
NETs and CF Lung Disease: Current Status and Future Prospects

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Investigating Innate Immunity in CF Using Animal Models
3. Airways Surface Liquid (ASL) Antimicrobial Peptides in CF
4. Neutrophils and Antimicrobial Defense in CF
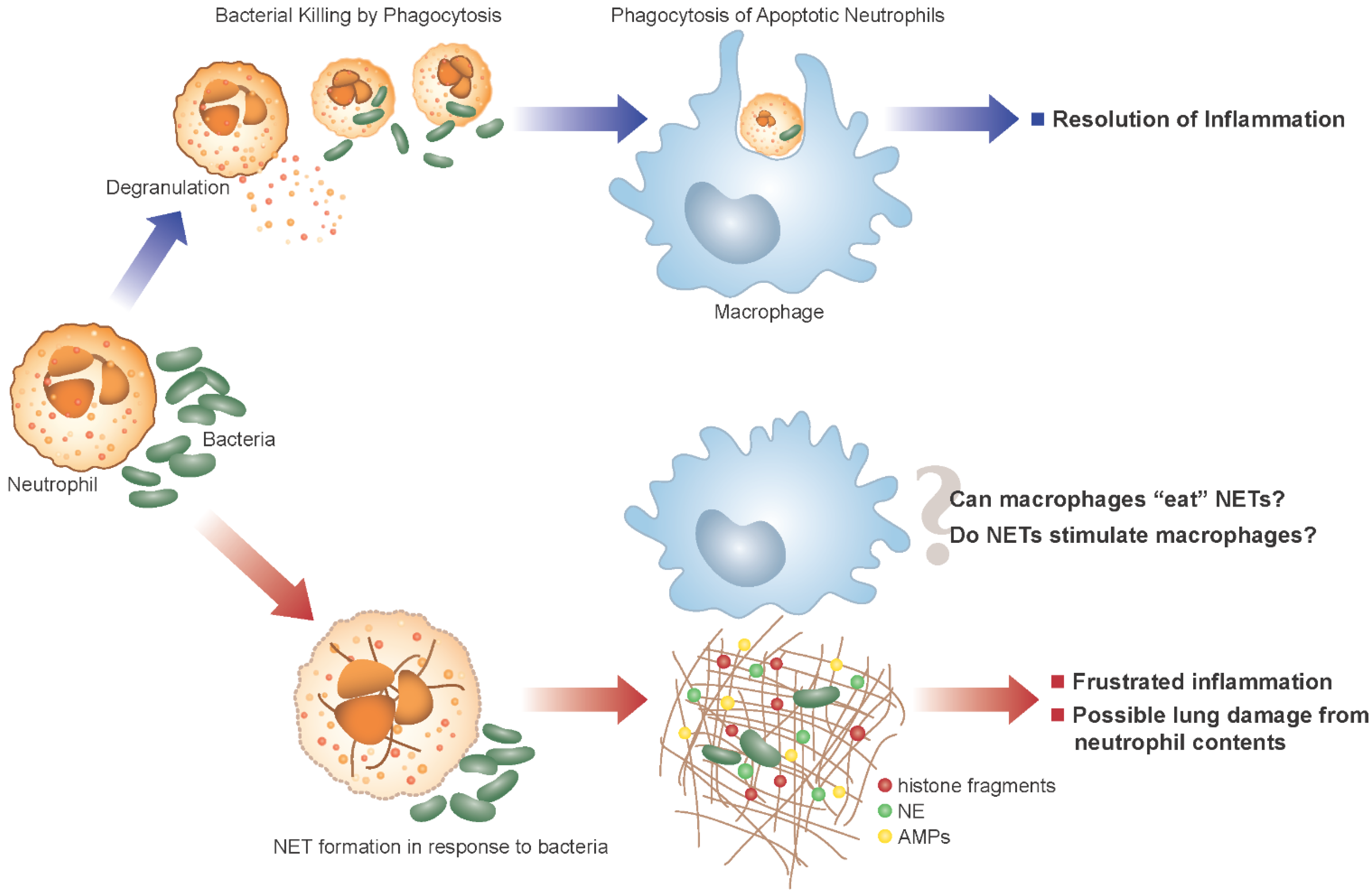
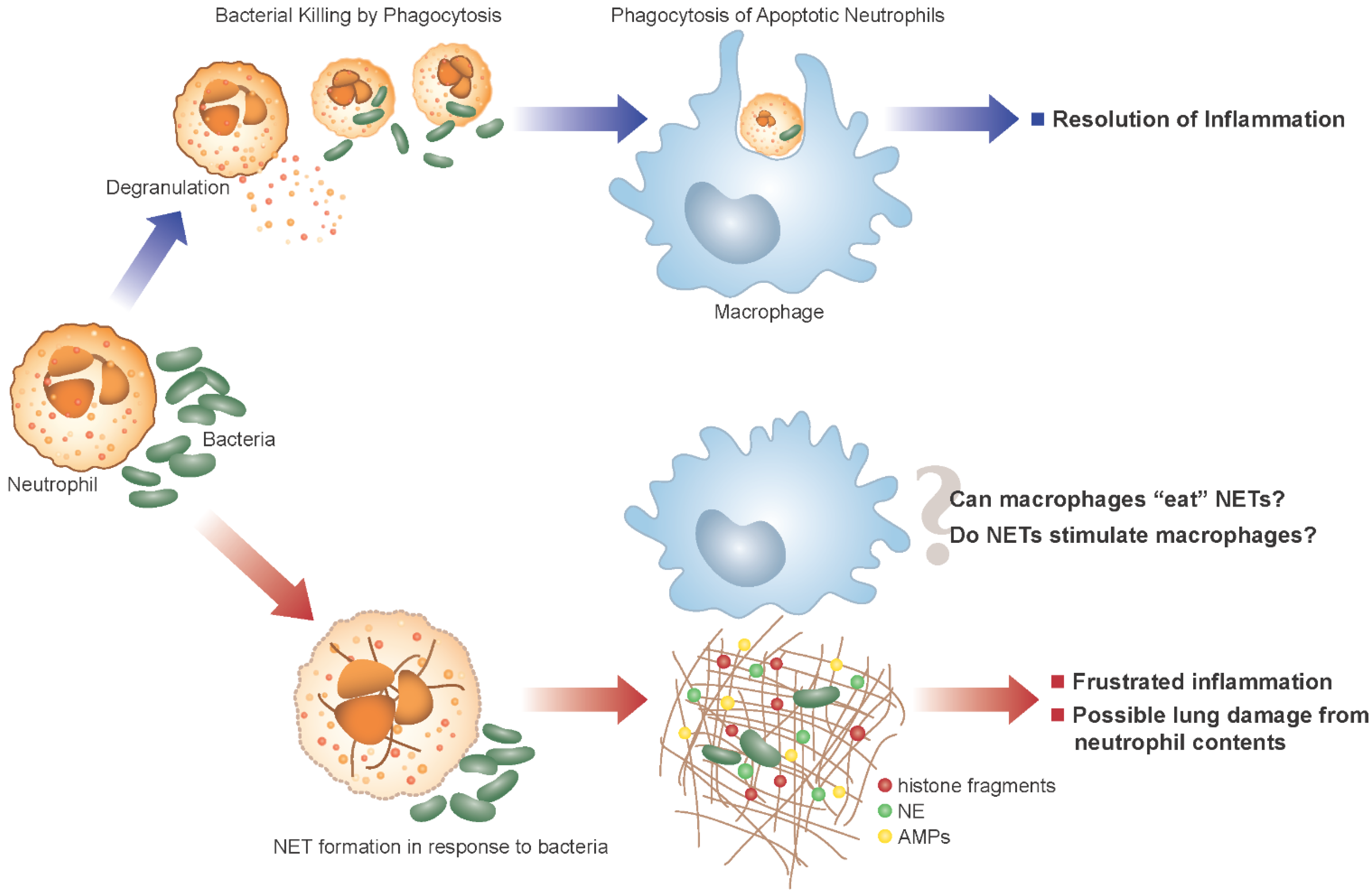
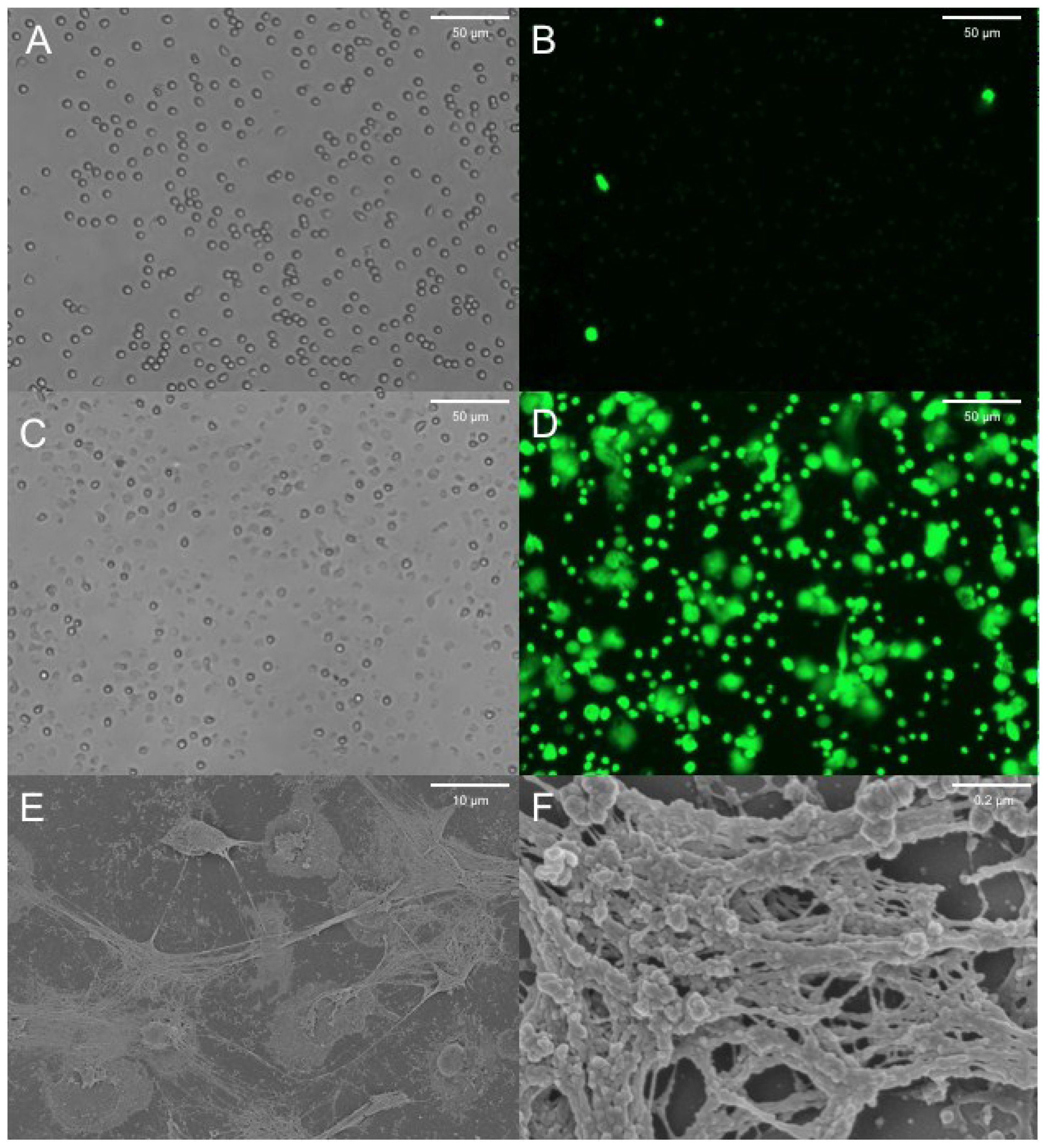
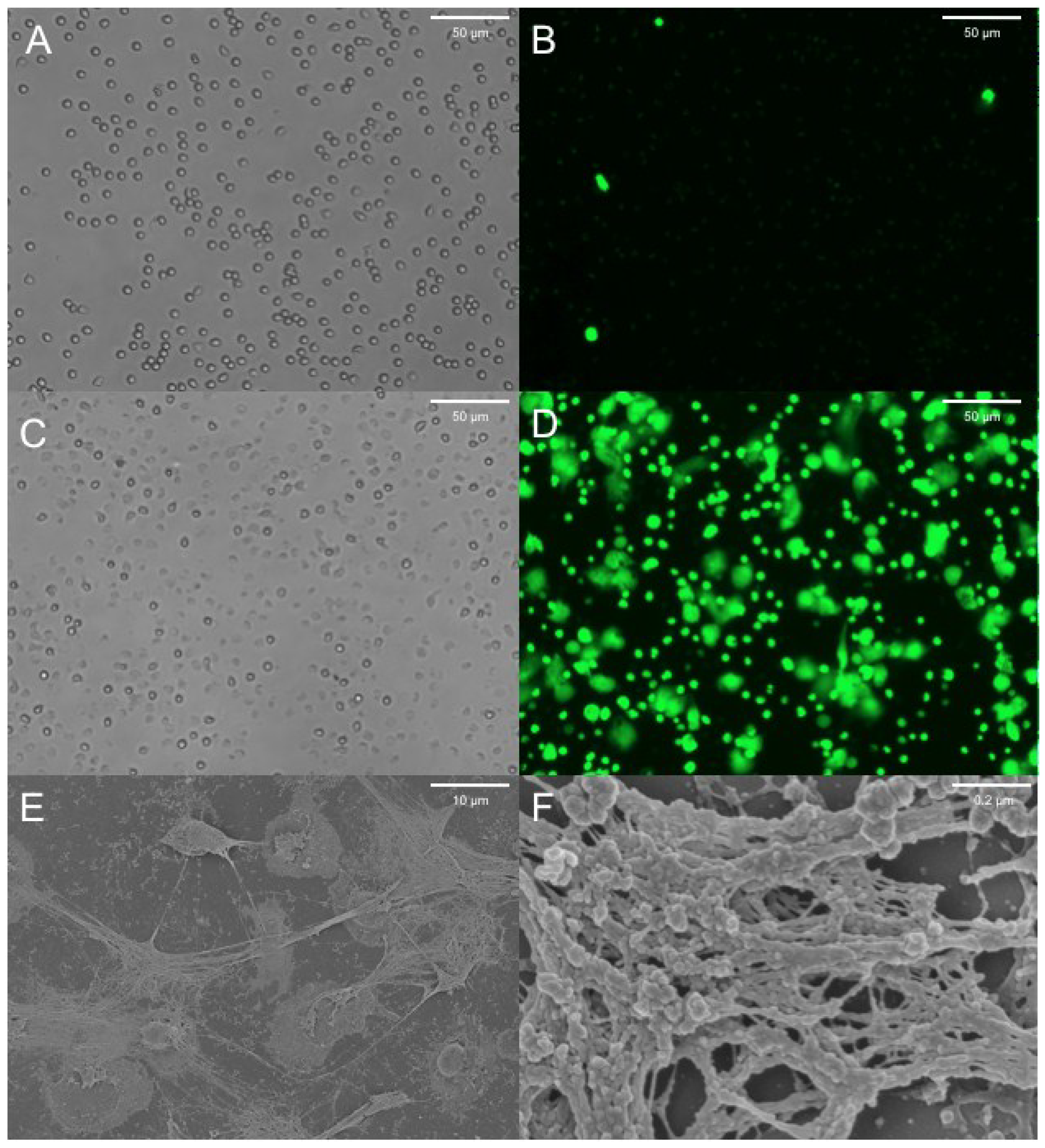
5. Neutrophil Extracellular Traps and CF
6. NET Formation and Killing in Response to Bacteria
7. Potential Damaging Consequences of NETs in CF
8. Outstanding Questions in CF
9. NETs Research in the CF Pig Model
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morgan, W.J.; Butler, S.M.; Johnson, C.A.; Colin, A.A.; FitzSimmons, S.C.; Geller, D.E.; Konstan, M.W.; Light, M.J.; Rabin, H.R.; Regelmann, W.E.; et al. Epidemiologic study of cystic fibrosis: Design and implementation of a prospective, multicenter, observational study of patients with cystic fibrosis in the U.S. and Canada. Pediatr. Pulmonol. 1999, 28, 231–241. [Google Scholar]
- Bartlett, J.A.; Fischer, A.J.; McCray, P.B., Jr. Innate immune functions of the airway epithelium. Contrib. Microbiol. 2008, 15, 147–163. [Google Scholar]
- Abman, S.H.; Ogle, J.W.; Harbeck, R.J.; Butler-Simon, N.; Hammond, K.B.; Accurso, F.J. Early bacteriologic, immunologic, and clinical courses of young infants with cystic fibrosis identified by neonatal screening. J. Pediatr. 1991, 119, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.J.; Dorin, J.R. The CF mouse: An important tool for studying cystic fibrosis. Expert Rev. Mol. Med. 2001, 2001, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the cftr gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2010, 2, 29ra31. [Google Scholar] [PubMed]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Cystic fibrosis. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Hoegger, M.J.; Awadalla, M.; Namati, E.; Itani, O.A.; Fischer, A.J.; Tucker, A.J.; Adam, R.J.; McLennan, G.; Hoffman, E.A.; Stoltz, D.A.; et al. Assessing mucociliary transport of single particles in vivo shows variable speed and preference for the ventral trachea in newborn pigs. Proc. Natl. Acad. Sci. USA 2014, 111, 2355–2360. [Google Scholar] [CrossRef]
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Invest. 2010, 120, 3149–3160. [Google Scholar] [CrossRef]
- Berkebile, A.R.; McCray, P.B., Jr. Effects of airway surface liquid ph on host defense in cystic fibrosis. Int. J. Biochem. Cell. Biol. 2014, 52, 124–129. [Google Scholar] [CrossRef]
- Singh, P.K.; Tack, B.F.; McCray, P.B., Jr.; Welsh, M.J. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L799–L805. [Google Scholar]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; OʼNeal, W.K.; Boucher, R.C. Abnormal surface liquid ph regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A.S. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.; MacGregor, G.; Davis, M.; Innes, J.A.; Greening, A.P. Airways in cystic fibrosis are acidified: Detection by exhaled breath condensate. Thorax 2002, 57, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface PH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Birrer, P.; McElvaney, N.G.; Rudeberg, A.; Sommer, C.W.; Liechti-Gallati, S.; Kraemer, R.; Hubbard, R.; Crystal, R.G. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1994, 150, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar] [PubMed]
- Konstan, M.W.; Hilliard, K.A.; Norvell, T.M.; Berger, M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am. J. Respir. Crit. Care Med. 1994, 150, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Brennan, S.; Gangell, C.; de Klerk, N.; Murray, C.; Mott, L.; Stick, S.M.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 2009, 180, 146–152. [Google Scholar] [CrossRef]
- Martino, M.E.; Olsen, J.C.; Fulcher, N.B.; Wolfgang, M.C.; OʼNeal, W.K.; Ribeiro, C.M. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 2009, 284, 14904–14913. [Google Scholar] [CrossRef]
- Weber, A.J.; Soong, G.; Bryan, R.; Saba, S.; Prince, A. Activation of NF-kappaB in airway epithelial cells is dependent on cftr trafficking and Cl− channel function. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L71–L78. [Google Scholar] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Bonvillain, R.W.; Valentine, V.G.; Lombard, G.A.; LaPlace, S.G.; Nauseef, W.M.; Wang, G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J. Leukoc. Biol. 2008, 83, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; DeLeo, F.R. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 2009, 43, 25–61. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Dransfield, I.; Gregory, C.; Haslett, C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002, 2, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Sontag, M.K.; Accurso, F.J. Relationship between antimicrobial proteins and airway inflammation and infection in cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Bergsson, G.; Reeves, E.P.; McNally, P.; Chotirmall, S.H.; Greene, C.M.; Greally, P.; Murphy, P.; OʼNeill, S.J.; McElvaney, N.G. Ll-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J. Immunol. 2009, 183, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Rogan, M.P.; Taggart, C.C.; Greene, C.M.; Murphy, P.G.; OʼNeill, S.J.; McElvaney, N.G. Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J. Infect. Dis. 2004, 190, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.J.; Weldon, S.; Taggart, C.C. Antiproteases as therapeutics to target inflammation in cystic fibrosis. Open Respir. Med. J. 2010, 4, 20–31. [Google Scholar] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; DʼOrtona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has netosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 2012, 11, 84–92. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLOS ONE 2011, 6, e28526. [Google Scholar] [CrossRef] [PubMed]
- Young, R.L.; Malcolm, K.C.; Kret, J.E.; Caceres, S.M.; Poch, K.R.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Randell, S.H.; Vasil, M.L.; et al. Neutrophil extracellular trap (net)-mediated killing of Pseudomonas aeruginosa: Evidence of acquired resistance within the CF airway, independent of CFTR. PLOS ONE 2011, 6, e23637. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. Pad4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during netosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Itakura, A.; McCarty, O.J. Pivotal role for the mtor pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am. J. Physiol. Cell Physiol. 2013, 305, C348–C354. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLOS ONE 2011, 6, e29318. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Touhami, J.; Herzenberg, L.A.; Conrad, C.; Taylor, N.; Battini, J.L.; Sitbon, M.; Tirouvanziam, R. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J. Immunol. 2013, 190, 6043–6050. [Google Scholar] [CrossRef] [PubMed]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Gernez, Y.; Conrad, C.K.; Moss, R.B.; Schrijver, I.; Dunn, C.E.; Davies, Z.A.; Herzenberg, L.A.; Herzenberg, L.A. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2008, 105, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make nets. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. Nets are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PLOS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.V.; Gauthier, A.; Brea, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am. J. Respir Cell Mol. Biol. 2012, 47, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. Net formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Winn, M.; Pang, L.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L.; Rada, B. Release of cystic fibrosis airway inflammatory markers from pseudomonas aeruginosa-stimulated human neutrophils involves nadph oxidase-dependent extracellular DNA trap formation. J. Immunol. 2010, 192, 4728–4738. [Google Scholar] [CrossRef]
- Rada, B.; Jendrysik, M.A.; Pang, L.; Hayes, C.P.; Yoo, D.G.; Park, J.J.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L. Pyocyanin-enhanced neutrophil extracellular trap formation requires the nadph oxidase. PLOS ONE 2013, 8, e54205. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; Dockrell, D.H.; Pattery, T.; Lee, D.G.; Cornelis, P.; Hellewell, P.G.; Whyte, M.K. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J. Immunol. 2005, 174, 3643–3649. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Menegazzi, R.; Decleva, E.; Dri, P. Killing by neutrophil extracellular traps: Fact or folklore? Blood 2012, 119, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 2011, 91, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Gadjeva, M. Does netosis contribute to the bacterial pathoadaptation in cystic fibrosis? Front. Immunol. 2014, 5, e378. [Google Scholar] [CrossRef]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Kockritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Kobayashi, S.D.; Freedman, B.; Dorward, D.W.; DeLeo, F.R. Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J. Immunol. 2013, 191, 6022–6029. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Hodson, M.E. Aerosolized dornase alfa (rhDNase) for therapy of cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, S70–S74. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The pulmozyme study group. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Gullstrand, B.; Jonsen, A.; Nilsson, J.A.; Martin, M.; Blom, A.M.; Bengtsson, A.A. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2013, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.T.; Dyrynda, E.A.; Gray, R.D.; Rossi, A.G.; Smith, V.J. Invertebrate extracellular phagocyte traps show that chromatin is an ancient defence weapon. Nat. Commun. 2014, 5, 4627. [Google Scholar] [CrossRef] [PubMed]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Brea, D.; Meurens, F.; Dubois, A.V.; Gaillard, J.; Chevaleyre, C.; Jourdan, M.L.; Winter, N.; Arbeille, B.; Si-Tahar, M.; Gauthier, F.; et al. The pig as a model for investigating the role of neutrophil serine proteases in human inflammatory lung diseases. Biochem. J. 2012, 447, 363–370. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gray, R.D.; McCullagh, B.N.; McCray, P.B., Jr. NETs and CF Lung Disease: Current Status and Future Prospects. Antibiotics 2015, 4, 62-75. https://doi.org/10.3390/antibiotics4010062
Gray RD, McCullagh BN, McCray PB Jr. NETs and CF Lung Disease: Current Status and Future Prospects. Antibiotics. 2015; 4(1):62-75. https://doi.org/10.3390/antibiotics4010062
Chicago/Turabian StyleGray, Robert D., Brian N. McCullagh, and Paul B. McCray, Jr. 2015. "NETs and CF Lung Disease: Current Status and Future Prospects" Antibiotics 4, no. 1: 62-75. https://doi.org/10.3390/antibiotics4010062
APA StyleGray, R. D., McCullagh, B. N., & McCray, P. B., Jr. (2015). NETs and CF Lung Disease: Current Status and Future Prospects. Antibiotics, 4(1), 62-75. https://doi.org/10.3390/antibiotics4010062