
Augmentation of Cationic Antimicrobial Peptide Production with Histone Deacetylase Inhibitors as a Novel Epigenetic Therapy for Bacterial Infections

Abstract
:1. Antimicrobial Peptides—An Innate Defense Against Microbial Pathogens
2. Pathogens Can Regulate HDAC-Mediated Expression of CAMPs
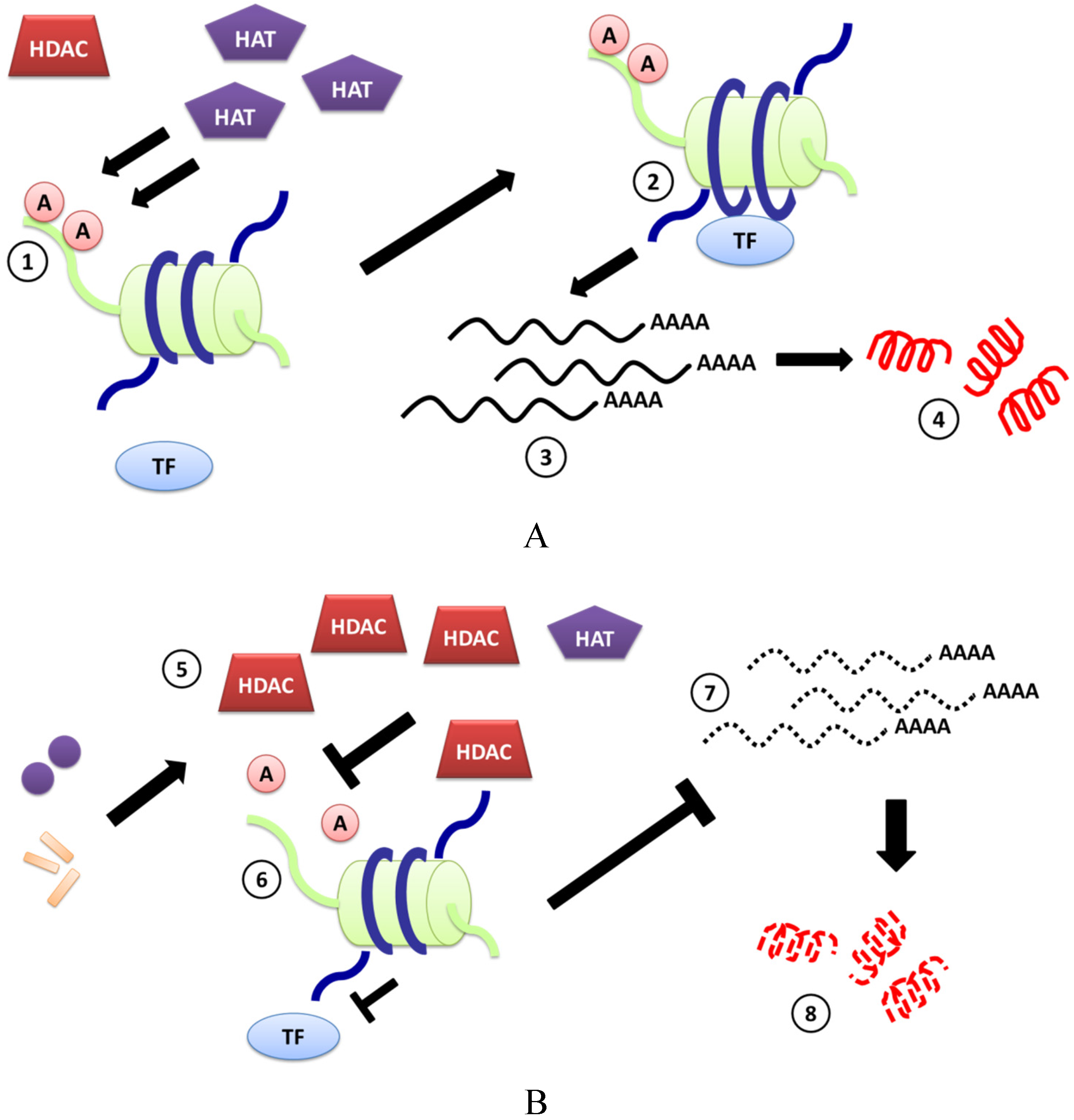
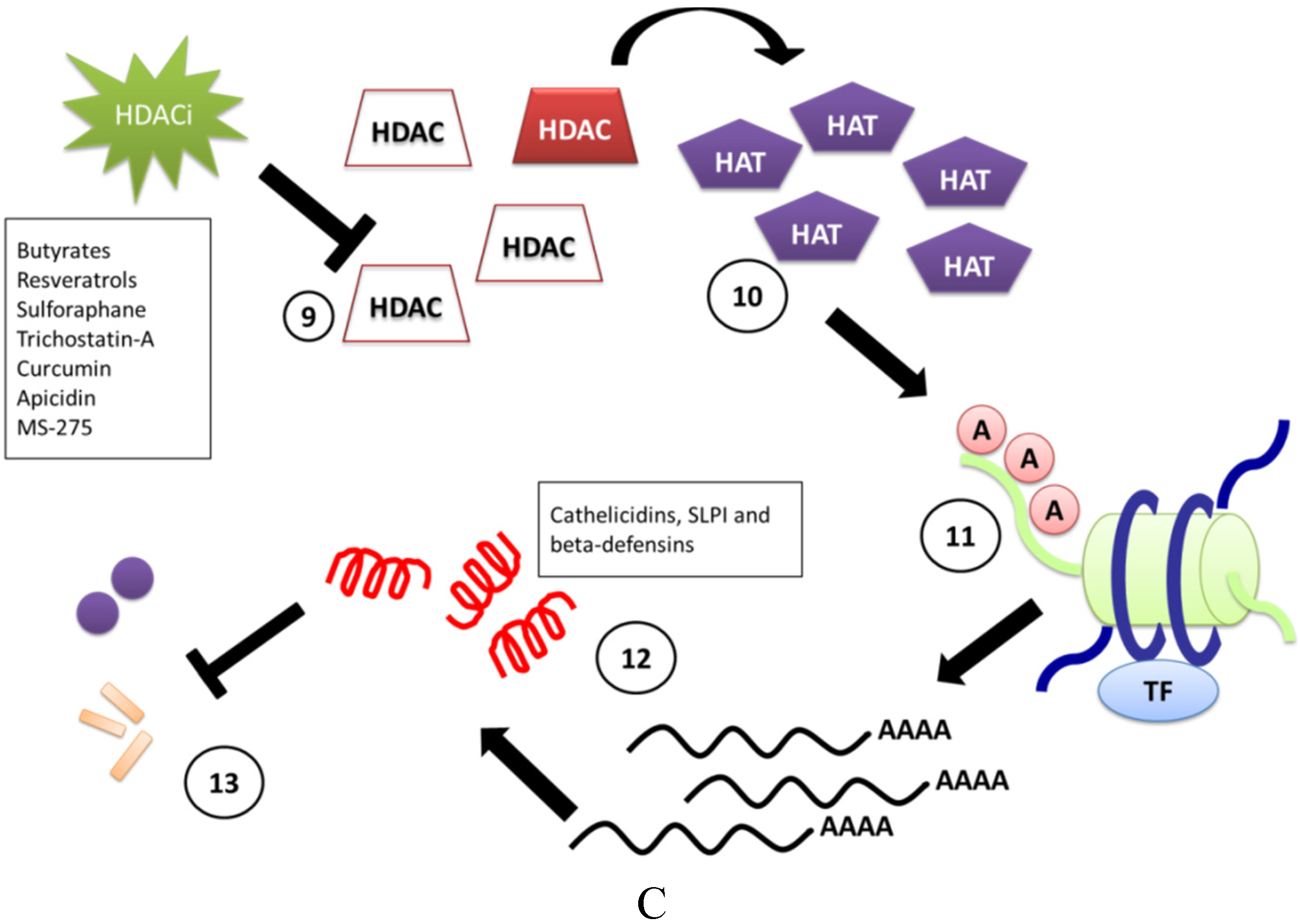
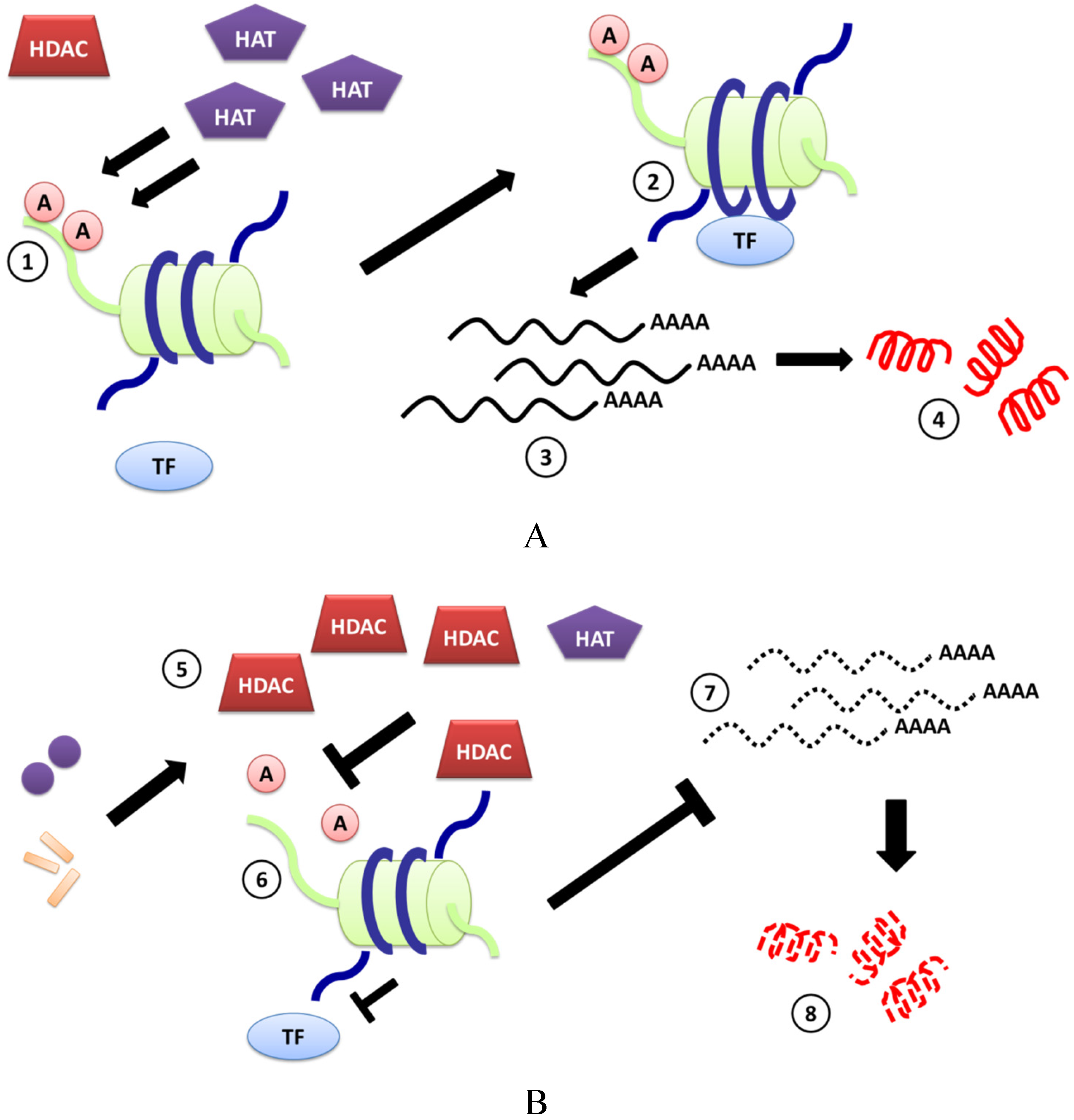
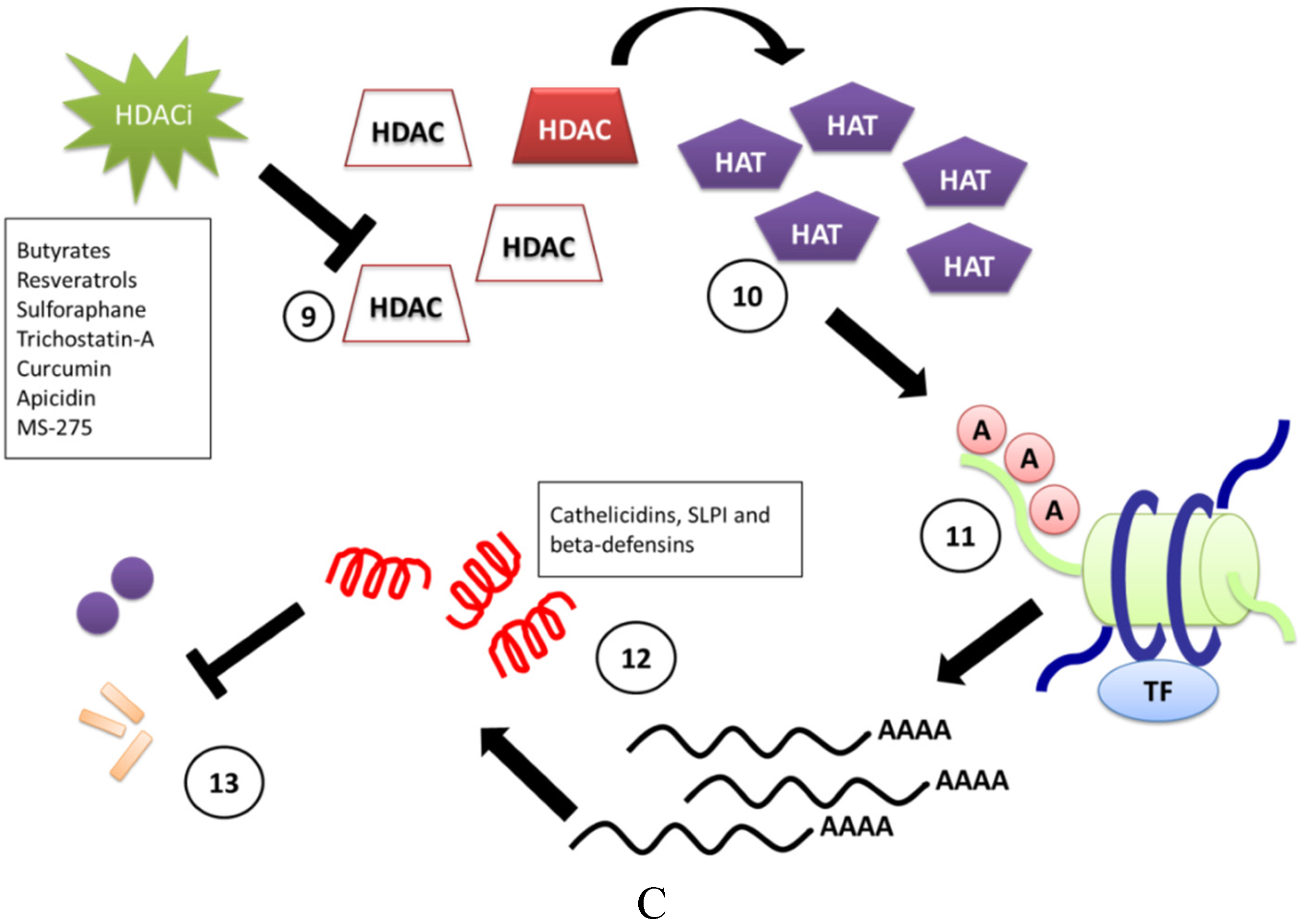
3. Induction of CAMPs by HDACi

{kind=link}
{kind=link}
| HDAC Inhibitor | System Tested | Effect on CAMP mRNA Expression | Reference |
|---|---|---|---|
| Butyrate | Human lung epithelial cell line EBC-1 | Cathelicidin ↑ | [25] |
| Human bronchial epithelial cell line VA10 | Cathelicidin ↑ HBD-1 ↑ | [26] | |
| Human airway epithelial cells NCI-H292 | Cathelicidin ↑ | [27] | |
| Human lung epithelial cell line A549 | HBD-1 ↑ | [17] | |
| Human primary gingival epithelial cells infected with P. gingivalis and F. nucleatum | HBD-2 ↑ | [16] | |
| Human monocyte cell line U937 | HBD-1 ↓ | [26] | |
| Adult patients with shigellosis | Cathelicidin ↑ | [28] | |
| Resveratrol | Human keratinocyte cell line HaCaT | Cathelicidin ↑ | [29] |
| Topical administration in female hairless mice | Cathelicidin ↑ | [30] | |
| Human monocyte cell line U937 | Cathelicidin ↑ | [29] | |
| Pseudomonas aeruginosa-infected A549 cells | HBD-2 ↓ | [31] | |
| Pterostilbene | Human monocyte cell line U937 | Cathelicidin ↑ | [29] |
| Polydatin | Human keratinocyte cell line HaCaT | HBD-2 ↑ | [32] |
| Sulforaphane | Liver tissue from SFN-treated C57BL/6 mice | MBD-10 ↑ | [33] |
| Human intestinal epithelial cell lines Caco-2, HT-29 and SW480 | HBD-2 ↑ | [34] | |
| Mouse monocyte macrophage cell line RAW 264.7 | SLPI ↑ | [35] | |
| Nasal lavage from healthy human adults who ingested SFN-containing broccoli shake homogenate | SLPI ↑ | [36] | |
| Trichostatin-A | Human primary gingival epithelial cells infected with P. gingivalis and F. nucleatum | HBD-2 ↑ | [16] |
| Human lung epithelial cell lines A549 and NCI-H727 | HBD-1 ↑ | [17] | |
| Human airway epithelial cells NCI-H292 | Cathelicidin ↑ | [27] | |
| Curcumin | Human cell lines: U937, HT-29 and HaCaT | Cathelicidin ↑ | [37] |
| Apicidin | Human lung epithelial cell line A549 | HBD-1 ↑ | [17] |
| MS-275 | Human lung epithelial cell line A549 | HBD-1 ↑ | [17] |
4. Butyrates
4.1. Butyrate-Induced Expression of Cathelicidins
4.2. Butyrate-Induced Expression of Beta-Defensins
5. Resveratrol and Structurally-Related Molecules
5.1. Pterostilbene
5.2. Polydatin
6. Sulforaphane
7. Trichostatin A
8. Curcumin
9. Apicidin
10. MS-275
11. Anti-Inflammatory Properties of HDACi
12. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cantas, L.; Shah, S.Q.; Cavaco, L.M.; Manaia, C.M.; Walsh, F.; Popowska, M.; Garelick, H.; Burgmann, H.; Sorum, H. A brief multi-disciplinary review on antimicrobial resistance in medicine and its linkage to the global environmental microbiota. Front. Microbiol. 2013, 4, e96. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial peptides: Old molecules with new ideas. J. Invest. Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.V.; Yedery, R.D.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Johansson, L.; Asp, V.; Plant, L.; Gudmundsson, G.H.; Jonsson, A.B.; Agerberth, B. Neisseria gonorrhoeae downregulates expression of the human antimicrobial peptide LL-37. Cell. Microbiol. 2005, 7, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Kim, Y.S.; Oh, J.E.; Min, B.M.; Choi, Y. Treponema denticola suppresses expression of human {beta}-defensin-3 in gingival epithelial cells through inhibition of the toll-like receptor 2 axis. Infect. Immun. 2010, 78, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Ghosh, S.; Koley, H.; Mukhopadhyay, A.K.; Ramamurthy, T.; Saha, D.R.; Mukhopadhyay, D.; Roychowdhury, S.; Hamabata, T.; Takeda, Y.; et al. Bacterial exotoxins downregulate cathelicidin (hCAP-18/LL-37) and human beta-defensin 1 (HBD-1) expression in the intestinal epithelial cells. Cell. Microbiol. 2008, 10, 2520–2537. [Google Scholar] [CrossRef]
- Draper, D.L.; Landers, D.V.; Krohn, M.A.; Hillier, S.L.; Wiesenfeld, H.C.; Heine, R.P. Levels of vaginal secretory leukocyte protease inhibitor are decreased in women with lower reproductive tract infections. Am. J. Obstet. Gynecol. 2000, 183, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G. Downregulation of bactericidal peptides in enteric infections: A novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 2001, 7, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.A.; Cossart, P. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 2008, 4, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.C.; Barat, N.C.; Trembley, S.J.; Dumler, J.S. Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLOS Pathog. 2009, 5, e1000488. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Chung, W.O. Epigenetic regulation of human beta-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 2011, 4, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Kallsen, K.; Andresen, E.; Heine, H. Histone deacetylase (HDAC) 1 controls the expression of beta defensin 1 in human lung epithelial cells. PLOS ONE 2012, 7, e50000. [Google Scholar] [CrossRef] [PubMed]
- Dashwood, R.H.; Ho, E. Dietary histone deacetylase inhibitors: From cells to mice to man. Semin. Cancer Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003, 2, 151–163. [Google Scholar] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Hideshima, T.; Akiyama, M.; Chauhan, D.; Munshi, N.; Gu, X.; et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: Biological and clinical implications. Proc. Natl. Acad. Sci. USA 2004, 101, 540–545. [Google Scholar] [CrossRef]
- Li, J.; Li, G.; Xu, W. Histone deacetylase inhibitors: An attractive strategy for cancer therapy. Curr. Med. Chem. 2013, 20, 1858–1886. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, P.V.; Ververis, K.; Tang, M.L.; El-Osta, A.; Karagiannis, T.C. Immunomodulatory effects of histone deacetylase inhibitors. Curr. Mol. Med. 2013, 13, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Miceli, M.; Altucci, L.; Nebbioso, A. Histone deacetylase inhibitors: A patent review (2009–2011). Expert Opin. Ther. Pat. 2013, 23, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y.; Shimizu, T.; Kuwano, K. Sodium butyrate up-regulates cathelicidin gene expression via activator protein-1 and histone acetylation at the promoter region in a human lung epithelial cell line, EBC-1. Mol. Immunol. 2006, 43, 1972–1981. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, J.; Halldorsson, S.; Agerberth, B.; Gudmundsson, G.H. Phenylbutyrate induces antimicrobial peptide expression. Antimicrob. Agents Chemother. 2009, 53, 5127–5133. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, J.; Roschmann, K.I.; van Egmond, D.; Golebski, K.; Fokkens, W.J.; Wang, D.; van Drunen, C.M. Histone deacetylase inhibitors up-regulate LL-37 expression independent of toll-like receptor mediated signalling in airway epithelial cells. J. Inflamm. (Lond.) 2013, 10, e15. [Google Scholar] [CrossRef]
- Raqib, R.; Sarker, P.; Mily, A.; Alam, N.H.; Arifuzzaman, A.S.; Rekha, R.S.; Andersson, J.; Gudmundsson, G.H.; Cravioto, A.; Agerberth, B. Efficacy of sodium butyrate adjunct therapy in shigellosis: A randomized, double-blind, placebo-controlled clinical trial. BMC Infect. Dis. 2012, 12, e111. [Google Scholar] [CrossRef]
- Guo, C.; Sinnott, B.; Niu, B.; Lowry, M.B.; Fantacone, M.L.; Gombart, A.F. Synergistic induction of human cathelicidin antimicrobial peptide gene expression by vitamin D and stilbenoids. Mol. Nutr. Food Res. 2014, 58, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Elias, P.M.; Hupe, M.; Borkowski, A.W.; Gallo, R.L.; Shin, K.O.; Lee, Y.M.; Holleran, W.M.; Uchida, Y. Resveratrol stimulates sphingosine-1-phosphate signaling of cathelicidin production. J. Investig. Dermatol. 2013, 133, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, A.M.; Khaper, N.; Lees, S.J.; Ulanova, M. The antioxidant resveratrol down-regulates inflammation in an in-vitro model of Pseudomonas aeruginosa infection of lung epithelial cells. Can. J. Physiol. Pharmacol. 2013, 91, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, G.; de Filippis, A.; Carteni, M.; de Maria, S.; Cozza, V.; Petrazzuolo, M.; Tufano, M.A.; Donnarumma, G. Polydatin, a natural precursor of resveratrol, induces beta-defensin production and reduces inflammatory response. Inflammation 2013, 36, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xu, C.; Shen, G.; Jain, M.R.; Khor, T.O.; Gopalkrishnan, A.; Lin, W.; Reddy, B.; Chan, J.Y.; Kong, A.N. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 2006, 243, 170–192. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Reynders, V.; Loitsch, S.; Steinhilber, D.; Schroder, O.; Stein, J. The dietary histone deacetylase inhibitor sulforaphane induces human beta-defensin-2 in intestinal epithelial cells. Immunology 2008, 125, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef]
- Meyer, M.; Kesic, M.J.; Clarke, J.; Ho, E.; Simmen, R.C.; Diaz-Sanchez, D.; Noah, T.L.; Jaspers, I. Sulforaphane induces SLPI secretion in the nasal mucosa. Respirat. Med. 2013, 107, 472–475. [Google Scholar] [CrossRef]
- Guo, C.; Rosoha, E.; Lowry, M.B.; Borregaard, N.; Gombart, A.F. Curcumin induces human cathelicidin antimicrobial peptide gene expression through a vitamin D receptor-independent pathway. J. Nutr. Biochem. 2013, 24, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Shin, H. Current trends in the development and application of molecular technologies for cancer epigenetics. World J. Gastroenterol. 2013, 19, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Di Costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenet. 2012, 4, e4. [Google Scholar] [CrossRef]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. N-butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [PubMed]
- Vernia, P.; Monteleone, G.; Grandinetti, G.; Villotti, G.; di Giulio, E.; Frieri, G.; Marcheggiano, A.; Pallone, F.; Caprilli, R.; Torsoli, A. Combined oral sodium butyrate and mesalazine treatment compared to oral mesalazine alone in ulcerative colitis: Randomized, double-blind, placebo-controlled pilot study. Dig. Dis. Sci. 2000, 45, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.C.; Zeitlin, P.L. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: Partial restoration of nasal epithelial CFTR function. Am. J. Respir. Crit. Care Med. 1998, 157, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.F., Jr.; Fransway, A.F.; Jackson, J.M.; Rohowsky, N. Hydrocortisone butyrate 0.1% cream in the treatment of chronic dermatitis. Cutis 2005, 75, 125–131. [Google Scholar] [PubMed]
- Soleas, G.J.; Diamandis, E.P.; Goldberg, D.M. Resveratrol: A molecule whose time has come? And gone? Clin. Biochem. 1997, 30, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Bencivenga, D.; Caldarelli, I.; Tramontano, A.; Borgia, A.; Zappia, V.; Ragione, F.D. Resveratrol: From basic studies to bedside. Cancer Treat. Res. 2014, 159, 167–184. [Google Scholar] [PubMed]
- Singh, C.K.; George, J.; Ahmad, N. Resveratrol-based combinatorial strategies for cancer management. Ann. N. Y. Acad. Sci. 2013, 1290, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Berger, A.; Bocker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLOS ONE 2013, 8, e73097. [Google Scholar] [CrossRef] [PubMed]
- McCormack, D.; McFadden, D. A review of pterostilbene antioxidant activity and disease modification. Oxid. Med. Cell. Longev. 2013, 2013. Article ID:575482. [Google Scholar] [CrossRef]
- Chen, R.H.H.; Sanchez, E.; Shen, J.; Li, M.J.; Wang, J.; Wong, E.; Adler, A.; Hu, M.Y.; Leung, C.; Wang, C.S.; et al. Pterostilbene: A novel histone deacetylase 1 inhibitor (HDAC1) demonstrating efficacy in multiple myeloma. In Proceedings of the 51st ASH Annual Meeting and Exposition, New Orleans, LA, USA, 5–8 December 2009.
- Du, Q.H.; Peng, C.; Zhang, H. Polydatin: A review of pharmacology and pharmacokinetics. Pharm. Biol. 2013, 51, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, e68. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Stephenson, K.K.; Wade, K.L.; Talalay, P. Urease from Helicobacter pylori is inactivated by sulforaphane and other isothiocyanates. Biochem. Biophys. Res. Commun. 2013, 435, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res. (Phila) 2009, 2, 353–360. [Google Scholar] [CrossRef]
- Roshan, D.; Yedery, A.M.; Shafer, W.; Jesre, A.E. Sulforaphane induces the expression of antimicrobial peptides that kill Neisseria gonorrhoeae and suppresses inflammation induced by gonococcal lipooligosaccharide. In Proceedings of the 18th International Pathogenic Neisseria Conference (IPNC), Würzburg, Germany, 9–14 September 2012.
- Unemo, M.; Nicholas, R.A. Emergence of multidrug-resistant, extensively drug-resistant and untreatable gonorrhea. Future Microbiol. 2012, 7, 1401–1422. [Google Scholar] [CrossRef] [PubMed]
- Codd, R.; Braich, N.; Liu, J.; Soe, C.Z.; Pakchung, A.A. Zn(II)-dependent histone deacetylase inhibitors: Suberoylanilide hydroxamic acid and trichostatin A. Int. J. Biochem. Cell Biol. 2009, 41, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [PubMed]
- Teiten, M.H.; Dicato, M.; Diederich, M. Curcumin as a regulator of epigenetic events. Mol. Nutr. Food Res. 2013, 57, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K.; Aggarwal, B.B. Curcumin, a component of golden spice: From bedside to bench and back. Biotechnol. Adv. 2014, 32, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shu, W.; Chen, W.; Wu, Q.; Liu, H.; Cui, G. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin. Pharmacol. Toxicol. 2007, 101, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A.; et al. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Altamura, S.; Chakravarty, P.K.; Cecchetti, O.; De Francesco, R.; Gallinari, P.; Ingenito, R.; Meinke, P.T.; Petrocchi, A.; Rowley, M.; et al. A series of novel, potent, and selective histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5948–5952. [Google Scholar] [CrossRef] [PubMed]
- Gallo, P.; Latronico, M.V.; Grimaldi, S.; Borgia, F.; Todaro, M.; Jones, P.; Gallinari, P.; de Francesco, R.; Ciliberto, G.; Steinkuhler, C.; et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc. Res. 2008, 80, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Hess-Stumpp, H.; Bracker, T.U.; Henderson, D.; Politz, O. MS-275, a potent orally available inhibitor of histone deacetylases—The development of an anticancer agent. Int. J. Biochem. Cell Biol. 2007, 39, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Segain, J.P.; Raingeard de la Bletiere, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottiere, H.M.; Galmiche, J.P. Butyrate inhibits inflammatory responses through NFkappaB inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [PubMed]
- Zhong, L.M.; Zong, Y.; Sun, L.; Guo, J.Z.; Zhang, W.; He, Y.; Song, R.; Wang, W.M.; Xiao, C.J.; Lu, D. Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLOS ONE 2012, 7, e32195. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Yan, C.; Deng, Q.; Gao, D.F.; Niu, X.L. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-kB pathways. Acta Pharmacol. Sin. 2013, 34, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2010, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yedery, R.D.; Jerse, A.E. Augmentation of Cationic Antimicrobial Peptide Production with Histone Deacetylase Inhibitors as a Novel Epigenetic Therapy for Bacterial Infections. Antibiotics 2015, 4, 44-61. https://doi.org/10.3390/antibiotics4010044
Yedery RD, Jerse AE. Augmentation of Cationic Antimicrobial Peptide Production with Histone Deacetylase Inhibitors as a Novel Epigenetic Therapy for Bacterial Infections. Antibiotics. 2015; 4(1):44-61. https://doi.org/10.3390/antibiotics4010044
Chicago/Turabian StyleYedery, Roshan D., and Ann E. Jerse. 2015. "Augmentation of Cationic Antimicrobial Peptide Production with Histone Deacetylase Inhibitors as a Novel Epigenetic Therapy for Bacterial Infections" Antibiotics 4, no. 1: 44-61. https://doi.org/10.3390/antibiotics4010044
APA StyleYedery, R. D., & Jerse, A. E. (2015). Augmentation of Cationic Antimicrobial Peptide Production with Histone Deacetylase Inhibitors as a Novel Epigenetic Therapy for Bacterial Infections. Antibiotics, 4(1), 44-61. https://doi.org/10.3390/antibiotics4010044