
Extraction, Isolation, Characterization, and Bioactivity of Polypropionates and Related Polyketide Metabolites from the Caribbean Region

 , ,
, ,  ,
, 
Abstract
1. Introduction
1.1. Extraction, Isolation, Structural Characterization, and Bioactivity
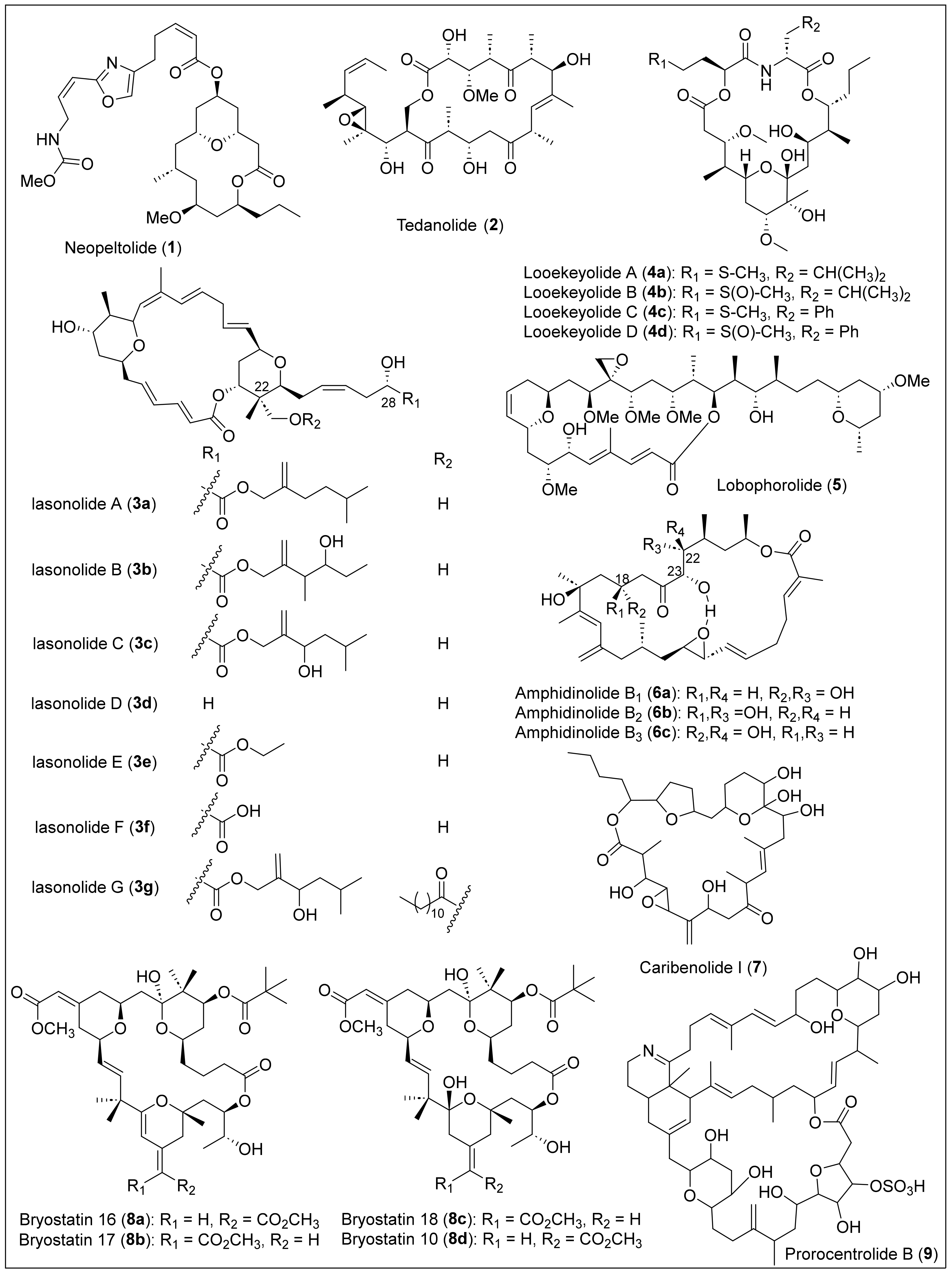
1.1.1. Macrolides
1.1.2. Polyene Macrolides
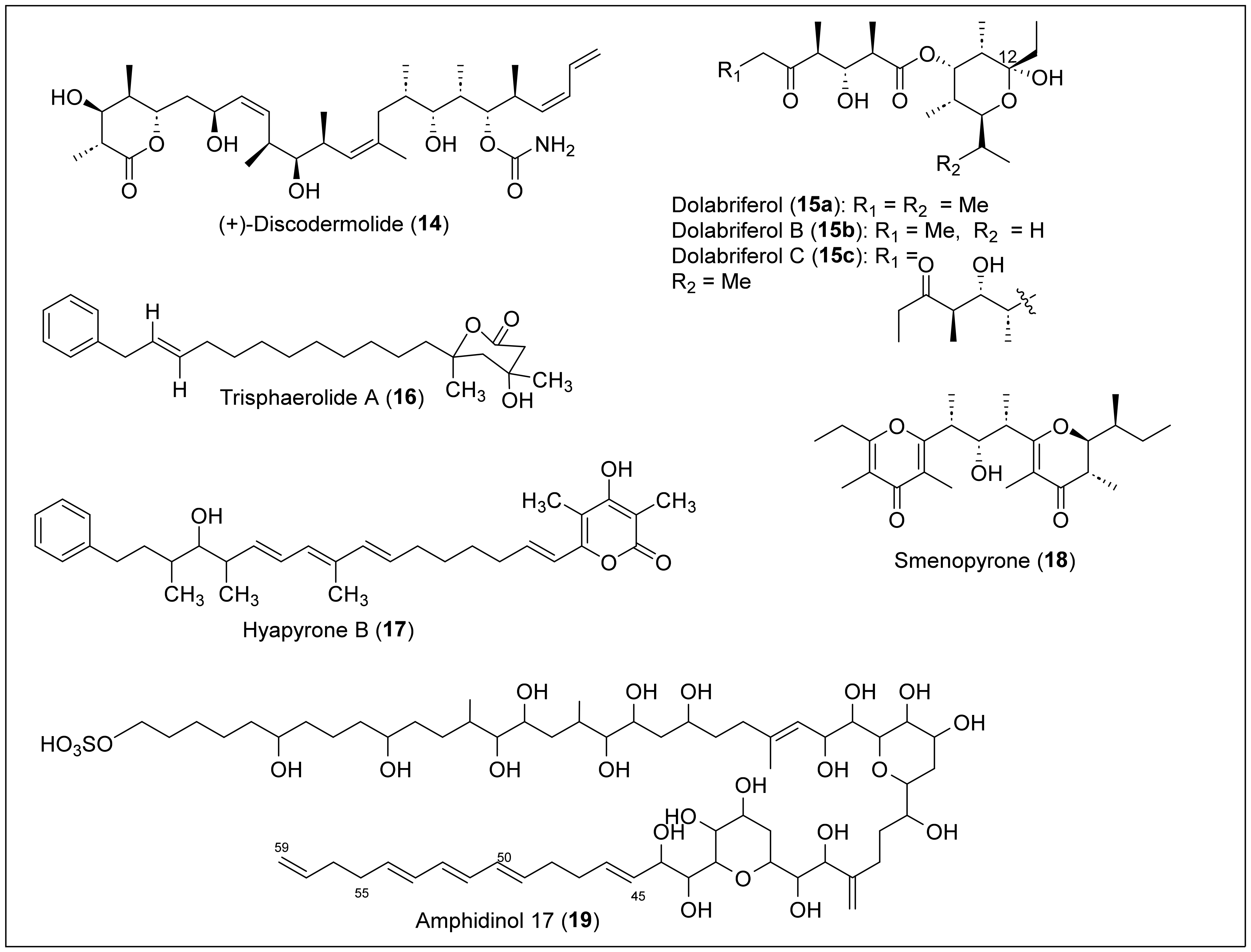
1.2. Linear Polypropionates
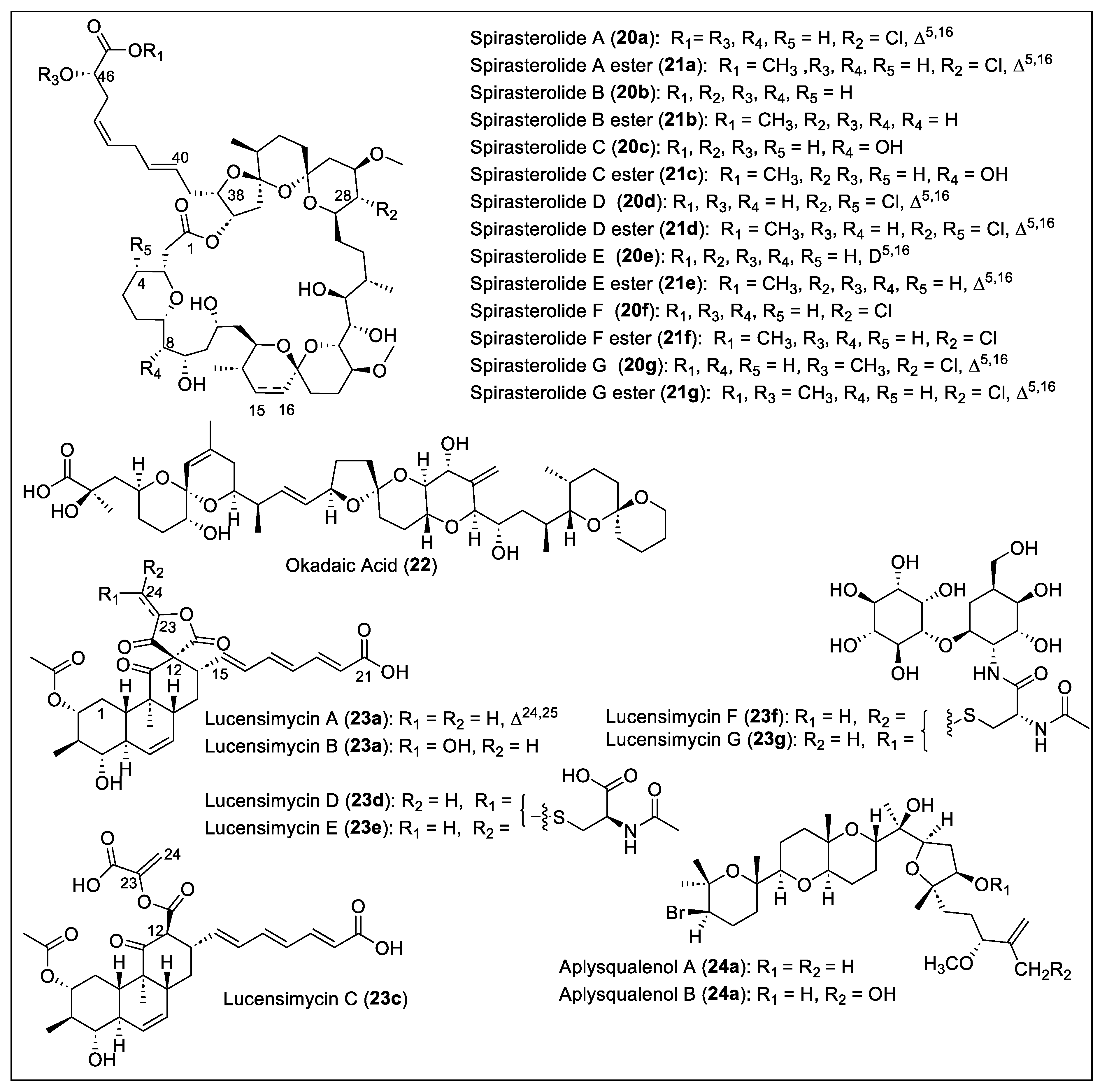
1.3. Cyclic Polyethers
1.4. Unusual Polyketides
1.4.1. Polyketides Containing Vinyl Chloride and Cyclopropane Moieties
1.4.2. Norpentaketides
1.4.3. Prenylated Polyketides
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Birch, A.J. Biosynthesis of Polyketides and Related Compounds. Science 1967, 156, 202–206. [Google Scholar] [CrossRef]
- Koskinen, A.M.; Karisalmi, K. Polyketide Stereotetrads in Natural Products. Chem. Soc. Rev. 2005, 34, 677–690. [Google Scholar] [CrossRef]
- Rodríguez-Berríos, R.R.; Isbel, S.R.; Bugarin, A. Epoxide-Based Synthetic Approaches toward Polypropionates and Related Bioactive Natural Products. Int. J. Mol. Sci. 2023, 24, 6195. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J. A New Role for Polyketides. Angew. Chem. Int. Ed. 2000, 39, 2847–2849. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Hamel, E. Antimitotic Natural Products and Their Interactions with Tubulin. Med. Res. Rev. 1996, 16, 207–231. [Google Scholar] [CrossRef]
- Davies-Coleman, M.T.; Garson, M.J. Marine Polypropionates. Nat. Prod. Rep. 1998, 15, 477–493. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Zhang, W. Natural Polypropionates in 1999–2020: An Overview of Chemical and Biological Diversity. Mar. Drugs 2020, 18, 569. [Google Scholar] [CrossRef]
- Demeritte, A.; Wuest, W.M. A Look around the West Indies: The Spices of Life Are Secondary Metabolites. Bioorg. Med. Chem. 2020, 28, 115792. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a Macrolide from a Lithistid Sponge of the Family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Youngsaye, W.; Lowe, J.T.; Pohlki, F.; Ralifo, P.; Panek, J.S. Total Synthesis and Stereochemical Reassignment of (+)-neopeltolide. Angew. Chem. 2007, 119, 9371–9374. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Scheidt, K.A. Total Synthesis and Structural Revision of the Marine Macrolide Neopeltolide. J. Am. Chem. Soc. 2008, 130, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, K.; Oda, M.; Fuwa, H. Total Synthesis of (+)-Neopeltolide by the Macrocyclization/Transannular Pyran Cyclization Strategy. Org. Lett. 2022, 24, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Gunasekera, S.P.; Yalamanchili, G.; Hossain, M.B.; Van der Helm, D. Tedanolide: A Potent Cytotoxic Macrolide from the Caribbean Sponge Tedania Ignis. J. Am. Chem. Soc. 1984, 106, 7251–7252. [Google Scholar] [CrossRef]
- McCarthy, M. Tedania Ignis. Available online: https://animaldiversity.org/accounts/Tedania_ignis/ (accessed on 23 December 2022).
- Smith, A.B.; Lee, D. Total Synthesis of (+)-Tedanolide. J. Am. Chem. Soc. 2007, 129, 10957–10962. [Google Scholar] [CrossRef]
- Lee, E.; Song, H.Y.; Joo, J.M.; Kang, J.W.; Kim, D.S.; Jung, C.K.; Hong, C.Y.; Jeong, S.; Jeon, K. Synthesis of (+)-Lasonolide A: (−)-Lasonolide A Is the Biologically Active Enantiomer. Bioorg. Med. Chem. Lett. 2002, 12, 3519–3520. [Google Scholar] [CrossRef]
- Horton, P.A.; Koehn, F.E.; Longley, R.E.; McConnell, O.J. Lasonolide A, A New Cytotoxic Macrolide from the Marine Sponge Forcepia sp. J. Am. Chem. Soc. 1994, 116, 6015–6016. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Ghosh, A.K.; Pommier, Y. Lasonolide A, a Potent and Reversible Inducer of Chromosome Condensation. Cell Cycle 2012, 11, 4424–4435. [Google Scholar] [CrossRef]
- Wright, A.E.; Chen, Y.; Winder, P.L.; Pitts, T.P.; Pomponi, S.A.; Longley, R.E. Lasonolides C-G, Five New Lasonolide Compounds from the Sponge Forcepia sp. J. Nat. Prod. 2004, 67, 1351–1355. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Meyer, J.L.; Ding, Y.; Abboud, K.A.; Luo, D.; Campbell, J.E.; Angerhofer, A.; Goodsell, J.L.; Raymundo, L.J.; Liu, J.; et al. Chemical and Metagenomic Studies of the Lethal Black Band Disease of Corals Reveal Two Broadly Distributed, Redox-Sensitive Mixed Polyketide/Peptide Macrocycles. J. Nat. Prod. 2019, 82, 111–121. [Google Scholar] [CrossRef]
- Meyer, J.L.; Gunasekera, S.P.; Brown, A.L.; Ding, Y.; Miller, S.; Teplitski, M.; Paul, V.J. Cryptic Diversity of Black Band Disease Cyanobacteria in Siderastrea Siderea Corals Revealed by Chemical Ecology and Comparative Genome-Resolved Metagenomics. Mar. Drugs 2023, 21, 76. [Google Scholar] [CrossRef]
- Kubanek, J.; Jensen, P.R.; Keifer, P.A.; Sullards, M.C.; Collins, D.O.; Fenical, W. Seaweed Resistance to Microbial Attack: A Targeted Chemical Defense against Marine Fungi. Proc. Natl. Acad. Sci. USA 2003, 100, 6916–6921. [Google Scholar] [CrossRef]
- Blain, J.C.; Mok, Y.-F.; Kubanek, J.; Allingham, J.S. Two Molecules of Lobophorolide Cooperate to Stabilize an Actin Dimer Using Both Their “Ring” and “Tail” Region. Chem. Biol. 2010, 17, 802–807. [Google Scholar] [CrossRef]
- Bauer, I.; Maranda, L.; Shimizu, Y.; Peterson, R.W.; Cornell, L.; Steiner, J.R.; Clardy, J. The Structures of Amphidinolide B Isomers: Strongly Cytotoxic Macrolides Produced by a Free-Swimming Dinoflagellate, Amphidinium sp. J. Am. Chem. Soc. 1994, 116, 2657–2658. [Google Scholar] [CrossRef]
- Ishibashi, M.; Ohizumi, Y.; Hamashima, M.; Nakamura, H.; Hirata, Y.; Sasaki, T.; Kobayashi, J. Amphidinolide-B, a Novel Macrolide with Potent Antineoplastic Activity from the Marine Dinoflagellate Amphidinium sp. J. Chem. Soc. Chem. Commun. 1987, 1127–1129. [Google Scholar] [CrossRef]
- Kobayashi, J.; Ishibashi, M.; Nakamura, H.; Ohizumi, Y.; Yamasu, T.; Hirata, Y.; Sasaki, T.; Ohta, T.; Nozoe, S. Cytotoxic Macrolides from a Cultured Marine Dinoflagellate of the Genus Amphidinium. J. Nat. Prod. 1989, 52, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Ishiyama, H.; Kobayashi, J. Absolute Stereochemistry of Amphidinolide B. Tetrahedron Lett. 1994, 35, 8241–8242. [Google Scholar] [CrossRef]
- Bauer, I.; Maranda, L.; Young, K.A.; Shimizu, Y.; Fairchild, C.; Cornell, L.; MacBeth, J.; Huang, S. Isolation and Structure of Caribenolide I, a Highly Potent Antitumor Macrolide from a Cultured Free-Swimming Caribbean Dinoflagellate, Amphidinium sp. S1-36-5. J. Org. Chem. 1995, 60, 1084–1086. [Google Scholar] [CrossRef]
- Jalce, G.; Franck, X.; Seon-Meniel, B.; Hocquemiller, R.; Figadère, B. Contribution to the Total Synthesis of Caribenolide I. Tetrahedron Lett. 2006, 47, 5905–5908. [Google Scholar] [CrossRef]
- Seck, M.; Seon-Meniel, B.; Jullian, J.-C.; Franck, X.; Hocquemiller, R.; Figadere, B. Synthesis of C1-C6 Fragment of Caribenolide I. Lett. Org. Chem. 2006, 3, 390–395. [Google Scholar] [CrossRef]
- Seck, M.; Franck, X.; Seon-Meniel, B.; Hocquemiller, R.; Figadère, B. A Baylis–Hillman Approach to the Synthesis of C1–C11 Fragment of Caribenolide I. Tetrahedron Lett. 2006, 47, 4175–4180. [Google Scholar] [CrossRef]
- Pettit, G.R.; Gao, F.; Blumberg, P.M.; Herald, C.L.; Coll, J.C.; Kamano, Y.; Lewin, N.E.; Schmidt, J.M.; Chapuis, J.-C. Antineoplastic Agents. 340. Isolation and Structural Elucidation of Bryostatins 16−18. J. Nat. Prod. 1996, 59, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Kamano, Y.; Herald, C.L. Antineoplastic Agents. 119. Isolation and Structure of Bryostatins 10 and 11. J. Org. Chem. 1987, 52, 2848–2854. [Google Scholar] [CrossRef]
- Hu, T.; deFreitas, A.S.W.; Curtis, J.M.; Oshima, Y.; Walter, J.A.; Wright, J.L.C. Isolation and Structure of Prorocentrolide B, a Fast-Acting Toxin from Prorocentrum maculosum. J. Nat. Prod. 1996, 59, 1010–1014. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Crouch, R.C.; Groweiss, A.; Pannell, L.K.; Van, Q.N.; Boyd, M.R. Application of High-Field NMR and Cryogenic Probe Technologies in the Structural Elucidation of Poecillastrin A, a New Antitumor Macrolide Lactam from the Sponge Poecillastra Species. Org. Lett. 2002, 4, 3293–3296. [Google Scholar] [CrossRef]
- Takada, K.; Choi, B.W.; Rashid, M.A.; Gamble, W.R.; Cardellina, J.H.; Van, Q.N.; Lloyd, J.R.; McMahon, J.B.; Gustafson, K.R. Structural Assignment of Poecillastrins B and C, Macrolide Lactams from the Deep-Water Caribbean Sponge Poecillastra Species. J. Nat. Prod. 2007, 70, 428–431. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, J.B.; Molinski, T.F. Caylobolide A, a Unique 36-Membered Macrolactone from a Bahamian Lyngbya m Ajuscula. Org. Lett. 2002, 4, 1535–1538. [Google Scholar] [CrossRef]
- Salvador, L.A.; Paul, V.J.; Luesch, H. Caylobolide B, a Macrolactone from Symplostatin 1-Producing Marine Cyanobacteria Phormidium Spp. from Florida. J. Nat. Prod. 2010, 73, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- De Joarder, D.; Jennings, M.P. Convergent Enantioselective Syntheses of Two Potential C25–C40 Subunits of (−)-Caylobolide A. Tetrahedron Lett. 2011, 52, 5124–5127. [Google Scholar] [CrossRef]
- Yadav, J.S.; Swapnil, N.; Venkatesh, M.; Prasad, A.R. Studies Directed toward the Synthesis of Caylobolide A: Convergent Synthesis of C21–C40 Subunit. Tetrahedron Lett. 2014, 55, 1164–1167. [Google Scholar] [CrossRef]
- Jiang, Z.-D.; Jensen, P.R.; Fenical, W. Lobophorins A and B, New Antiinflammatory Macrolides Produced by a Tropical Marine Bacterium. Bioorg. Med. Chem. Lett. 1999, 9, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Niu, J.; Liu, N.; Lü, Y.; Liu, M.; Li, Y. Cloning and Identification of the Lobophorin Biosynthetic Gene Cluster from Marine Streptomyces Olivaceus Strain FXJ7.023. Pak. J. Pharm. Sci. 2016, 29, 287–293. [Google Scholar]
- Cereghetti, D.M.; Carreira, E.M. Amphotericin B: 50 Years of Chemistry and Biochemistry. Synthesis 2006, 2006, 914–942. [Google Scholar] [CrossRef]
- Waugh, C.D. Amphotericin B. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. ISBN 978-0-08-055232-3. [Google Scholar]
- Ganis, P.; Avitabile, G.; Mechlinski, W.; Schaffner, C.P. Polyene Macrolide Antibiotic Amphotericin B. Crystal Structure of the N-Iodoacetyl Derivative. J. Am. Chem. Soc. 1971, 93, 4560–4564. [Google Scholar] [CrossRef]
- Zotchev, S.B. Polyene Macrolide Antibiotics and Their Applications in Human Therapy. Curr. Med. Chem. 2003, 10, 211–223. [Google Scholar] [CrossRef]
- Kim, D.-G.; Moon, K.; Kim, S.-H.; Park, S.-H.; Park, S.; Lee, S.K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Bahamaolides A and B, Antifungal Polyene Polyol Macrolides from the Marine Actinomycete Streptomyces sp. J. Nat. Prod. 2012, 75, 959–967. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Schulte, G.K. Discodermolide: A New Bioactive Polyhydroxylated Lactone from the Marine Sponge Discodermia dissoluta. J. Org. Chem. 1990, 55, 4912–4915. [Google Scholar] [CrossRef]
- Ruiz, C.; Valderrama, K.; Zea, S.; Castellanos, L. Mariculture and Natural Production of the Antitumoural (+)-Discodermolide by the Caribbean Marine Sponge Discodermia dissoluta. Mar. Biotechnol. 2013, 15, 571–583. [Google Scholar] [CrossRef]
- Smith, A.B.; Freeze, B.S. (+)-Discodermolide: Total Synthesis, Construction of Novel Analogues, and Biological Evaluation. Tetrahedron 2007, 64, 261–298. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Gavagnin, M.; Puliti, R.; Cimino, G.; Martinez, E.; Ortea, J.; Mattia, C.A. Dolabriferol: A New Polypropionate from the Skin of the Anaspidean Mollusc Dolabrifera dolabrifera. Tetrahedron 1996, 52, 12831–12838. [Google Scholar] [CrossRef]
- Jiménez-Romero, C.; González, K.; Rodríguez, A.D. Dolabriferols B and C, Non-Contiguous Polypropionate Esters from the Tropical Sea Hare Dolabrifera dolabrifera. Tetrahedron Lett. 2012, 53, 6641–6645. [Google Scholar] [CrossRef] [PubMed]
- Van Altena, I.; van Soest, R.; Roberge, M.; Andersen, R.J. Trisphaerolide A, a Novel Polyketide from the Dominican Sponge Erylus trisphaerus. J. Nat. Prod. 2003, 66, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Okanya, P.W.; Mohr, K.I.; Gerth, K.; Kessler, W.; Jansen, R.; Stadler, M.; Müller, R. Hyafurones, Hyapyrrolines, and Hyapyrones: Polyketides from Hyalangium minutum. J. Nat. Prod. 2014, 77, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Teta, R.; Della Sala, G.; Pawlik, J.; Mangoni, A.; Costantino, V. Isolation of Smenopyrone, a Bis-γ-Pyrone Polypropionate from the Caribbean Sponge Smenospongia aurea. Mar. Drugs 2018, 16, 285. [Google Scholar] [CrossRef]
- Meng, Y.; Van Wagoner, R.M.; Misner, I.; Tomas, C.; Wright, J.L.C. Structure and Biosynthesis of Amphidinol 17, a Hemolytic Compound from Amphidinium carterae. J. Nat. Prod. 2010, 73, 409–415. [Google Scholar] [CrossRef]
- Rutkowski, J.; Brzezinski, B. Structures and Properties of Naturally Occurring Polyether Antibiotics. BioMed Res. Int. 2013, 2013, 162513. [Google Scholar] [CrossRef]
- Williams, D.E.; Roberge, M.; Van Soest, R.; Andersen, R.J. Spirastrellolide A, An Antimitotic Macrolide Isolated from the Caribbean Marine Sponge Spirastrella coccinea. J. Am. Chem. Soc. 2003, 125, 5296–5297. [Google Scholar] [CrossRef]
- Williams, D.E.; Lapawa, M.; Feng, X.; Tarling, T.; Roberge, M.; Andersen, R.J. Spirastrellolide A: Revised Structure, Progress toward the Relative Configuration, and Inhibition of Protein Phosphatase 2A. Org. Lett. 2004, 6, 2607–2610. [Google Scholar] [CrossRef]
- Warabi, K.; Williams, D.E.; Patrick, B.O.; Roberge, M.; Andersen, R.J. Spirastrellolide B Reveals the Absolute Configuration of the Spirastrellolide Macrolide Core. J. Am. Chem. Soc. 2007, 129, 508–509. [Google Scholar] [CrossRef]
- Williams, D.E.; Keyzers, R.A.; Warabi, K.; Desjardine, K.; Riffell, J.L.; Roberge, M.; Andersen, R.J. Spirastrellolides C to G: Macrolides Obtained from the Marine Sponge Spirastrella coccinea. J. Org. Chem. 2007, 72, 9842–9845. [Google Scholar] [CrossRef]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic Acid, a Cytotoxic Polyether from Two Marine Sponges of the Genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Dickey, R.W.; Bobzin, S.C.; Faulkner, D.J.; Bencsath, F.A.; Andrzejewski, D. Identification of Okadaic Acid from a Caribbean Dinoflagellate, Prorocentrum convavum. Toxicon 1990, 28, 371–377. [Google Scholar] [CrossRef]
- Singh, S.B.; Zink, D.L.; Huber, J.; Genilloud, O.; Salazar, O.; Diez, M.T.; Basilio, A.; Vicente, F.; Byrne, K.M. Discovery of Lucensimycins A and B from Streptomyces lucensis MA7349 Using an Antisense Strategy. Org. Lett. 2006, 8, 5449–5452. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Zink, D.L.; Herath, K.B.; Salazar, O.; Genilloud, O. Discovery and Antibacterial Activity of Lucensimycin C from Streptomyces lucensis. Tetrahedron Lett. 2008, 49, 2616–2619. [Google Scholar] [CrossRef]
- Singh, S.B.; Zink, D.L.; Dorso, K.; Motyl, M.; Salazar, O.; Basilio, A.; Vicente, F.; Byrne, K.M.; Ha, S.; Genilloud, O. Isolation, Structure, and Antibacterial Activities of Lucensimycins D−G, Discovered from Streptomyces lucensis MA7349 Using an Antisense Strategy. J. Nat. Prod. 2009, 72, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Vera, B.; Rodríguez, A.D.; Avilés, E.; Ishikawa, Y. Aplysqualenols A and B: Squalene-Derived Polyethers with Antitumoral and Antiviral Activity from the Caribbean Sea Slug Aplysia dactylomela. Eur. J. Org. Chem. 2009, 2009, 5327–5336. [Google Scholar] [CrossRef]
- Shimizu, Y.; Chou, H.N.; Bando, H.; Van Duyne, G.; Clardy, J. Structure of Brevetoxin A (GB-1 Toxin), the Most Potent Toxin in the Florida Red Tide Organism Gymnodinium Breve (Ptychodiscus Brevis). J. Am. Chem. Soc. 1986, 108, 514–515. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Risk, M.; Ray, S.M.; Van Engen, D.; Clardy, J.; Golik, J.; James, J.C.; Nakanishi, K. Isolation and Structure of Brevetoxin B from the “Red Tide” Dinoflagellate Ptychodiscus Brevis (Gymnodinium Breve). J. Am. Chem. Soc. 1981, 103, 6773–6775. [Google Scholar] [CrossRef]
- Konoki, K.; Baden, D.G.; Scheuer, T.; Catterall, W.A. Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels. Toxins 2019, 11, 513. [Google Scholar] [CrossRef]
- Vernoux, J.-P.; Lewis, R.J. Isolation and Characterisation of Caribbean Ciguatoxins from the Horse-Eye Jack (Caranx latus). Toxicon 1997, 35, 889–900. [Google Scholar] [CrossRef]
- Lewis, R.J.; Vernoux, J.-P.; Brereton, I.M. Structure of Caribbean Ciguatoxin Isolated from Caranx latus. J. Am. Chem. Soc. 1998, 120, 5914–5920. [Google Scholar] [CrossRef]
- Marquais, M.; Sauviat, M.-P. Effet Des Ciguatoxines Sur Le Système Cardio-Circulatoire. J. De La Société De Biol. 1999, 193, 495–504. [Google Scholar] [CrossRef]
- Sauviat, M.-P.; Marquais, M.; Vernoux, J.-P. Muscarinic Effects of the Caribbean Ciguatoxin C-CTX-1 on Frog Atrial Heart Muscle. Toxicon 2002, 40, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Kryuchkov, F.; Robertson, A.; Miles, C.O.; Mudge, E.M.; Uhlig, S. LC–HRMS and Chemical Derivatization Strategies for the Structure Elucidation of Caribbean Ciguatoxins: Identification of C-CTX-3 and -4. Mar. Drugs 2020, 18, 182. [Google Scholar] [CrossRef] [PubMed]
- Bertin, M.; Wahome, P.; Zimba, P.; He, H.; Moeller, P. Trichophycin A, a Cytotoxic Linear Polyketide Isolated from a Trichodesmium thiebautii Bloom. Mar. Drugs 2017, 15, 10. [Google Scholar] [CrossRef]
- Bertin, M.J.; Saurí, J.; Liu, Y.; Via, C.W.; Roduit, A.F.; Williamson, R.T. Trichophycins B–F, Chlorovinylidene-Containing Polyketides Isolated from a Cyanobacterial Bloom. J. Org. Chem. 2018, 83, 13256–13266. [Google Scholar] [CrossRef]
- Balunas, M.J.; Grosso, M.F.; Villa, F.A.; Engene, N.; McPhail, K.L.; Tidgewell, K.; Pineda, L.M.; Gerwick, L.; Spadafora, C.; Kyle, D.E.; et al. Coibacins A–D, Antileishmanial Marine Cyanobacterial Polyketides with Intriguing Biosynthetic Origins. Org. Lett. 2012, 14, 3878–3881. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Proteau, P.J.; Nagle, D.G.; Hamel, E.; Blokhin, A.; Slate, D.L. Structure of Curacin A, a Novel Antimitotic, Antiproliferative and Brine Shrimp Toxic Natural Product from the Marine Cyanobacterium Lyngbya majuscula. J. Org. Chem. 1994, 59, 1243–1245. [Google Scholar] [CrossRef]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and Biosynthesis of the Jamaicamides, New Mixed Polyketide-Peptide Neurotoxins from the Marine Cyanobacterium Lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef]
- Nagle, D.G.; Geralds, R.S.; Yoo, H.-D.; Gerwick, W.H.; Kim, T.-S.; Nambu, M.; White, J.D. Absolute Configuration of Curacin A, a Novel Antimitotic Agent from the Tropical Marine Cyanobacterium Lyngbya majuscula. Tetrahedron Lett. 1995, 36, 1189–1192. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Lai, J.-Y.; Yoo, H.-D.; Yu, J.; Marquez, B.; Nagle, D.G.; Nambu, M.; White, J.D.; Falck, J.R.; Gerwick, W.H. Structure-Activity Analysis of the Interaction of Curacin A, the Potent Colchicine Site Antimitotic Agent, with Tubulin and Effects of Analogs on the Growth of MCF-7 Breast Cancer Cells. Mol. Pharmacol. 1998, 53, 62–76. [Google Scholar] [CrossRef]
- Graf, K.M.; Tabor, M.G.; Brown, M.L.; Paige, M. Synthesis of (S)-Jamaicamide C Carboxylic Acid. Org. Lett. 2009, 11, 5382–5385. [Google Scholar] [CrossRef]
- Watanabe, S.; Watanabe, S.; Aoki, N.; Usuki, T. Synthesis of the Polyketide (E)-Olefin of the Jamaicamides. Synth. Commun. 2013, 43, 1397–1403. [Google Scholar] [CrossRef]
- Teta, R.; Irollo, E.; Della Sala, G.; Pirozzi, G.; Mangoni, A.; Costantino, V. Smenamides A and B, Chlorinated Peptide/Polyketide Hybrids Containing a Dolapyrrolidinone Unit from the Caribbean Sponge Smenospongia aurea. Evaluation of Their Role as Leads in Antitumor Drug Research. Mar. Drugs 2013, 11, 4451–4463. [Google Scholar] [CrossRef] [PubMed]
- Amegadzie, A.K.; Ayer, W.A.; Sigler, L. Unusual Polyketides from the Wood-Decay Fungus Sistotrema raduloides. Can. J. Chem. 1995, 73, 2119–2125. [Google Scholar] [CrossRef]
- Mohamed, I.E.; Gross, H.; Pontius, A.; Kehraus, S.; Krick, A.; Kelter, G.; Maier, A.; Fiebig, H.-H.; König, G.M. Epoxyphomalin A and B, Prenylated Polyketides with Potent Cytotoxicity from the Marine-Derived Fungus Phoma sp. Org. Lett. 2009, 11, 5014–5017. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caribbean Region | Natural Product | Reference |
|---|---|---|
| Summerland Key, Florida | Tedanolide (2) | [16] |
| Key West, Florida | Caylobolide B (11b) | [41] |
| Naples, Florida, Gulf of Mexico | Lasonolide C–G (3c–g) | [22] |
| Florida, Gulf of Mexico | Bryostatin 16–18 (8a–c) | [31,32] |
| Florida, Gulf of Mexico | Brevetoxin A (25a) | [71] |
| Florida, Gulf of Mexico | Brevetoxin B (25b) | [72] |
| Padre Island, Corpus Christi, Texas, Gulf of Mexico | Trichophycin A (27a) | [79] |
| Padre Island, Corpus Christi, Texas, Gulf of Mexico | Trichophycin B–F (27b–f) and Tricholactone (28) | [80] |
| Florida, Belize, and Honduras | Looekeyolides A–B (4a–b) | [23] |
| Florida and Belize | Looekeyolide C–D (4c–d) | [24] |
| Belize | Lobophorin A–B (12a–b) | [38,39] |
| Dauphin Island, Alabama | Prorocentrolide B (9) | [37] |
| Bahamas | Lobophorolide (5) | [25] |
| Grand Bahama Island, Bahamas | Poecillastrin A–C (10a–c) | [34,35] |
| Bahamas | Caylobolide A (11a) | [40] |
| Bahamas | Bahamaolides A–B (13a–b) | [50] |
| Bahamas | Smenopyrone (18) | [58] |
| Little San Salvador Island, Bahamas | Amphidinol 17 (19) | [59] |
| Little Inagua, Bahamas | Smenamides A–B (32a–b) | [88] |
| St. Thomas, U.S. Virgin Islands | Amphidinolide B1–3 (6a–c) | [25,26,27] |
| St. Thomas, U.S. Virgin Islands | Caribenolide I (7) | [31] |
| St. Thomas, U.S. Virgin Islands | Ciguatoxins-3 (26c) and -4 (26d) | [78] |
| British Virgin Islands | Lasonolide A (3a) | [18,19] |
| British Virgin Islands and Florida Keys | Okadaic Acid (22) | [59,60] |
| Cuba | Dolabriferol (15a) | [54] |
| Puerto Rico | Dolabriferol B–C (15b) | [55] |
| Puerto Rico | Aplysqualenol A–B (24a–b) | [70] |
| Puerto Rico | Sistodiolynne (33), Sistolynone (34) and Sistopyrone (35) | [89] |
| Jamaica | (+)-Neopeltolide (1) | [12] |
| Jamaica | Jamaicamides A–C (31a–c) | [83] |
| Dominica | Trisphaerolide A (16) | [56] |
| Dominica | Spirastrellolide A–G (20a–g) | [55,56,57,58] |
| Dominica | Epoxyphomalin A–B (36a–b) | [90] |
| Windward Islands | Hyapyrone B (17) | [57] |
| Martinique | Lucensimycin A–G (23a–g) | [61,62,63] |
| St. Barthelemy | Ciguatoxins-1 (26a) and -2 (26b) | [68,69] |
| Curacao | Curacin A (30) | [82] |
| Santa Marta, Colombia | (+)-Discodermolide (14) | [51] |
| Panama | Coibacin A–D (29a–d) | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Berríos, R.R.; Ríos-Delgado, A.M.; Perdomo-Lizardo, A.P.; Cardona-Rivera, A.E.; Vidal-Rosado, Á.G.; Narváez-Lozano, G.A.; Nieves-Quiñones, I.A.; Rodríguez-Vargas, J.A.; Álamo-Diverse, K.Y.; Lebrón-Acosta, N.; et al. Extraction, Isolation, Characterization, and Bioactivity of Polypropionates and Related Polyketide Metabolites from the Caribbean Region. Antibiotics 2023, 12, 1087. https://doi.org/10.3390/antibiotics12071087
Rodríguez-Berríos RR, Ríos-Delgado AM, Perdomo-Lizardo AP, Cardona-Rivera AE, Vidal-Rosado ÁG, Narváez-Lozano GA, Nieves-Quiñones IA, Rodríguez-Vargas JA, Álamo-Diverse KY, Lebrón-Acosta N, et al. Extraction, Isolation, Characterization, and Bioactivity of Polypropionates and Related Polyketide Metabolites from the Caribbean Region. Antibiotics. 2023; 12(7):1087. https://doi.org/10.3390/antibiotics12071087
Chicago/Turabian StyleRodríguez-Berríos, Raúl R., Agnes M. Ríos-Delgado, Amanda P. Perdomo-Lizardo, Andrés E. Cardona-Rivera, Ángel G. Vidal-Rosado, Guillermo A. Narváez-Lozano, Iván A. Nieves-Quiñones, Jeremy A. Rodríguez-Vargas, Keiry Y. Álamo-Diverse, Naiara Lebrón-Acosta, and et al. 2023. "Extraction, Isolation, Characterization, and Bioactivity of Polypropionates and Related Polyketide Metabolites from the Caribbean Region" Antibiotics 12, no. 7: 1087. https://doi.org/10.3390/antibiotics12071087
APA StyleRodríguez-Berríos, R. R., Ríos-Delgado, A. M., Perdomo-Lizardo, A. P., Cardona-Rivera, A. E., Vidal-Rosado, Á. G., Narváez-Lozano, G. A., Nieves-Quiñones, I. A., Rodríguez-Vargas, J. A., Álamo-Diverse, K. Y., Lebrón-Acosta, N., Medina-Berríos, N., Rivera-Lugo, P. S., Avellanet-Crespo, Y. A., & Ortiz-Colón, Y. W. (2023). Extraction, Isolation, Characterization, and Bioactivity of Polypropionates and Related Polyketide Metabolites from the Caribbean Region. Antibiotics, 12(7), 1087. https://doi.org/10.3390/antibiotics12071087