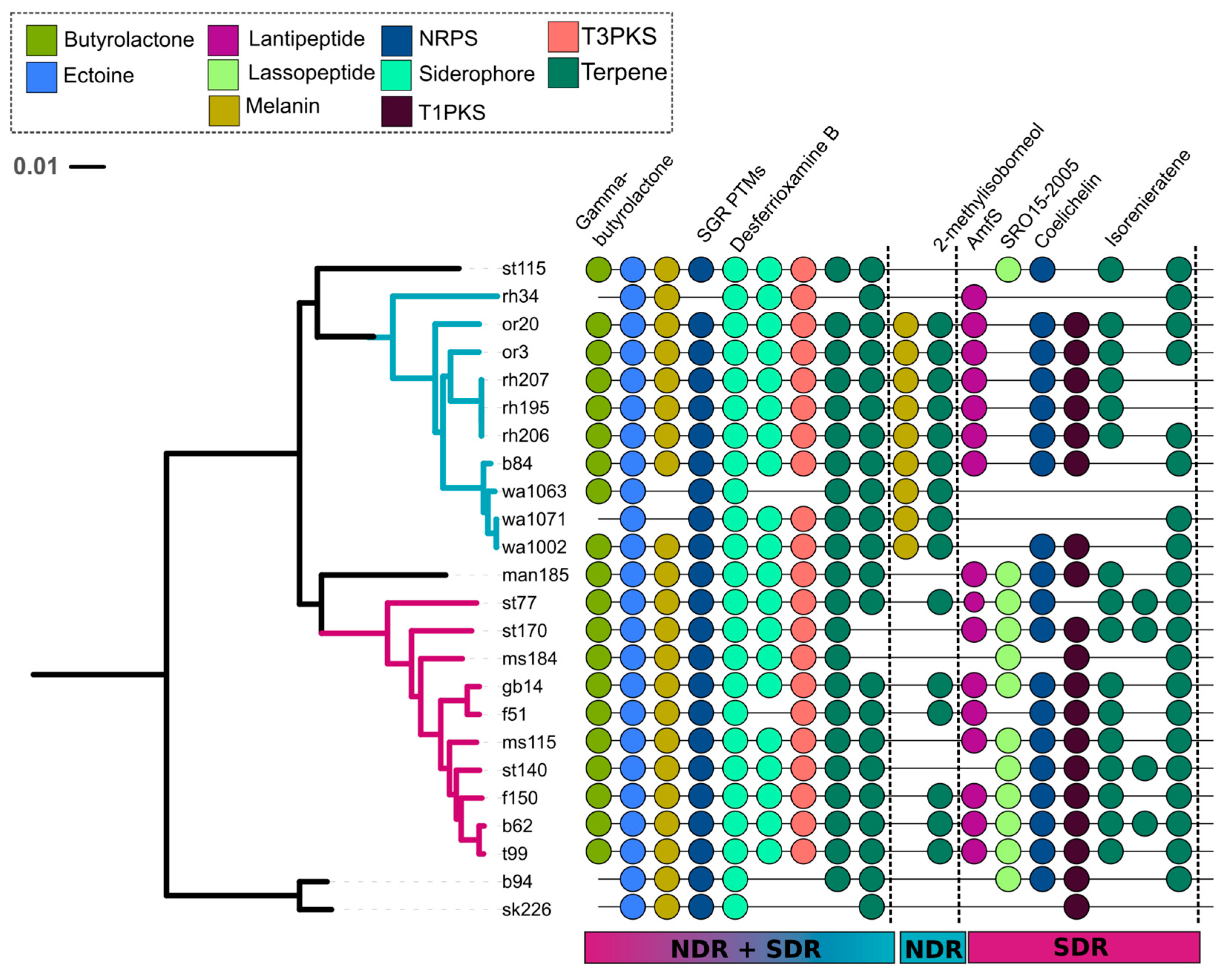
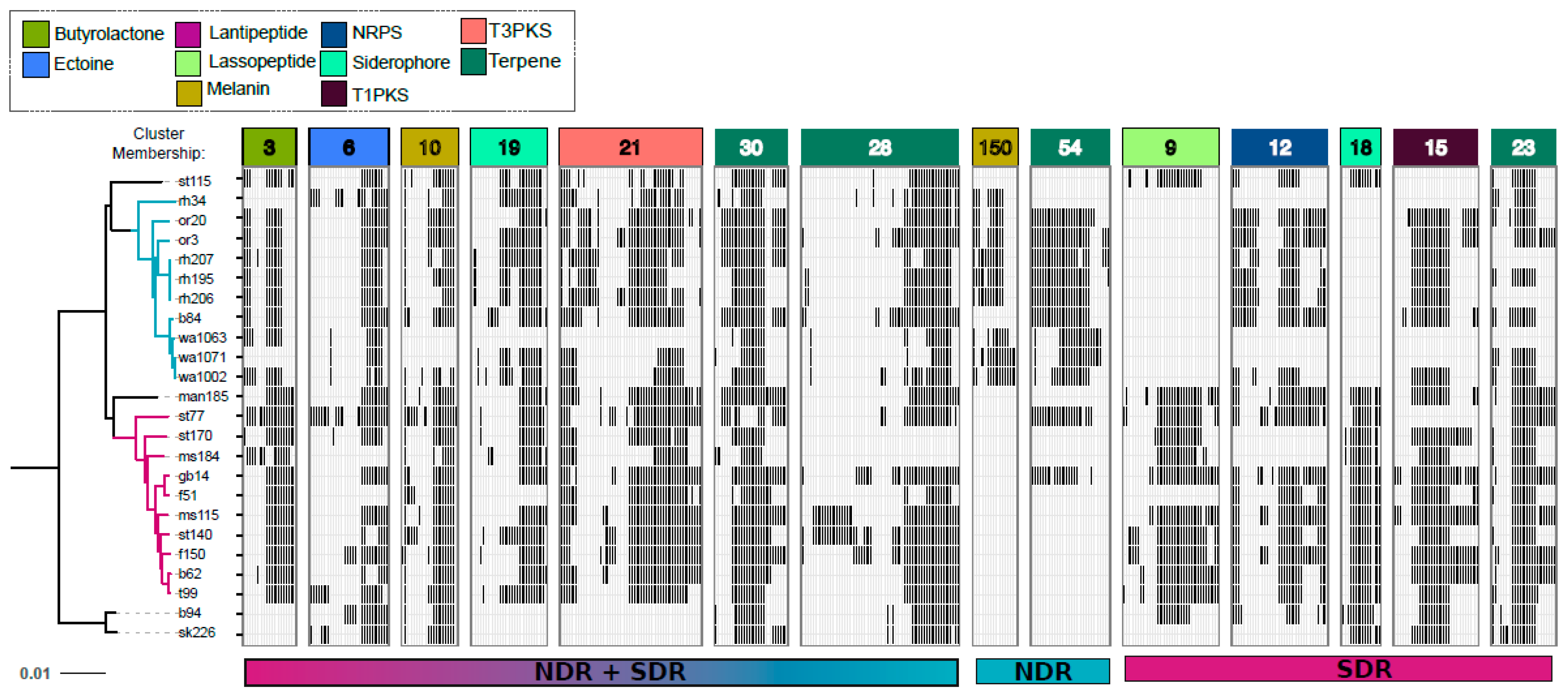
2.3. Core and Accessory SMGCs of Streptomyces Sister-Taxa
2.4. Evolutionary Dynamics of Core and Accessory SMGCs
3. Materials and Methods
3.1. Streptomyces Isolation and DNA Extraction
3.2. Whole Genome Sequencing, Assembly, and Annotation
3.3. Phylogenetic Reconstruction
3.4. Secondary Metabolite Biosynthetic Gene Cluster (SMGC) Identification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kossel, A. Ueber die chemische zusammensetzung der zelle. Arch. Physiol. 1891, 4, 181–186. [Google Scholar]
- Doroghazi, J.R.; Metcalf, W.W. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genom. 2013, 14, 611. [Google Scholar] [CrossRef] [PubMed]
- Karlovsky, P. Secondary Metabolites in Soil Ecology; Springer: New York, NY, USA, 2008; pp. 1–19. [Google Scholar]
- Davies, J. Where have all the antibiotics gone? Can. J. Infect. Dis. Med. Microbiol. 2006, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Charter, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Bérdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Watve, M.; Tickoo, R.; Jog, M.; Bhole, B. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Wright, G.D. An ecological perspective of microbial secondary metabolism. Curr. Opin. Biotechnol. 2011, 22, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Vaz Jauri, P.; Bakker, M.G.; Salomon, C.E.; Kinkel, L.L. Subinhibitory antibiotic concentrations mediate nutrient use and competition among soil Streptomyces. PLoS ONE 2013, 8, e81064. [Google Scholar] [CrossRef] [PubMed]
- Traxler, M.F.; Kolter, R. Natural products in soil microbe interactions and evolution. Nat. Prod. Rep. 2015, 32, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, D.A. Soil to genomics: The Streptomyces chromosome. Annu. Rev. Genet. 2006, 40, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, P. The family Streptomycetaceae Part I: Taxonomy. In The Prokaryotes; Springer: New York, NY, USA, 2006; pp. 538–604. [Google Scholar]
- Van der Meij, A.; Worsley, S.F.; Hutchings, M.I.; van Wezel, G.P. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol. Rev. 2017, 41, 392–416. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kazempour, D.; Fischbach, M.A.; Breitling, R.; Takano, E.; Weber, T. AntiSMASH 2.0—A versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41, W204–W212. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Williams, P.G.; Oh, D.-C.; Zeigler, L.; Fenical, W. Species-specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl. Environ. Microbiol. 2007, 73, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millán-Aguiñaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA 2014, 111, E1130–E1139. [Google Scholar] [CrossRef] [PubMed]
- Wawrik, B.; Kerkhof, L.; Zylstra, G.J.; Kukor, J.J. Identification of unique type II polyketide synthase genes in soil. Appl. Environ. Microbiol. 2005, 71, 2232–2238. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Brady, S.F. Chemical-biogeographic survey of secondary metabolism in soil. Proc. Natl. Acad. Sci. USA 2014, 111, 3757–3762. [Google Scholar] [CrossRef] [PubMed]
- Davelos, A.L.; Kinkel, L.L.; Samac, D.A. Spatial variation in frequency and intensity of antibiotic interactions among Streptomycetes from prairie soil. Appl. Environ. Microbiol. 2004, 70, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Davelos Baines, A.L.; Xiao, K.; Kinkel, L.L. Lack of correspondence between genetic and phenotypic groups amongst soil-borne Streptomycetes. FEMS Microbiol. Ecol. 2007, 59, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Guimarães, D.O.; de Frias, U.A.; Pupo, M.T.; Seepe, P.; Feng, Z.; Brady, S.F. Global biogeographic sampling of bacterial secondary metabolism. Elife 2015, 4, e05048. [Google Scholar] [CrossRef] [PubMed]
- Wawrik, B.; Kutliev, D.; Abdivasievna, U.A.; Kukor, J.J.; Zylstra, G.J.; Kerkhof, L. Biogeography of actinomycete communities and type II polyketide synthase genes in soils collected in New Jersey and Central Asia. Appl. Environ. Microbiol. 2007, 73, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.B.; Kallifidas, D.; Kim, J.H.; Charlop-Powers, Z.; Feng, Z.; Brady, S.F. Natural product biosynthetic gene diversity in geographically distinct soil microbiomes. Appl. Environ. Microbiol. 2012, 78, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Choudoir, M.J.; Campbell, A.N.; Buckley, D.H. Grappling with Proteus: Population level approaches to understanding microbial diversity. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Andam, C.P.; Doroghazi, J.R.; Campbell, A.N.; Kelly, P.J.; Choudoir, M.J.; Buckley, D.H. A Latitudinal diversity gradient in terrestrial bacteria of the genus Streptomyces. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Choudoir, M.J.; Doroghazi, J.R.; Buckley, D.H. Latitude delineates patterns of biogeography in terrestrial Streptomyces. Environ. Microbiol. 2016, 18, 4931–4945. [Google Scholar] [CrossRef] [PubMed]
- Choudoir, M.J.; Buckley, D.H. Phylogenetic conservatism of thermal traits explains dispersal limitation and genomic differentiation of Streptomyces sister-taxa. ISME J. 2018. under review. [Google Scholar]
- Rong, X.; Huang, Y. Taxonomic evaluation of the Streptomyces griseus clade using multilocus sequence analysis and DNA-DNA hybridization, with proposal to combine 29 species and three subspecies as 11 genomic species. Int. J. Syst. Evol. Microbiol. 2010, 60, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Ramette, A.; Tiedje, J.M. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. AntiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Ōmura, S.; Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Takahashi, C.; Shinose, M.; Takahashi, Y.; Horikawa, H.; Nakazawa, H.; Osonoe, T.; et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: Deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. USA 2001, 98, 12215–12220. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Ishikawa, J.; Hara, H.; Suzuki, H.; Ikenoya, M.; Ikeda, H.; Yamashita, A.; Hattori, M.; Horinouchi, S. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 2008, 190, 4050–4060. [Google Scholar] [CrossRef] [PubMed]
- Aigle, B.; Lautru, S.; Spiteller, D.; Dickschat, J.S.; Challis, G.L.; Leblond, P.; Pernodet, J.L. Genome mining of Streptomyces ambofaciens. J. Ind. Microbiol. Biotechnol. 2014, 41, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Seipke, R.F. Strain-level diversity of secondary metabolism in Streptomyces albus. PLoS ONE 2015, 10, e0116457. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Sánchez, C.; Shen, B. Hybrid peptide–polyketide natural products: Biosynthesis and prospects toward engineering novel molecules. Metab. Eng. 2001, 3, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Olano, C.; García, I.; González, A.; Rodriguez, M.; Rozas, D.; Rubio, J.; Sánchez-Hidalgo, M.; Braña, A.F.; Méndez, C.; Salas, J.A. Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microb. Biotechnol. 2014, 7, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Claesen, J.; Bibb, M.J. Biosynthesis and regulation of grisemycin, a new member of the linaridin family of ribosomally synthesized peptides produced by Streptomyces griseus IFO 13350. J. Bacteriol. 2011, 193, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Zetterström, R. Selman, A. Waksman (1888–1973) Nobel Prize in 1952 for the discovery of streptomycin, the first antibiotic effective against tuberculosis. Acta Paediatr. 2007, 96, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Waksman, S.A.; Reilly, H.C.; Johnstone, D.B. Isolation of streptomycin-producing strains of Streptomyces griseus. J. Bacteriol. 1946, 52, 393–397. [Google Scholar] [PubMed]
- Hotta, K.; Ishikawa, J. Strain- and species-specific distribution of the streptomycin gene cluster and kan-related sequences in Streptomyces griseus. J. Antibiot. 1988, 41, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Lefébure, T.; Bitar, P.D.P.; Suzuki, H.; Stanhope, M.J. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol. Evol. 2010, 2, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Imbert, M.; Béchet, M.; Blondeau, R. Comparison of the main siderophores produced by some species of Streptomyces. Curr. Microbiol. 1995, 31, 129–133. [Google Scholar] [CrossRef]
- Roberts, A.A.; Schultz, A.W.; Kersten, R.D.; Dorrestein, P.C.; Moore, B.S. Iron acquisition in the marine actinomycete genus Salinispora is controlled by the desferrioxamine family of siderophores. FEMS Microbiol. Lett. 2012, 335, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Antony-Babu, S.; Stien, D.; Eparvier, V.; Parrot, D.; Tomasi, S.; Suzuki, M.T. Multiple Streptomyces species with distinct secondary metabolomes have identical 16S rRNA gene sequences. Sci. Rep. 2017, 7, 11089. [Google Scholar] [CrossRef] [PubMed]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Marri, P.R.; Hao, W.; Golding, G.B. Gene gain and gene loss in Streptococcus: Is it driven by habitat? Mol. Biol. Evol. 2006, 23, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Reno, M.L.; Held, N.L.; Fields, C.J.; Burke, P.V.; Whitaker, R.J. Biogeography of the Sulfolobus islandicus pan-genome. Proc. Natl. Acad. Sci. USA 2009, 106, 8605–8610. [Google Scholar] [CrossRef] [PubMed]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Homma, K.; Fukuchi, S.; Nakamura, Y.; Gojobori, T.; Nishikawa, K. Gene cluster analysis method identifies horizontally transferred genes with high reliability and indicates that they provide the main mechanism of operon gain in 8 species of γ-Proteobacteria. Mol. Biol. Evol. 2006, 24, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Penn, K.; Jenkins, C.; Nett, M.; Udwary, D.W.; Gontang, E.A.; McGlinchey, R.P.; Foster, B.; Lapidus, A.; Podell, S.; Allen, E.E.; et al. Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. ISME J. 2009, 3, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, E.; Hershberg, R. Gene loss dominates as a source of genetic variation within clonal pathogenic bacterial species. Genome Biol. Evol. 2015, 7, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Choudoir, M.J.; Panke-Buisse, K.; Andam, C.P.; Buckley, D.H. Genome surfing as driver of microbial genomic diversity. Trends Microbiol. 2017, 25, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Buckley, D.H. Widespread homologous recombination within and between Streptomyces species. ISME J. 2010, 4, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Tritt, A.; Eisen, J.A.; Facciotti, M.T.; Darling, A.E. An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 2012, 7, e42304. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.N.; Henriksen, J.R.; Metcalf, W.W.; Whitaker, R.J.; Price, N.D. ITEP: An integrated toolkit for exploration of microbial pan-genomes. BMC Genom. 2014, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. In Lectures on Mathematics in the Life Sciences; American Mathematical Society: Providence, RI, USA, 1986; Volume 17, pp. 57–86. [Google Scholar]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J.; Renner, S. A rapid bootstrap algorithm for the RAxML Web Servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Daily, J. Parasail: SIMD C library for global, semi-global, and local pairwise sequence alignments. BMC Bioinform. 2016, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. 2006. Available online: http://www.interjournal.org/manuscript_abstract.php?361100992 (accessed on 6 February 2018).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choudoir, M.J.; Pepe-Ranney, C.; Buckley, D.H. Diversification of Secondary Metabolite Biosynthetic Gene Clusters Coincides with Lineage Divergence in Streptomyces. Antibiotics 2018, 7, 12. https://doi.org/10.3390/antibiotics7010012
Choudoir MJ, Pepe-Ranney C, Buckley DH. Diversification of Secondary Metabolite Biosynthetic Gene Clusters Coincides with Lineage Divergence in Streptomyces. Antibiotics. 2018; 7(1):12. https://doi.org/10.3390/antibiotics7010012
Chicago/Turabian StyleChoudoir, Mallory J., Charles Pepe-Ranney, and Daniel H. Buckley. 2018. "Diversification of Secondary Metabolite Biosynthetic Gene Clusters Coincides with Lineage Divergence in Streptomyces" Antibiotics 7, no. 1: 12. https://doi.org/10.3390/antibiotics7010012
APA StyleChoudoir, M. J., Pepe-Ranney, C., & Buckley, D. H. (2018). Diversification of Secondary Metabolite Biosynthetic Gene Clusters Coincides with Lineage Divergence in Streptomyces. Antibiotics, 7(1), 12. https://doi.org/10.3390/antibiotics7010012