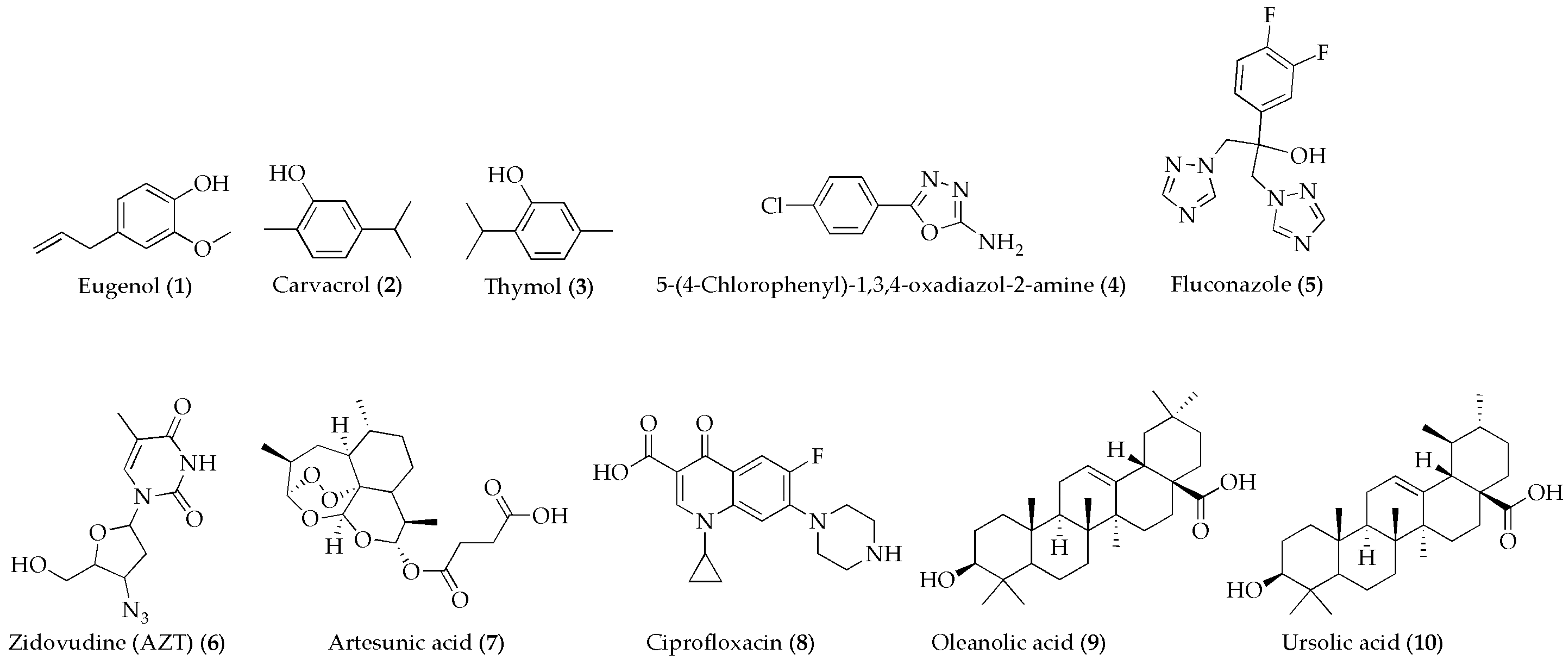
3.2.1. General Synthetic Procedure for Hybrid Compounds 12–16
Bulleted compounds
12–16 were synthesized by following the procedure outlined in
Scheme 1 using a modified version of the synthetic procedures established by Anthwa et al. [
50], Salama et al. [
51], and Ayyash et al. [
52]. 5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-amine (
4) (1 g, 5.11 mmol, 1 equivalent) was reacted with chloroacetyl chloride (0.577 g, 5.11 mmol, 1 equivalent) in the presence of TEA (0.85 mL, 6.13 mmol, 1.2 equivalents) using anhydrous DCM (25 mL) as a solvent to form N-(chloroacetyl)-5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (
11). The base (TEA) neutralized the hydrochloric acid formed during the reaction. The resulting chloroacetylated intermediate (
11) was then reacted with compounds
1–4 and
8 in the presence of TEA to form the final products
12–16 through nucleophilic attack by the hydroxyl groups of eugenol (
1), thymol (
3), carvacrol (
2) or by the NH group of 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine or ciprofloxacin on the electrophilic chloroacetyl group. After each reaction step, the organic phase was washed with brine and water, dried using Na
2SO
4, and evaporated under low pressure. The final products were then purified by column chromatography.
2-(4-Allyl-2-methoxyphenoxy)-N-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)acetamide (12): N-(chloroacetyl)-5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (11) (0.200 g, 0.732 mmol, 1.0 equivalent), eugenol (1) (0.120 g, 0.732 mmol, 1.0 equivalent), TEA (0.12 mL, 6.13 mmol, 1.2 equivalent), DCM(20 mL). Column eluent: EtOAc/Hex(4:10). Yield: 57%. Rf = 0.56.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—171.16 (C-1, 157.07 (C-10), 150.86 (C-8), 136.49 (C-23), 135.37 (C-18), 129.81 (C-1, C-3), 127.22 (C-4, C-5, C-6), 123.75 (C-21), 109.96 (C-20), 84.49 (C-19), 83.96 (C-28), 61.32 (C-22), 60.67 (C-15), 36.68 (C-25), 12.62 (C-26) (
Figure S1).
1H-NMR (500-MHz, recorded in DMSO-d
6): ppm(δ) = 7.80 (H-6, doublet, 1H,
J = 10.0 Hz), 7.80 (H-4, doublet, 1H,
J = 10.0 Hz), 7.59 (H-1, doublet, 1H,
J = 10.0 Hz), 7.59 (H-3, doublet, 1H,
J = 10.0 Hz), 7.28 (H-22, singlet, 1H), 7.21 (H-20, doublet, 1H,
J = 5.0 Hz), 6.80 (H-19, doublet, 1H,
J = 10.0), 6.68–6.58 (H-27, multiplet, 1H), 5.33 (H-13, singlet, 1H), 4,85 (H-15, singlet, 1H), 4.42–4.39 (H-28, doublet of a doublet, 2H), 3.32 (H-25, singlet, 3H), 3.10 (H-26, doublet, 1H,
J = 10.0 Hz) (
Figure S1).
FT–IR (KBr ν in cm
−1): 3554 (N–H stretching), 2931–2855 (C–H stretching from alkanes), 1710 (C=O stretching), 1626–1481 (C=C stretching from aromatic or alkene), 1381–1156 (C–H bending (alkanes or aromatics)), 1000–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S11).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
20H
18ClN
3O
4]
+: calculated 399.0986, observed 399.0958 (
Figure S21).
2-(2-Isopropyl-5-methylphenoxy)-N-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)acetamide (13): N-(chloroacetyl)-5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (11) (0.200 g, 0.732 mmol, 1.0 equivalent), thymol (3) (131.9 mg, 0.878 mmol, 1.2 equivalents), TEA (0.153 mL, 1.098 mmol, 1.5 equivalents). Column eluent: EtOAc/Hex (7:3). Yield: 55%. Rf = 0.58.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—170.31 (C-14), 164.46 (C-18), 157.08 (C-10), 155.09 (C-8), 136.50 (C-2), 135.39 (C-20), 134.46 (C-23), 129.79 (C-3), 129.05 (C-4), 127.23 (C-22), 123.69 (C-21), 115.86 (C-19), 68.09 (C-15), 25.97 (C-24), 23.06 (C-27), 22.36 (C-25), 21.72 (C-26) (
Figure S2).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—7.80 (H-6, doublet, 1H,
J = 10.0 Hz), 7.80 (H-4, doublet, 1H,
J = 10.0 Hz), 7.58 (H-1, doublet, 1H,
J = 10.0 Hz), 7.58 (H-3, doublet, 1H,
J = 10.0 Hz), 7.28 (H-19, singlet, 1H), 7.19 (H-22, doublet, 1H,
J = 5.0 Hz), 6.37 (H-21, doublet, 1H,
J = 10.0 Hz), 5.73 (H-13, singlet, 1H), 4.62 (H-15, singlet, 2H), 3.69–3.58 (H-24, multiplet, 1H), 3.39 (H-25, singlet, 3H), 1.10 (H-27, doublet, 6H,
J = 10.0 Hz) (
Figure S2).
FT–IR (KBr, ν in cm
−1): 3354 (N–H stretching), 2933–2872 (C–H stretching from alkanes), 1735 (C=O stretching), 1500 (C=C stretching from aromatic or alkene), 1388 (C–H bending of alkanes or aromatics), 1157–500 (C–O stretching, C–Cl bond, or skeletal vibrations from aromatic rings) (
Figure S12).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
20H
20ClN
3O
3]
+: calculated 385.1193, observed 385.1185 (
Figure S22).
2-(5-Isopropyl-2-methylphenoxy)-N-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)acetamide (14): N-(chloroacetyl)-5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (11) (0.200 g, 0.732 mmol, 1.0 equivalent), carvacrol (2) (131.9 mg, 0.878 mmol, 1.2 equivalents), TEA (0.153 mL, 1.098 mmol, 1.5 equivalents). Column eluent: EtOAc/Hex (3:7). Yield: 52%. Rf = 0.63.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—171.21 (C-14), 164.48 (C-18), 157.07 (C-10), 154.61 (C-8), 135.71 (C-3), 135.38 (C-1), 131.68 (C-22), 129.82 (C-5), 127.23 (C-4), 126.08 (C-6), 123.75 (C-23), 120.10 (C-21), 116.06 (C-19), 71.02 (C-15), 26.51 (C-25), 23.05 (C-26), 23.05 (C-27), 21.10 (C-24) (
Figure S3).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—7.81 (H-6, doublet, 1H,
J = 5.0 Hz), 7.81 (H-4, doublet, 1H,
J = 5.0 Hz), 7.60 (H-1, doublet, 1H,
J = 5.0 Hz), 7.60 (H-3, doublet, 1H,
J = 5 Hz), 6.97–6.95 (H-22, doublet, 1H,
J = 10.0 Hz), 6.56 (H-19, singlet, 1H), 6.55 (H-21, doublet, 1H,
J = 10.0 Hz), 4.88 (H-13, singlet, 1H), 3.33 (H-15, singlet, 1H), 3.18–3.12 (H-25, multiplet, 1H), 2.17 (H-24, doublet, 3H,
J = 5.0 Hz), 1.13 (H-26, doublet, 3H,
J = 10.0 Hz), 1.13 (H-27, doublet, 3H,
J = 10.0 Hz) (
Figure S3).
FT–IR (KBr, ν in cm
−1): 3451 (N–H stretching), 2942–2890 (C–H stretching from alkanes), 1694 (C=O stretching), 1468–1391 (C=C stretching from aromatic or alkene). 1389–1033 (C–H bending (alkanes or aromatics)), 1000–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S13).
HRMS (ESI) mass spectrometry analysis analysis (
m/
z) of [C
20H
20ClN
3O
3]
+: calculated 385.1193, observed 385.1179 (
Figure S23).
2-(5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-ylamino)-N-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)acetamide (15): N-(chloroacetyl)-5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (11) (0.300 g, 1.098 mmol, 1.0 equivalent), 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (260.5 mg, 1.318 mmol, 1.2 equivalents), TEA (0.23 mL, 1.647 mmol, 1.5 equivalents). Column eluent: EtOAc/Hex (3:5). Yield: 29%. Rf = 0.25.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—172.21 (C-14), 151.27 (C-26), 147.97 (C-10), 145.58 (C-8), 145.24 (C-24), 138.66 (C-19), 130.95 (C-21), 121.02 (C-5), 115.90 (C-6), 115.67 (C-22), 113.22 (C-20), 56.09 (C-15) (
Figure S4).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm) = 6.73 (H-4, doublet, 1H,
J = 10.0 Hz), 6.73 (H-6, doublet, 1H,
J = 10.0 Hz), 6.70 (H-22, doublet, 1H,
J = 5.0 Hz), 6.70 (H-20, doublet, 1H,
J = 5.0 Hz), 6.58 (H-1, doublet, 1H,
J = 10.0 Hz), 6.58 (H-3, doublet, 1H,
J = 10.0 Hz), 6.58 (H-17, doublet, 1H,
J = 10.0 Hz), 6.58 (H-19, doublet, 1H,
J = 10.0 Hz), 5.08 (H-13, singlet, 1H), 3.75 (H-15, doublet, 2H,
J = 10.0 Hz), 3.32 (H-29, singlet, 1H) (
Figure S4).
FT–IR (KBr, ν in cm
−1): 3544–3352 (N–H stretching) 2930–2856 (C–H stretching from alkanes), 1708 (C=O stretching), 1623–1493 (C=C stretching (aromatic or alkene)), 1455–1361 (C–H bending (alkanes or aromatics)), 1156–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S14).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
18H
12Cl
2N
6O
3]
+: calculated 430.0348, observed 430.0329 (
Figure S24).
7-(4-(2-(5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-ylamino)acetyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid (16): N-(chloroacetyl)-5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (200 mg, 0.732 mmol, 1.0 equivalent), ciprofloxacin (290.7 mg, 0.878 mmol, 1.2 equivalents), TEA (0.153 mL, 1.098 mmol, 1.5 equivalents). Column eluent: EtOAc/Hex 2:8. Yield: 60%. Rf = 0.62.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—172.81 (C-22), 164.48 (C-25), 157.07 (C-2), 154.61 (C29), 148.72 (C-31), 147.30 (C-12), 145.79 (C-14), 135.71 (C-4), 135.38 (C-37), 131.68 (C-9), 129.82 (C38), 127.23 (C-38), 126.08 (C-36), 123.75 (C-34), 120.10 (C-39), 116.06 (C-35), 110.99 (C-10), 107.99 (C-11), 104.98 (C-3), 53.79 (C-21), 52.89 (C-16), 47.45 (C-20), 46.33 (C-19), 43.14 (C-17), 26.51 (C-26), 23.95 (C-6), 23.95 (C-7), 21.10 (C-8) (
Figure S5).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—8.33 (H-4, singlet, 1H), 7.49 (H-34, doublet, 1H,
J = 5.0 Hz), 7.49 (H-38, doublet, 1H,
J = 5.0 Hz), 7.45 (H-35, doublet, 1H,
J = 10.0 Hz), 7.43 (H-37, singlet, 1H), 6.35 (H-21, singlet, 1H,
J = 5.0 Hz), 5.36 (H-27, singlet, 1H), 3.73 (H-20, triplet, 2H,
J = 5.0 Hz), 3.51 (H-16, triplet, 2H,
J = 5.0 Hz), 3.23 (H-25, singlet, 2H), 3.01–2.94 (H-6, multiplet, 1H), 2.79 (H-19, triplet, 2H,
J = 5.0 Hz), 2.66 (H-17, triplet, 2H,
J = 5.0 Hz), 0.88–0.77 (H-7, multiplet, 2H), 0.77–0.67 (H-8, multiplet, 2H) (
Figure S5).
FT-IR (KBr, ν in cm
−1): 3302 (OH stretching), 2942–2891 (C–H stretching from alkanes), 1688 (C=O stretching), 1667–1557 (C=C stretching from aromatic or alkene), 1465–1373 (C–H bending (alkanes or aromatics)), 1043–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S15).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
27H
24ClFN
6O
5]
+: calculated 566.1481, observed 566.1521 (
Figure S25).
3.2.2. General Synthetic Procedure for Hybrid Compounds 17 and 18
Two ursolic acid or oleanolic acid derivatives containing the 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine moiety were synthesized as outlined in
Scheme 1 by following a modified procedure [
53]. UA or OA was dissolved in anhydrous DMF, and then DCC and DMAP were added to activate their carboxyl groups. 5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-amine (
4) was then introduced, allowing the amine group to react with the activated carboxyl group of ursolic acid or oleanolic acid, forming an amide bond. The reaction mixture was stirred at room temperature for 1 h and refluxed at 80 °C overnight. After the reaction, the solvent was removed, and the product was purified using column chromatography.
Hybrid 17: ursolic acid (300 mg, 0.657 mmol, 1.0 equivalent), 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (155.2 mg, 0.789 mmol, 1.2 equivalents), DCC (149.2 mg, 0.723 mmol, 1.1 equivalents), DMAP (8.1 mg, 0.066 mmol, 0.1 equivalent), DMF (10 mL). Column eluent: EtOAc/Hex (3:7). Yield: 42%. Rf = 0.72.
13C-NMR (500 MHz, recorded in CDCl-d
3): chemical shifts (δ, ppm)—172.31 (C-1), 157.09 (C-36), 156.02 (C-39), 138.33 (C-27), 136.07 (C-44), 132.15 (C-45), 130.67 (C-43), 128.77 (C-41), 127.60 (C-46), 125.96 (C-42), 124.54 (C-28), 77.29 (C-2), 55.22 (C-9), 52.41 (C-25), 49.65 (C-30), 47.98 (C-18), 47.35 (C-14), 42.13 (C-12), 39.08 (C-23), 38.85 (C-6), 38.66 (C-21), 36.95 (C-5), 33.81 (C-31), 31.62 (C-11), 30.31 (C-20), 29.45 (C-4), 28.70 (C-15), 25.80 (C-29), 24.92 (C-7), 23.76 (C-8), 21.24 (C-10), 17.46 (C-22), 17.37 (C-24), 16.52 (C-13), 15.50 (C-32) (
Figure S6).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—7.97 (H-33, singlet, 1H), 7.71 (H-41, doublet, 1H,
J = 5.0 Hz), 7.71 (H-45, doublet, 1H,
J = 5.0 Hz), 7.66 (H-42, doublet, 1H,
J = 5.0 Hz), 7.66 (H-44, doublet, 1H,
J = 10.0 Hz), 5.40 (H-48, triplet, 1H,
J = 5.0 Hz), 3.01 (H-2, triplet, 1H,
J = 5.0 Hz), 2.12 (H-44, doublet, 1H,
J = 10.0 Hz), 1.93–0.69 (aliphatic protons from the ursolic acid moiety) (
Figure S6).
FT-IR (KBr, ν in cm
−1): 3357 (NH stretching), 3265 (OH stretching), 2941–2871 (C–H stretching from alkanes), 1581 (C=O stretching), 1557–1436 (C=C stretching from aromatic or alkene), 1375–1138 (C–H bending (alkanes or aromatics)), 902–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S16).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
38H
52Cl
2N
3O
3]
+: calculated 633.3697, observed 633. 3665 (
Figure S26).
Hybrid 18: oleanolic acid (300 mg, 0.657 mmol, 1.0 equivalent), 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (155.2 mg, 0.789 mmol, 1.2 equivalents), DCC (149.2 mg, 0.723 mmol, 1.1 equivalents), DMAP (8.1 mg, 0.066 mmol, 0.1 equivalent). Column eluent: EtOAc/Hex (4:6). Yield: 38%. Rf = 0.3.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—177.81 (C-1), 157.13 (C-34), 154.08 (C-2), 140.33 (C-25), 136.56 (C-42), 134.34 (C-43), 134.08 (C-41), 129.25 (C-39), 126.93 (C-44), 126.93 (C-40), 80.39 (C-2), 52.53 (C-9), 51.58 (C-28), 45.47 (C-22), 44.37 (C-18), 40.85 (C-23), 37.47 (C-14), 37.36 (C-12), 36.39 (C-6), 36.30 (C-5), 34.73 (C-29), 34.22 (C-20), 30.81 (C-11), 26.04 (C-21), 25.92 (C-46), 25.92 (C-47), 24.72 (C-19), 24.55 (C-19), 20.35 (C16), 13.17 (C-13), 12.72 (C-30) (
Figure S7).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—7.95 (H-36, singlet, 1H), 7.03 (H-43, doublet, 1H,
J = 10.0 Hz), 7.03 (H-47, doublet, 1H,
J = 10.0 Hz), 6.67 (H-44, doublet, 1H,
J = 10.0 Hz), 6.67 (H-46, doublet, 1H,
J = 10.0 Hz), 5.14 (H-29, triplet, 1H,
J = 5.0 Hz), 3.0 1 (H-27, triplet, 1H,
J = 5.0 Hz), 2.48–2.47 (H-2, doublet of a doublet, 1H,
J = 5.0 Hz), 2.21–0.69 (aliphatic protons from the oleanolic acid moiety) (
Figure S7).
FT-IR (KBr, ν in cm
−1): 3357 (NH stretching), 3265 (OH stretching), 2941–2871 (C–H stretching from alkanes), 1581 (C=O stretching), 1557–1436 (C=C stretching from aromatic or alkene), 1375–1138 (C–H bending (alkanes or aromatics)), 902–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S17).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
38H
52Cl2N
3O
3]
+: calculated 633.3697, observed 633.3635 (
Figure S27).
3.2.3. Synthetic Procedure for Hybrid Compound 19
The synthesis of a 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine-artesunate hybrid (19) was carried out by activating artesunic acid (7) to react with the amine group of 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine. First, artesunic acid (7) was dissolved in anhydrous DMF and activated using NHS to form an ester. Next, the activated artesunate was reacted with the amine group of 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine in DMF at room temperature, and the reaction was allowed to proceed for 24 h. After completion, the solvent was removed, and the crude product was extracted with dichloromethane and purified with column chromatography.
Artesunic acid (250 mg, 0.621 mmol, 1.0 equivalent), 5-(4-chlorophenyl)-1,3,4-oxadiazol-2-amine (122.1 mg, 0.261 mmol, 1.0 equivalent), NHS (78.6 mg, 0.683 mmol, 1.1 equivalent). Column eluent: EtOAc/Hex (3:7). Yield: 48%. Rf = 0.47.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—178.67 (C-26), 173.18 (C-22), 166.56 (C-32), 164.30 (C-29), 138.67 (C-37), 132.25 (C-38), 129.85 (C-36), 126.83 (C-34), 125.05 (C-39), 123.97 (C-35), 77.34 (C-5), 55.27 (C-6), 52.89 (C-11), 47.32 (C-18), 42.32 (C-1), 38.97 (C-13), 38.91 (C-14), 36.79 (C-25), 30.38 (C-7), 28.73 (C-24), 23.74 (C-12), 21.50 (C-15), 16.51 (C-20), 15.68 (C-8) (
Figure S8).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—8.00 (H-27, singlet, 1H), 7.99 (H-35, doublet, 1H,
J = 5.0 Hz), 7.99 (H-39, doublet, 1H,
J = 5.0 Hz), 7.63 (H-36, doublet, 1H,
J = 5.0 Hz), 7.60 (H-38, doublet, 1H,
J = 5.0 Hz), 6.04 (H-19, doublet, 1H,
J = 5.0 Hz), 5.14 (H-2, doublet, 1H,
J = 5.0 Hz), 4.26 (H-41, singlet, 1H), 3.15 (H-24, triplet, 2H,
J = 5.0 Hz), 2.82 (H-25, triplet, 2H,
J = 5.0 Hz), 2.13–0.69 (aliphatic protons from artesunate) (
Figure S8).
FT–IR (KBr, ν in cm
−1): 3357 (NH stretching), 3550–3371 (NH stretching), 2930–2858 (C–H stretching from alkanes), 1709 (C=O stretching), 1627 (C=C stretch from aromatic or alkene), 1383–1209 (C–H bending (alkanes or aromatics)), 1035–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S18).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
27H
32ClN
3O
8]
+: calculated 561.1878, observed 561.1902 (
Figure S28).
3.2.4. Synthetic Procedure for Hybrid Compound 21
5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-amine (250 mg, 1.272 mmol, 1.0 equivalent) was reacted with succinic anhydride (152.7 mg, 1.527 mmol, 1.2 equivalents) in pyridine under reflux. After 4 h, the product was precipitated in cold water, filtered, and dried to yield the N-succinylated intermediate (20). The N-succinylated intermediate was dissolved in dichloromethane, followed by the addition of zidovudine (6), coupling reagents, and DCC and DMAP as catalysts. The mixture was stirred at 0 °C for 15 min and left overnight at room temperature. After workup and purification, the final succinylated oxadiazole-zidovudine (21) hybrid was obtained.
(3-Azido-tetrahydro-5-(3,4-dihydro-5-methyl-2,4-dioxopyrimidin-1(2H)-yl)furan-2-yl)methyl 3-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-ylcarbamoyl)propanoate (21): 3-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-ylcarbamoyl)propanoic acid (20) (152 mg, 0.475 mmol, 1.0 equivalent), zidovudine (6) (127.0 mg, 0.475 mmol, 1.0 equivalent), DCC (107 mg, 0.522 mmol, 1.1 equivalents), DMAP (5.8 mg, 0.0475 mmol, 0.1 equivalent). Column eluent: DCM/EtOAc (15:3). Yield: 51%. Rf = 0.53.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—178.67 (C-14), 169.28 (C-17), 155.60 (C-10), 154.44 (C-8), 151.02 (C-28), 138.67 (C-32), 134.42 (C-2), 130.36 (C-3), 129.59 (C-1), 128.36 (C-5), 125.06 (C-4), 107.85 (C-31), 77.34 (C-25), 55.28 (C-22), 52.89 (C-21), 47.52 (C-15), 47.32 (C-16), 30.67 (C2-4), 27.48 (C-23), 15.80 (C-33) (
Figure S9).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—8.67 (H-29, singlet, 1H), 8.33 (H-13, singlet, 1H), 7.80 (H-32, singlet, 1H), 7.53 (H-6, doublet, 1H,
J = 10.0 Hz), 7.51 (H-4, doublet, 1H,
J = 10.0 Hz), 7.43 (H-1, doublet, 1H,
J = 10.0 Hz), 7.43 (H-3, doublet, 1H,
J = 10.0 Hz), 6.73–6.69 (H-24, doublet of a doublet, 2H), 6.57 (H-21, doublet, 2H,
J = 10.0 Hz), 5.98–5.90 (H-22, multiplet, 1H), 5.08–5.01 (H-24, multiplet, 1H), 3.32 (H-33, singlet, 3H), 3.27 (H-25, doublet, 1H,
J = 10.0 Hz), 2.72 (H-16, triplet, 2H,
J = 5.0 Hz), 2.63 (H-15, triplet, 2H,
J = 5.0 Hz) (
Figure S9).
FT–IR (KBr, ν in cm
−1): 3535–3393 (NH stretching), 2932–2855 (C–H stretching from alkanes). 1704–1626 (C=O stretching), 1494–1452 (C=C stretching (aromatic or alkene)), 1379–1253 (C–H bending (alkanes or aromatics)), 1038–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S19).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
22H
21ClN
8O
7]
+: calculated 544.1222, found 544.1254 (
Figure S29).
3.2.5. Synthetic Procedure for Hybrid Compound 23
Compound
23 was synthesized by following a modified procedure based on the method reported by Hou et al. [
54]. Eugenol (
1) (500 mg, 3.056 mmol, 1.0 equiv.) was first esterified with succinic anhydride (365.9 mg, 3.655 mmol, 1.2 equiv.) in anhydrous DMF (15 mL) using a catalytic amount of DMAP (55.9 mg, 0.457 mmol, 0.15 equiv.). The reaction mixture was stirred at room temperature under an inert atmosphere overnight. The progress of the reaction was monitored by TLC, and the product was purified by column chromatography. The obtained eugenol–succinic acid intermediate (
22) was then dissolved in anhydrous DMF, and DCC and TEA were added. The mixture was stirred in an ice bath for 15 min before adding fluconazole (
5), followed by overnight stirring at room temperature. After completion, the reaction mixture was filtered to remove by-products, and the crude product was extracted with ethyl acetate, washed with brine and water, dried over Na
2SO
4, and purified by column chromatography.
4-Allyl-2-methoxyphenyl 2-(3,4-difluorophenyl)-1,3-di(1H-1,2,4-triazol-1-yl)propan-2-yl succinate(23): fluconazole (150 mg, 0.4900 mmol, 1.0 equivalent), 3-(4-allyl-2-methoxyphenoxy)carbonyl)propanoic acid (22) (129.05 mg, 0.4900 mmol, 1.00 equivalent), DCC (101.0 mg, 0.4900 mmol, 1.00 equivalent), TEA (49.6 mg, 0.4900 mmol, 1.00 equivalent), DMF (10 mL). Column eluent: EtOAc/Hex (6:10). Yield: 53%. Rf = 0.67.
13C-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—172.86 (C-15), 170.54 (C-11), 154.59 (C-37), 154.44 (C-32), 152.74 (C-6), 151.27 (C-5), 145.58 (C-23), 145.58 (C-27), 135.70 (C-35), 131.66 (C-34), 130.28 (C-21), 126.08 (C-18), 123.94 (C-27), 120.09 (C-2), 120.09 (C-19), 116.05 (C-20), 111.42 (C-4), 111.28 (C-28), 104.60 (C-1), 104.39 (C-22), 74.15 (C-7), 55.42 (C-29), 29.93 (C-8), 26.50 (C-26), 23.06 (C-14), 21.10 (C-13) (
Figure S10).
1H-NMR (500 MHz, recorded in DMSO-d
6): chemical shifts (δ, ppm)—8.33 (H-32, singlet, 1H), 8.33 (H-37, singlet, 1H), 7.80 (H-34, singlet, 1H), 7.80 (H-35, singlet, 1H), 6.73 (H-4, singlet, 1H), 6.70 (H-2, doublet, 1H,
J = 5.0 Hz), 6.70 (H-19, doublet, 1H,
J = 5.0 Hz), 6.58 (H-22, singlet, 1H), 6.57 (H-1, doublet, 1H,
J = 10.0 Hz), 6.57 (H-20, doublet, 1H,
J = 10.0 Hz,), 5.98–5.90 (H-27, multiplet, 1H), 5.04–5.03 (H-28, doublet, 2H,
J = 5.0 Hz), 3.75 (H-8, singlet, 2H), 3.75 (H-29, singlet, 2H), 3.32 (H-25, singlet, 3H), 3.26 (H-26, doublet, 1H,
J = 10.0 Hz), 2.89 (H-14, triplet, 2H,
J = 5.0 Hz), 2.73 (H-13, triplet, 2H,
J = 5.0 Hz) (
Figure S10).
FT–IR (KBr, ν in cm
−1): 2945–2905 (C–H stretching from alkanes). 1710 (C=O stretching), 1471 (C=C stretching (aromatic or alkene)), 1431–1258 (C–H bending (alkanes or aromatics)), 1180–500 (C–O stretching, C–Cl bonds, or skeletal vibrations from aromatic rings) (
Figure S20).
HRMS (ESI) mass spectrometry analysis (
m/
z) of [C
27H
26F
2N
6O
5]
+: calculated 552.1933, observed 552.1919 (
Figure S30).


 ,
, 


{kind=link}
{kind=link}