




Comparative Genomics of Pseudomonas aeruginosa Strains Isolated from Different Ecological Niches

 ,
,  , ,
, ,  , and
, and 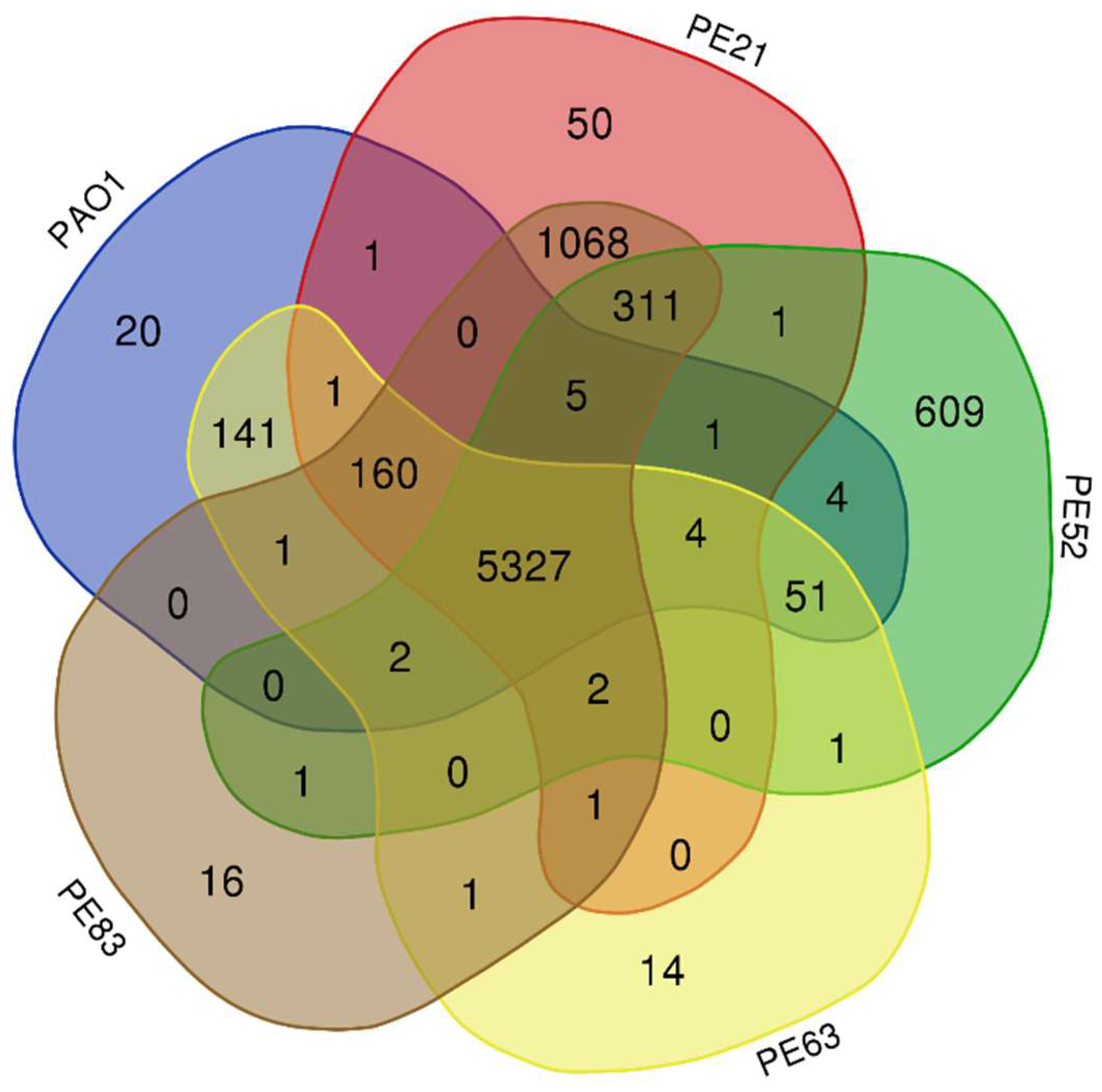{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. General Features of the Genomes
2.2. Multilocus Sequence Typing (MLST)
2.3. Genomic Comparison of Strains Recovered from Mexican Hospital
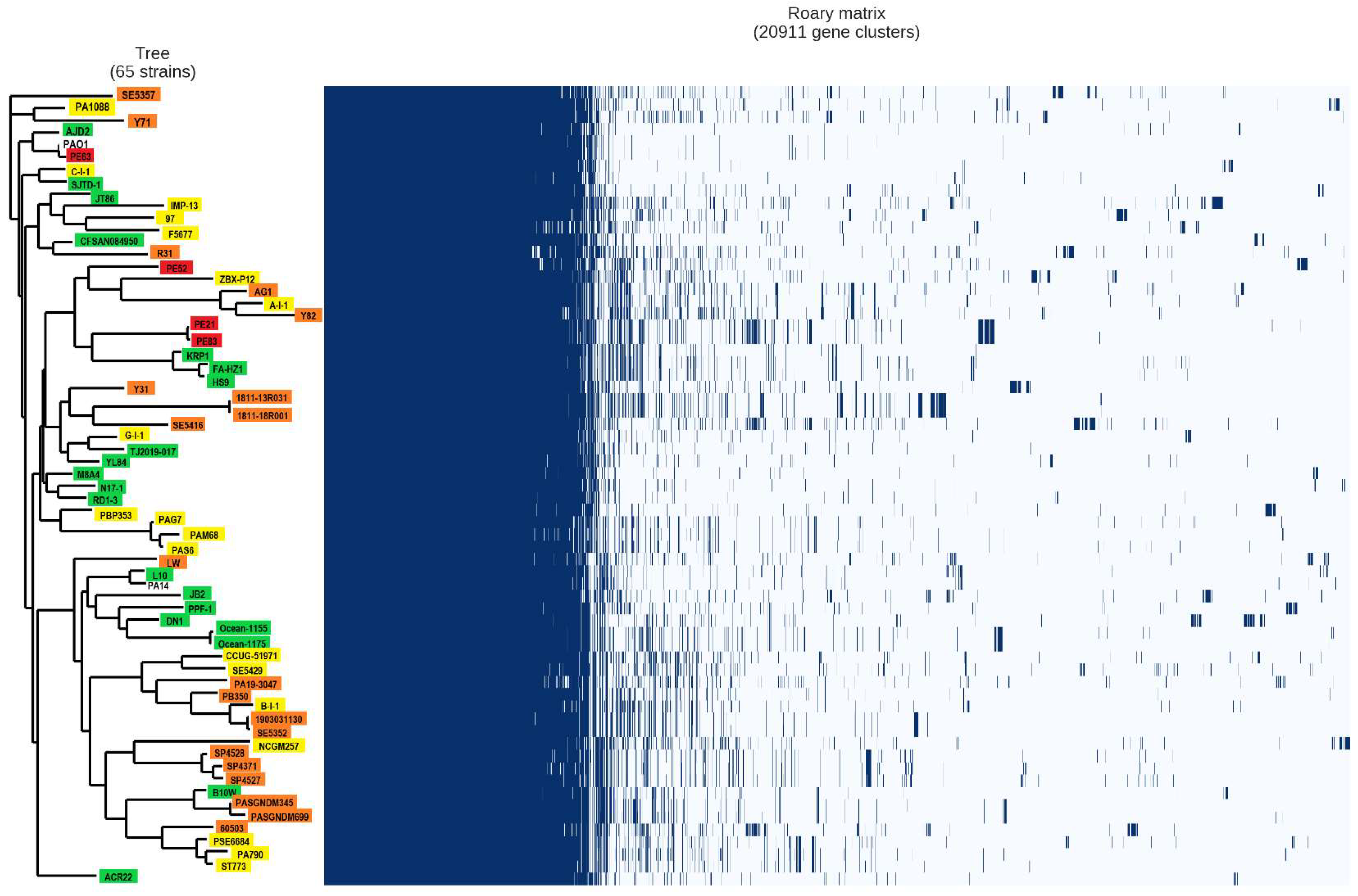
2.4. Analysis of the Pan-Genome of the Strains from the Mexican Hospital and of the GenBank
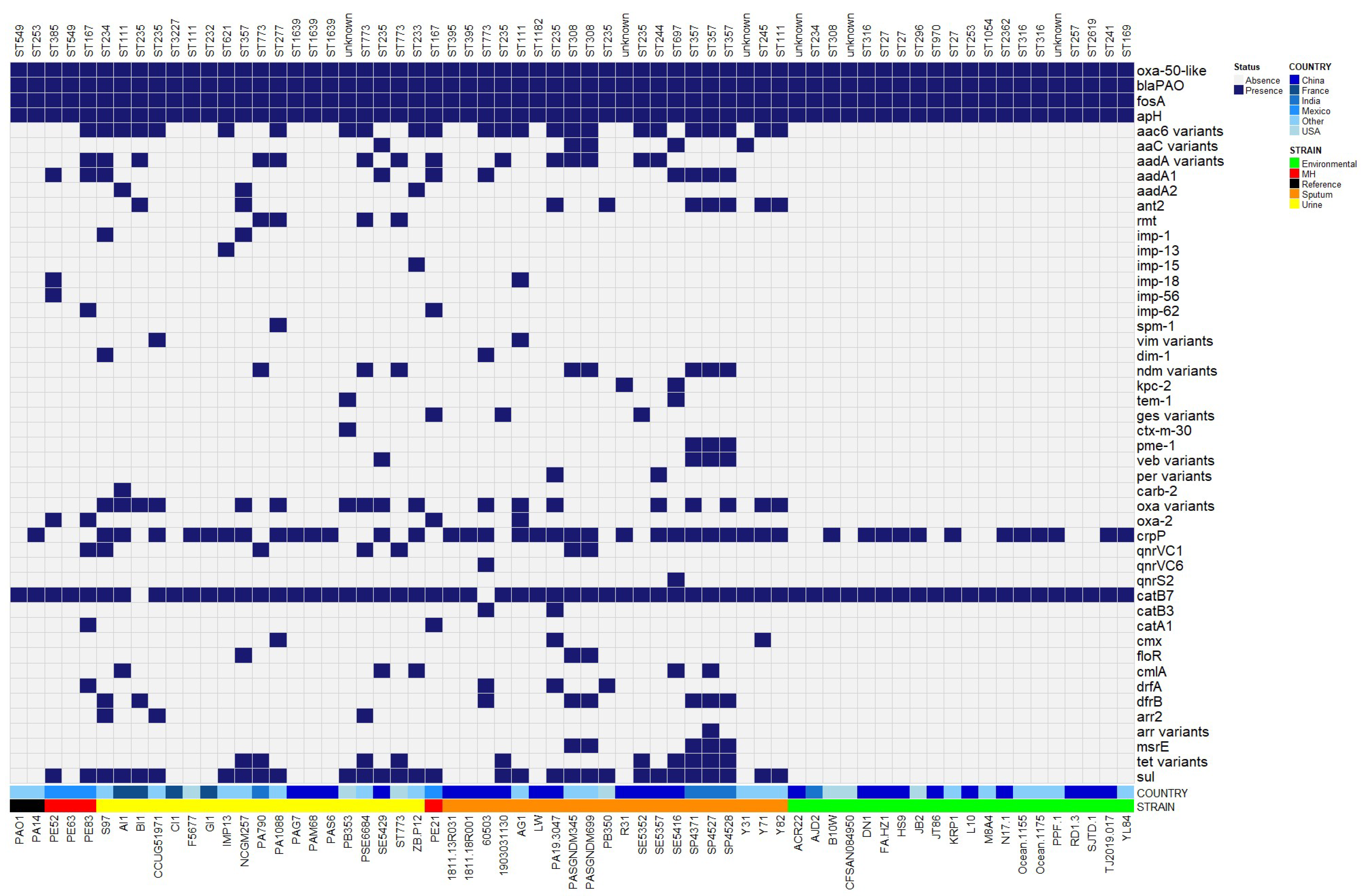
2.5. Antibiotic Resistance Genes
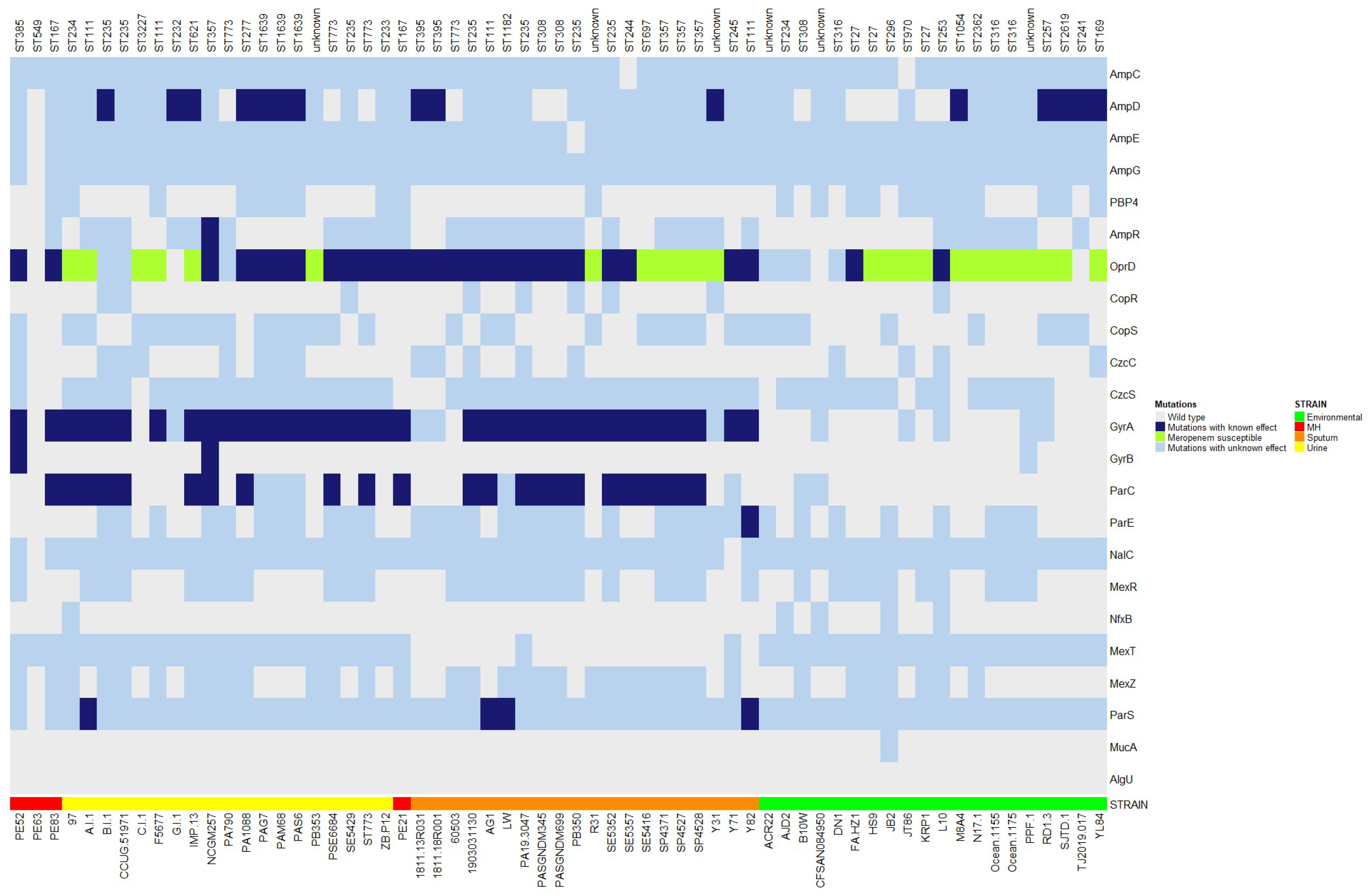
2.6. Gene Mutations Associated with Drug Resistance
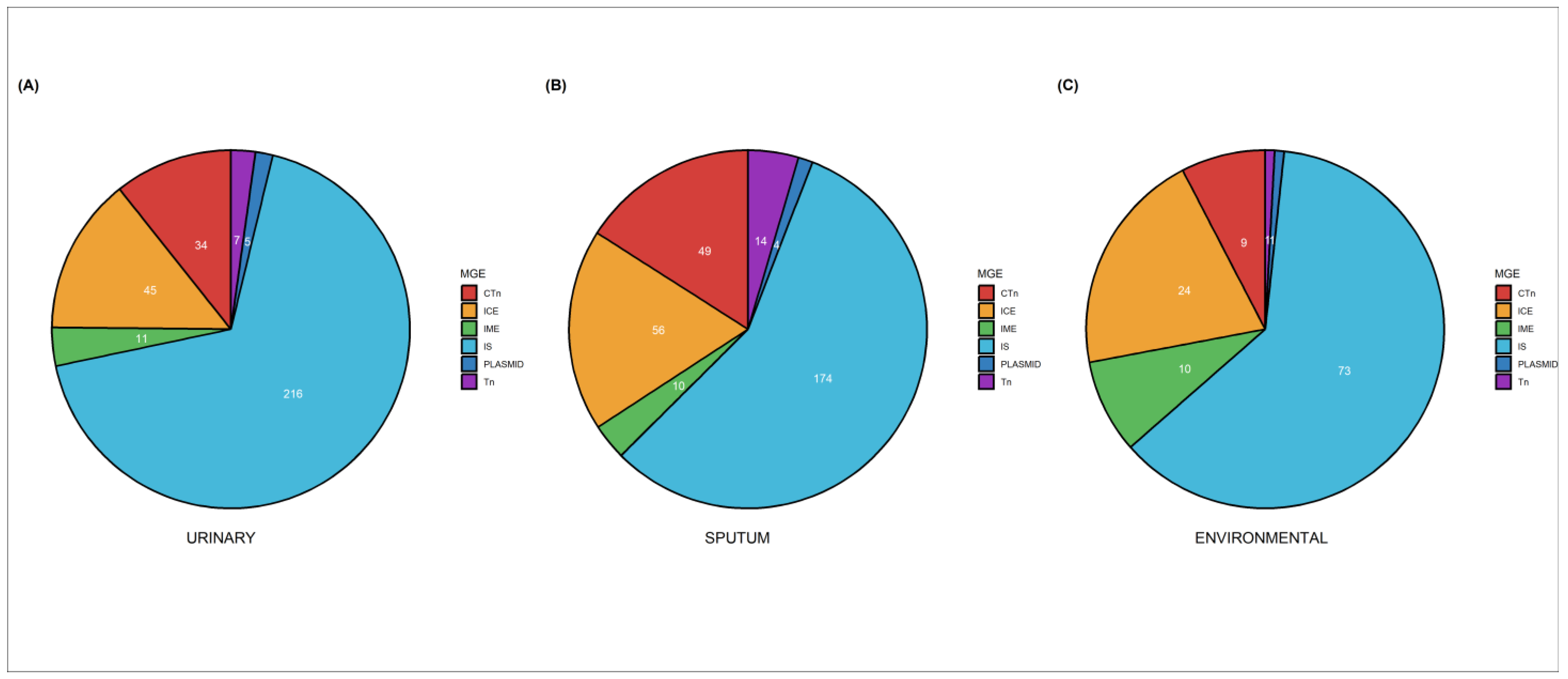
2.7. Mobilome
2.8. Correlation between the Presence of Plasmids, Relaxases MOB, CRISPR-Cas, and Anti-CRISPR Systems
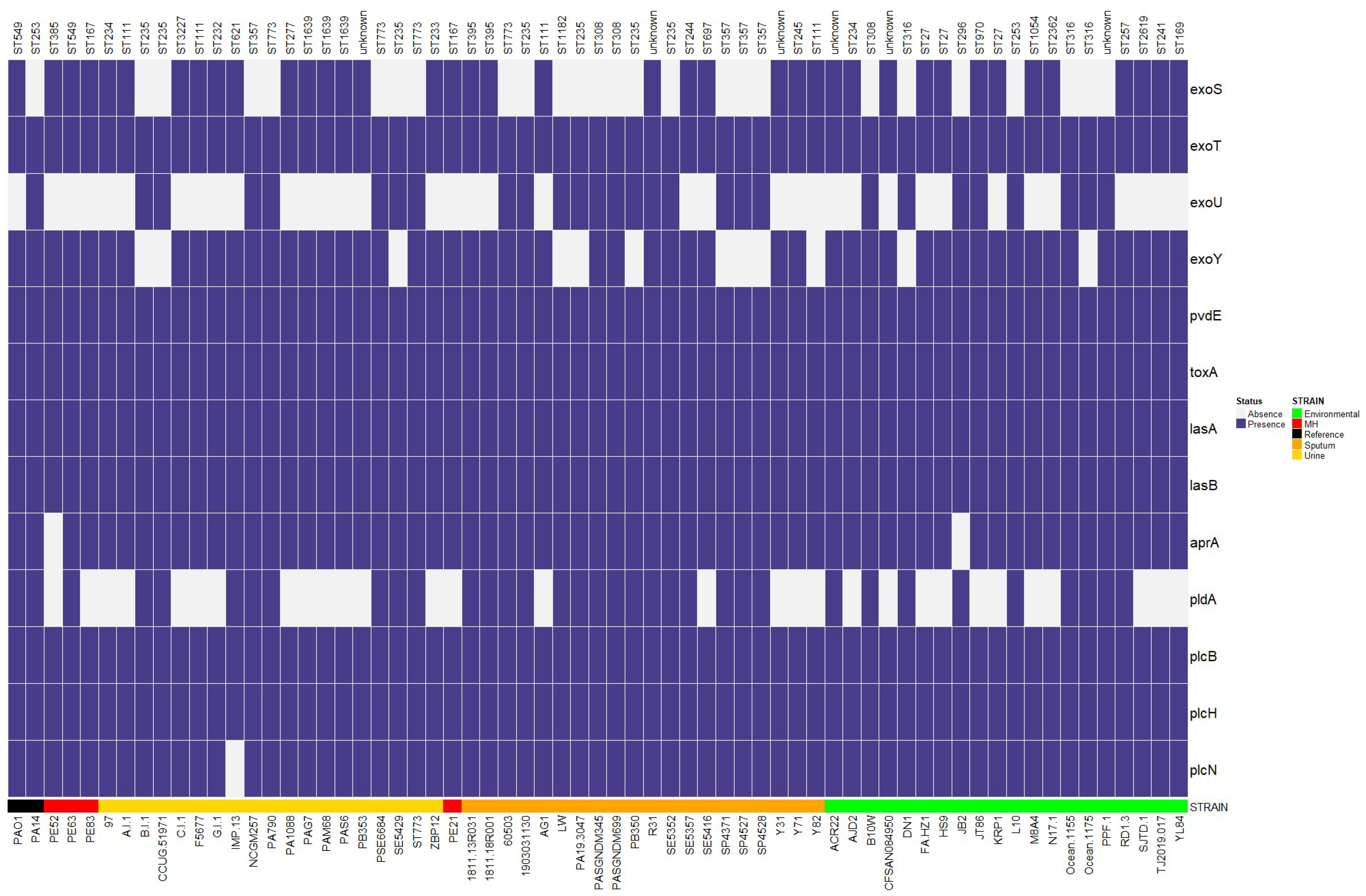
2.9. Virulence Genes
3. Discussion
4. Materials and Methods
4.1. Bacterial Genomes
4.2. MLST (Multilocus Sequence Typing)
4.3. Genome Comparison of Strains Isolated from the Mexican Hospital
4.4. Comparative Analysis of Strains from Three Isolation Sources
4.5. Resistome Analysis
4.6. Virulence Analysis
4.7. Mobile Genetic Elements (MGEs)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zarei, O.; Shokoohizadeh, L.; Hossainpour, H.; Alikhani, M.Y. Molecular analysis of Pseudomonas aeruginosa isolated from clinical, environmental and cockroach sources by ERIC-PCR. BMC Res. Notes 2018, 11, 668. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Wilson, M.G.; Pandey, S. Pseudomonas aeruginosa; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Breidenstein, E.B.M.; de la Fuente-Núñez, C.; Hancock, R.E.W. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic resistance in Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist. Updat. 2019, 44, 100640. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Huang, X.; Wang, Q.; Yao, D.; Lu, W. Virulence Factors of Pseudomonas aeruginosa and Antivirulence Strategies to Combat Its Drug Resistance. Front. Cell. Infect. Microbiol. 2022, 12, 926758. [Google Scholar] [CrossRef]
- Shehabi, A.A.; Kamal, A.M. Pseudomonas aeruginosa a common opportunistic pathogen in Jordan: A review article. Int. Arab. J. Antimicrob. Agents 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Miyada, C.G.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar] [CrossRef]
- Sawa, T.; Shimizu, M.; Moriyama, K.; Wiener-Kronish, J.P. Association between Pseudomonas aeruginosa Type III Secretion, Antibiotic Resistance, and Clinical Outcome: A Review. Crit. Care 2014, 18, 668. [Google Scholar] [CrossRef]
- Sawa, T.; Momiyama, K.; Mihara, T.; Kainuma, A.; Kinoshita, M.; Moriyama, K. Molecular epidemiology of clinically high-risk Pseudomonas aeruginosa strains: Practical overview. Microbiol. Immunol. 2020, 64, 331–344. [Google Scholar] [CrossRef]
- Kung, V.L.; Ozer, E.A.; Hauser, A.R. The Accessory Genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2010, 74, 621–641. [Google Scholar] [CrossRef]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed]
- Mathee, K.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; Zeng, E.; et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 2007, 105, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Subedi, D.; Vijay, A.K.; Kohli, G.S.; Rice, S.A.; Willcox, M. Comparative genomics of clinical strains of Pseudomonas aeruginosa strains isolated from different geographic sites. Sci. Rep. 2018, 8, 15668. [Google Scholar] [CrossRef]
- Subedi, D.; Kohli, G.S.; Vijay, A.K.; Willcox, M.; Rice, S.A. Accessory genome of the multi-drug resistant ocular isolate of Pseudomonas aeruginosa PA34. PLoS ONE 2019, 14, e0215038. [Google Scholar] [CrossRef] [PubMed]
- Dettman, J.R.; Kassen, R. Evolutionary genomics of niche-specific adaptation to the cystic fibrosis lung in Pseudomonas aeruginosa. Mol. Biol. Evol. 2021, 38, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.E.; Higgs, P.G. Testing the infinitely many genes model for the evolution of the bacterial core genome and pangenome. Mol. Biol. Evol. 2012, 29, 3413–3425. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An audacious pathogen with an adaptable arsenal of virulence factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef]
- Curran, B.; Jonas, D.; Grundmann, H.; Pitt, T.; Dowson, C.G. Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 2004, 42, 5644–5649. [Google Scholar] [CrossRef]
- Oliver, A.; Mulet, X.; López-Causapé, C.; Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2015, 21–22, 41–59. [Google Scholar] [CrossRef]
- del Barrio-Tofiño, E.; López-Causapé, C.; Oliver, A. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int. J. Antimicrob. Agents 2020, 56, 106196. [Google Scholar] [CrossRef] [PubMed]
- Guzvinec, M.; Izdebski, R.; Butic, I.; Jelic, M.; Abram, M.; Koscak, I.; Baraniak, A.; Hryniewicz, W.; Gniadkowski, M.; Andrasevic, A.T. Sequence types 235, 111, and 132 Predominate among multidrug-resistant Pseudomonas aeruginosa clinical isolates in croatia. Antimicrob. Agents Chemother. 2014, 58, 6277–6283. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.L.; Turton, J.F.; Livermore, D.M.; Hopkins, K.L.; Woodford, N. Dominance of international “high-risk clones” among metallo-β-lactamase-producing Pseudomonas aeruginosa in the UK. J. Antimicrob. Chemother. 2015, 70, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.S.; Song, W.; Park, M.J.; Jeong, S.; Lee, N.; Jeong, S.H. Molecular Characterization of the First Emerged NDM-1-Producing Pseudomonas aeruginosa Isolates in South Korea. Microbial. Drug Resist. 2021, 27, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Kidd, T.J.; Ritchie, S.R.; Ramsay, K.A.; Grimwood, K.; Bell, S.C.; Rainey, P.B. Pseudomonas aeruginosa Exhibits Frequent Recombination, but Only a Limited Association between Genotype and Ecological Setting. PLoS ONE 2012, 7, e44199. [Google Scholar] [CrossRef]
- Moloney, E.M.; Deasy, E.C.; Swan, J.S.; Brennan, G.I.; O’Donnell, M.J.; Coleman, D.C. Whole-genome sequencing identifies highly related Pseudomonas aeruginosa strains in multiple washbasin U-bends at several locations in one hospital: Evidence for trafficking of potential pathogens via wastewater pipes. J. Hosp. Infect. 2020, 104, 484–491. [Google Scholar] [CrossRef]
- Pseudomonas aeruginosa|PubMLST. Available online: https://pubmlst.org/organisms/pseudomonas-aeruginosa (accessed on 5 March 2023).
- Haenni, M.; Hocquet, D.; Ponsin, C.; Cholley, P.; Guyeux, C.; Madec, J.Y.; Bertrand, X. Population structure and antimicrobial susceptibility of Pseudomonas aeruginosa from animal infections in France. BMC Vet. Res. 2015, 11, 9. [Google Scholar] [CrossRef]
- Ruiz-Roldán, L.; Bellés, A.; Bueno, J.; Azcona-Gutiérrez, J.M.; Rojo-Bezares, B.; Torres, C.; Castillo, F.J.; Sáenz, Y.; Seral, C. Pseudomonas aeruginosa Isolates from Spanish Children: Occurrence in Faecal Samples, Antimicrobial Resistance, Virulence, and Molecular Typing. Biomed Res. Int. 2018, 2018, 8060178. [Google Scholar] [CrossRef] [PubMed]
- Seidl, K.; Leimer, N.; Palheiros Marques, M.; Furrer, A.; Holzmann-Bürgel, A.; Senn, G.; Zbinden, R.; Zinkernagel, A.S. Clonality and antimicrobial susceptibility of methicillin-resistant Staphylococcus aureus at the University Hospital Zurich, Switzerland between 2012 and 2014. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Tedim, A.P.; Coque, T.M. Antibiotic resistance shaping multi-level population biology of bacteria. Front. Microbiol. 2013, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Guimarães, L.C.; Silva, A.; Soares, S.C.; Baraúna, R.A. First Steps in the Analysis of Prokaryotic Pan-Genomes. Bioinform. Biol. Insights 2020, 14, 1177932220938064. [Google Scholar] [CrossRef] [PubMed]
- Gautreau, G.; Bazin, A.; Gachet, M.; Planel, R.; Burlot, L.; Dubois, M.; Perrin, A.; Médigue, C.; Calteau, A.; Cruveiller, S.; et al. PPanGGOLiN: Depicting microbial diversity via a partitioned pangenome graph. PLoS Comput. Biol. 2020, 16, e1007732. [Google Scholar] [CrossRef] [PubMed]
- Valot, B.; Guyeux, C.; Rolland, J.Y.; Mazouzi, K.; Bertrand, X.; Hocquet, D. What it takes to be a Pseudomonas aeruginosa? The core genome of the opportunistic pathogen updated. PLoS ONE 2015, 10, e0126468. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, K.; Thirumalmuthu, K.; Prajna, N.V.; Lalitha, P.; Mohankumar, V.; Devarajan, B. Comparative genomics of ocular Pseudomonas aeruginosa strains from keratitis patients with different clinical outcomes. Genomics 2020, 112, 4769–4776. [Google Scholar] [CrossRef] [PubMed]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.G.; Kukavica-Ibrulj, I.; Dupont, M.J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef]
- Poulsen, B.E.; Yang, R.; Clatworthy, A.E.; White, T.; Osmulski, S.J.; Li, L.; Penaranda, C.; Lander, E.S.; Shoresh, N.; Hung, D.T. Defining the core essential genome of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2019, 116, 10072–10080. [Google Scholar] [CrossRef]
- Bianconi, I.; Jeukens, J.; Freschi, L.; Alcalá-Franco, B.; Facchini, M.; Boyle, B.; Molinaro, A.; Kukavica-Ibrulj, I.; Tümmler, B.; Levesque, R.C.; et al. Comparative genomics and biological characterization of sequential Pseudomonas aeruginosa isolates from persistent airways infection. BMC Genom. 2015, 16, 1105. [Google Scholar] [CrossRef]
- Muthukumarasamy, U.; Preusse, M.; Kordes, A.; Koska, M.; Schniederjans, M.; Khaledi, A.; Häussler, S. Single-nucleotide polymorphism-based genetic diversity analysis of clinical Pseudomonas aeruginosa isolates. Genome Biol. Evol. 2020, 12, 396–406. [Google Scholar] [CrossRef]
- Jackson, R.W.; Vinatzer, B.; Arnold, D.L.; Dorus, S.; Murillo, J. The influence of the accessory genome on bacterial pathogen evolution. Mob. Genet. Elem. 2011, 1, 55–65. [Google Scholar] [CrossRef]
- Janice, J.; Agyepong, N.; Owusu-Ofori, A.; Govinden, U.; Essack, S.Y.; Samuelsen, Ø.; Sundsfjord, A.; Pedersen, T. Carbapenem resistance determinants acquired through novel chromosomal integrations in extensively drug-resistant pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2021, 65, e00289-21. [Google Scholar] [CrossRef]
- Guimarães, L.C.; Benevides De Jesus, L.; Vinícius, M.; Viana, C.; Silva, A.; Thiago, R.; Ramos, J.; De, S.; Soares, C.; Azevedo, V. Inside the Pan-genome-Methods and Software Overview. Curr. Genom. 2015, 16, 245–252. [Google Scholar] [CrossRef] [PubMed]
- López-García, A.; del Carmen Rocha-Gracia, R.; Bello-López, E.; Juárez-Zelocualtecalt, C.; Sáenz, Y.; Castañeda-Lucio, M.; López-Pliego, L.; Cristina González-Vázquez, M.; Torres, C.; Ayala-Nuñez, T.; et al. Characterization of antimicrobial resistance mechanisms in carbapenem-resistant Pseudomonas aeruginosa carrying IMP variants recovered from a Mexican hospital. Infect. Drug Resist. 2018, 11, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.J.; Jeong, S.H. Mobile carbapenemase genes in Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 614058. [Google Scholar] [CrossRef]
- Khan, M.; Stapleton, F.; Summers, S.; Rice, S.A.; Willcox, M.D.P. Antibiotic resistance characteristics of Pseudomonas aeruginosa isolated from keratitis in Australia and India. Antibiotics 2020, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Irum, S.; Naz, K.; Ullah, N.; Mustafa, Z.; Ali, A.; Arslan, M.; Khalid, K.; Andleeb, S. Antimicrobial resistance and genomic characterization of six new sequence types in multidrug-resistant Pseudomonas aeruginosa clinical isolates from pakistan. Antibiotics 2021, 10, 1386. [Google Scholar] [CrossRef]
- Diorio-Toth, L.; Irum, S.; Potter, R.F.; Wallace, M.A.; Arslan, M.; Munir, T.; Andleeb, S.; Burnham, C.-A.D.; Dantas, G. Genomic Surveillance of Clinical Pseudomonas aeruginosa Isolates Reveals an Additive Effect of Carbapenemase Production on Carbapenem Resistance. Microbiol. Spectr. 2022, 10, e00766-22. [Google Scholar] [CrossRef]
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048. [Google Scholar] [CrossRef]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-lactamase database (BLDB)–structure and function. J. Enzyme Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Streling, A.P.; Cayô, R.; Nodari, C.S.; Almeida, L.G.P.; Bronze, F.; Siqueira, A.V.; Matos, A.P.; Oliveira, V.; Vasconcelos, A.T.R.; Marcondes, M.F.M.; et al. Kinetics Analysis of β-Lactams Hydrolysis by OXA-50 Variants of Pseudomonas aeruginosa. Microb. Drug Resist. 2022, 28, 849–852. [Google Scholar] [CrossRef]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M enzymes: Origin and diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef]
- Bathoorn, E.; Tsioutis, C.; da Silva Voorham, J.M.; Scoulica, E.V.; Ioannidou, E.; Zhou, K.; Rossen, J.W.; Gikas, A.; Friedrich, A.W.; Grundmann, H. Emergence of pan-resistance in KPC-2 carbapenemase-producing Klebsiella pneumoniae in Crete, Greece: A close call. J. Antimicrob. Chemother. 2016, 71, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Falco, A.; Ramos, Y.; Franco, E.; Guzmán, A.; Takiff, H. A cluster of KPC-2 and VIM-2-producing Klebsiella pneumoniae ST833 isolates from the pediatric service of a Venezuelan Hospital. BMC Infect. Dis. 2016, 16, 595. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Baishya, J.; Wakeman, C.A. Pseudomonas aeruginosa polymicrobial interactions during lung infection. Curr. Opin. Microbiol. 2020, 53, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gaston, J.R.; Johnson, A.O.; Bair, K.L.; White, A.N.; Armbruster, C.E. Polymicrobial interactions in the urinary tract: Is the enemy of my enemy my friend? Infect. Immun. 2021, 89, e00652-20. [Google Scholar] [CrossRef]
- Ramsay, K.A.; Wardell, S.J.T.; Patrick, W.M.; Brockway, B.; Reid, D.W.; Winstanley, C.; Bell, S.C.; Lamont, I.L. Genomic and phenotypic comparison of environmental and patient-derived isolates of Pseudomonas aeruginosa suggest that antimicrobial resistance is rare within the environment. J. Med. Microbiol. 2019, 68, 1591–1595. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Jacobo, V.M.; Hernández-Ramírez, K.C.; Romo-Rodríguez, P.; Viridiana Pérez-Gallardo, R.; Campos-García, J.; Félix Gutiérrez-Corona, J.; Pablo García-Merinos, J.; Meza-Carmen, V.; Silva-Sánchez, J.; Ramírez-Díaz, M.I. CrpP Is a Novel Ciprofloxacin-Modifying Enzyme Encoded by the Pseudomonas aeruginosa pUM505 Plasmid. Antimicrob. Agents Chemother. 2018, 62, e02629-17. [Google Scholar] [CrossRef]
- Zubyk, H.L.; Wright, G.D. Crpp is not a fluoroquinolone-inactivating enzyme. Antimicrob. Agents Chemother. 2021, 65, e00773-21. [Google Scholar] [CrossRef]
- Sánchez-Martinez, G.; Garza-Ramos, U.J.; Reyna-Flores, F.L.; Gaytán-Martínez, J.; Lorenzo-Bautista, I.G.; Silva-Sanchez, J. In169, A New Class 1 Integron that Encoded blaIMP-18 in a Multidrug-Resistant Pseudomonas aeruginosa Isolate from Mexico. Arch. Med. Res. 2010, 41, 235–239. [Google Scholar] [CrossRef]
- Touati, M.; Diene, S.M.; Dekhil, M.; Djahoudi, A.; Racherache, A.; Rolain, J.M. Dissemination of a class I integron carrying VIM-2 carbapenemase in Pseudomonas aeruginosa clinical isolates from a hospital intensive care unit in annaba Algeria. Antimicrob. Agents Chemother. 2013, 57, 2426–2427. [Google Scholar] [CrossRef]
- el Salabi, A.; Toleman, M.A.; Weeks, J.; Bruderer, T.; Frei, R.; Walsh, T.R. First report of the metallo-β-lactamase SPM-1 in Europe. Antimicrob. Agents Chemother. 2010, 54, 582. [Google Scholar] [CrossRef]
- Ramírez, D.G.; Nicola, F.; Zarate, S.; Relloso, S.; Smayevsky, J.; Arduino, S. Emergence of Pseudomonas aeruginosa with KPC-type carbapenemase in a teaching hospital: An 8-year study. J. Med. Microbiol. 2013, 62, 1565–1570. [Google Scholar] [CrossRef]
- Tohya, M.; Tada, T.; Watanabe, S.; Kuwahara-Arai, K.; Zin, K.N.; Zaw, N.N.; Aung, M.Y.; Mya, S.; Zan, K.N.; Kirikae, T.; et al. Emergence of carbapenem-resistant Pseudomonas asiatica producing NDM-1 and VIM-2 Metallo-Lactamases in Myanmar. Antimicrob. Agents Chemother. 2019, 63, e00475-19. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Rojo-Bezares, B.; Chichón, G.; Sáenz, Y. Resistance to Fluoroquinolones in Pseudomonas aeruginosa from Human, Animal, Food and Environmental Origin: The Role of CrpP and Mobilizable ICEs. Antibiotics 2022, 11, 1271. [Google Scholar] [CrossRef] [PubMed]
- Ramsamy, Y.; Mlisana, K.P.; Amoako, D.G.; Abia, A.L.K.; Ismail, A.; Allam, M.; Mbanga, J.; Singh, R.; Essack, S.Y. Mobile genetic elements-mediated Enterobacterales-associated carbapenemase antibiotic resistance genes propagation between the environment and humans: A One Health South African study. Sci. Total Environ. 2022, 806, 150641. [Google Scholar] [CrossRef]
- Shintani, M.; Sanchez, Z.K.; Kimbara, K. Genomics of microbial plasmids: Classification and identification based on replication and transfer systems and host taxonomy. Front. Microbiol. 2015, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- van Belkum, A.; Soriaga, L.B.; LaFave, M.C.; Akella, S.; Veyrieras, J.B.; Barbu, E.M.; Shortridge, D.; Blanc, B.; Hannum, G.; Zambardi, G.; et al. Phylogenetic distribution of CRISPR-Cas systems in antibiotic- resistant Pseudomonas aeruginosa. MBio 2015, 6, e01796-15. [Google Scholar] [CrossRef]
- Shehreen, S.; Chyou, T.Y.; Fineran, P.C.; Brown, C.M. Genome-wide correlation analysis suggests different roles of CRISPR-Cas systems in the acquisition of antibiotic resistance genes in diverse species. Phil. Trans. R. Soc. B 2019, 374, 20180384. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Garcia, B.; Strum, S.; Du, M.; Rollins, M.F.; Hidalgo-Reyes, Y.; Wiedenheft, B.; Maxwell, K.L.; Davidson, A.R. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 2015, 526, 136–139. [Google Scholar] [CrossRef]
- Wheatley, R.M.; MacLean, R.C. CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa. ISME J. 2021, 15, 1420–1433. [Google Scholar] [CrossRef]
- Pinilla-Redondo, R.; Mayo-Muñoz, D.; Russel, J.; Garrett, R.A.; Randau, L.; Sørensen, S.J.; Shah, S.A. Type IV CRISPR-Cas systems are highly diverse and involved in competition between plasmids. Nucleic Acids Res. 2020, 48, 2000–2012. [Google Scholar] [CrossRef] [PubMed]
- Pinilla-Redondo, R.; Russel, J.; Mayo-Muñoz, D.; Shah, S.A.; Garrett, R.A.; Nesme, J.; Madsen, J.S.; Fineran, P.C.; Sørensen, S.J. CRISPR-Cas systems are widespread accessory elements across bacterial and archaeal plasmids. Nucleic Acids Res. 2022, 50, 4315–4328. [Google Scholar] [CrossRef] [PubMed]
- Kamruzzaman, M.; Iredell, J.R. CRISPR-Cas System in Antibiotic Resistance Plasmids in Klebsiella pneumoniae. Front. Microbiol. 2020, 10, 2934. [Google Scholar] [CrossRef]
- Almendros, C.; Guzmán, N.M.; García-Martínez, J.; Mojica, F.J.M. Anti-cas spacers in orphan CRISPR4 arrays prevent uptake of active CRISPR-Cas I-F systems. Nat. Microbiol. 2016, 1, 16081. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, Y.F.; Williams, B.J.; Blackwell, T.S.; Xie, C.M. Structure and function of OprD protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 2012, 302, 63–68. [Google Scholar] [CrossRef]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [PubMed]
- González-Vázquez, M.C.; Rocha-Gracia, R.d.C.; Carabarín-Lima, A.; Bello-López, E.; Huerta-Romano, F.; Martínez-Laguna, Y.; Lozano-Zarain, P. Location of OprD porin in Pseudomonas aeruginosa clinical isolates. APMIS 2021, 129, 213–224. [Google Scholar] [CrossRef]
- Epp, S.F.; Köhler, T.; Plésiat, P.; Michéa-Hamzehpour, M.; Frey, J.; Pechère, J.C. C-terminal region of Pseudomonas aeruginosa outer membrane porin OprD modulates susceptibility to meropenem. Antimicrob. Agents Chemother 2001, 45, 1780–1787. [Google Scholar] [CrossRef]
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S.; et al. Evolution of the Pseudomonas aeruginosa mutational resistome in an international Cystic Fibrosis clone. Sci. Rep. 2017, 7, 5555. [Google Scholar] [CrossRef]
- Martinez, J.L. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc. Biol. Sci. 2009, 276, 2521–2530. [Google Scholar] [CrossRef]
- Berrazeg, M.; Jeannot, K.; Ntsogo Enguéné, V.Y.; Broutin, I.; Loeffert, S.; Fournier, D.; Plésiat, P. Mutations in β-lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255. [Google Scholar] [CrossRef] [PubMed]
- Jouault, A.; Saliba, A.M.; Touqui, L. Modulation of the immune response by the Pseudomonas aeruginosa type-III secretion system. Front. Cell. Infect. Microbiol. 2022, 12, 1806. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, D.M.; McLean, K.; Haneef, A.S.; Fernig, D.G.; Winstanley, C.; Berry, N.; Kaye, S.B. Pseudomonas aeruginosa toxin ExoU as a therapeutic target in the treatment of bacterial infections. Microorganisms 2019, 7, 707. [Google Scholar] [CrossRef]
- Diaz, M.H.; Shaver, C.M.; King, J.D.; Musunuri, S.; Kazzaz, J.A.; Hauser, A.R. Pseudomonas aeruginosa induces localized immunosuppression during pneumonia. Infect. Immun. 2008, 76, 4414–4421. [Google Scholar] [CrossRef]
- Hauser, A.R.; Cobb, E.; Bodí, M.; Mariscal, D.; Vallés, J.; Engel, J.N.; Rello, J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit. Care Med. 2002, 30, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.M.K.; Wiehlmann, L.; Ashelford, K.E.; Preston, S.J.; Frimmersdorf, E.; Campbell, B.J.; Neal, T.J.; Hall, N.; Tuft, S.; Kaye, S.B.; et al. Genetic characterization indicates that a specific subpopulation of Pseudomonas aeruginosa is associated with keratitis infections. J. Clin. Microbiol. 2011, 49, 993–1003. [Google Scholar] [CrossRef]
- Yousefi-Avarvand, A.; Khashei, R.; Sedigh Ebrahim-Saraie, H.; Emami, A.; Zomorodian, K.; Motamedifar, M. The Frequency of Exotoxin A and Exoenzymes S and U Genes Among Clinical Isolates of Pseudomonas aeruginosa in Shiraz, Iran. Int. J. Mol. Cell Med. 2015, 4, 167–173. [Google Scholar]
- Hassuna, N.A.; Mandour, S.A.; Mohamed, E.S. Virulence constitution of multi-drug-resistant Pseudomonas aeruginosa in upper Egypt. Infect. Drug Resist. 2020, 13, 587–595. [Google Scholar] [CrossRef]
- Horna, G.; Amaro, C.; Palacios, A.; Guerra, H.; Ruiz, J. High frequency of the exoU+/exoS+ genotype associated with multidrug-resistant “high-risk clones” of Pseudomonas aeruginosa clinical isolates from Peruvian hospitals. Sci. Rep. 2019, 9, 10874. [Google Scholar] [CrossRef]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef]
- Feltman, H.; Schulert, G.; Khan, S.; Jain, M.; Peterson, L.; Hauser, A.R. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology 2001, 147, 2659–2669. [Google Scholar] [CrossRef] [PubMed]
- Elmouaden, C.; Laglaoui, A.; Ennanei, L.; Bakkali, M.; Abid, M. Virulence genes and antibiotic resistance of Pseudomonas aeruginosa isolated from patients in the Northwestern of Morocco. J. Infect. Dev. Ctries 2019, 13, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Boulant, T.; Boudehen, Y.M.; Filloux, A.; Plesiat, P.; Naas, T.; Dortet, L. Higher Prevalence of PldA, a Pseudomonas aeruginosa Trans-Kingdom H2-Type VI Secretion System Effector, in Clinical Isolates Responsible for Acute Infections and in Multidrug Resistant Strains. Front. Microbiol. 2018, 9, 2578. [Google Scholar] [CrossRef] [PubMed]
- Wilderman, P.J.; Vasil, A.I.; Johnson, Z.; Vasil, M.L. Genetic and biochemical analyses of a eukaryotic-like phospholipase D of Pseudomonas aeruginosa suggest horizontal acquisition and a role for persistence in a chronic pulmonary infection model. Mol. Microbiol. 2001, 39, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Alva, P.P.; Raj, J.M.; Karunasagar, I.; Premanath, R. Environmental and clinical Pseudomonas aeruginosa isolates with pooled presence of exo S, exo U, exo T and exo Y genes. J. Pure Appl. Microbiol. 2018, 12, 1119–1124. [Google Scholar] [CrossRef]
- Radó, J.; Kaszab, E.; Petrovics, T.; Pászti, J.; Kriszt, B.; Szoboszlay, S. Characterization of environmental Pseudomonas aeruginosa using multilocus sequence typing scheme. J. Med. Microbiol. 2017, 66, 1457–1466. [Google Scholar] [CrossRef]
- Andrews, S. FastQC—A Quality Control Tool for High Throughput Sequence Data [Online]. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 February 2021).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Larsen, M.v.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. SOFTWARE Open Access PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Draw Venn Diagram. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 5 March 2023).
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- RStudio Team RStudio: Integrated Development for R. Available online: http://www.rstudio.com/ (accessed on 21 February 2023).
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Vielva, L.; de Toro, M.; Lanza, V.F.; de La Cruz, F. PLACNETw: A web-based tool for plasmid reconstruction from bacterial genomes. Bioinformatics 2017, 33, 3796–3798. [Google Scholar] [CrossRef]
- Garcillán-Barcia, M.P.; Redondo-Salvo, S.; Vielva, L.; de la Cruz, F. MOBscan: Automated Annotation of MOB Relaxases. Methods Mol. Biol. 2020, 2075, 295–308. [Google Scholar] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Huang, L.; Yang, B.; Gomez, J.; Zhang, H.; Yin, Y. AcrFinder: Genome mining anti-CRISPR operons in prokaryotes and their viruses. Nucleic Acids Res. 2020, 48, W358–W365. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Martínez, J.; Rocha-Gracia, R.d.C.; Bello-López, E.; Cevallos, M.A.; Castañeda-Lucio, M.; Sáenz, Y.; Jiménez-Flores, G.; Cortés-Cortés, G.; López-García, A.; Lozano-Zarain, P. Comparative Genomics of Pseudomonas aeruginosa Strains Isolated from Different Ecological Niches. Antibiotics 2023, 12, 866. https://doi.org/10.3390/antibiotics12050866
Gómez-Martínez J, Rocha-Gracia RdC, Bello-López E, Cevallos MA, Castañeda-Lucio M, Sáenz Y, Jiménez-Flores G, Cortés-Cortés G, López-García A, Lozano-Zarain P. Comparative Genomics of Pseudomonas aeruginosa Strains Isolated from Different Ecological Niches. Antibiotics. 2023; 12(5):866. https://doi.org/10.3390/antibiotics12050866
Chicago/Turabian StyleGómez-Martínez, Jessica, Rosa del Carmen Rocha-Gracia, Elena Bello-López, Miguel Angel Cevallos, Miguel Castañeda-Lucio, Yolanda Sáenz, Guadalupe Jiménez-Flores, Gerardo Cortés-Cortés, Alma López-García, and Patricia Lozano-Zarain. 2023. "Comparative Genomics of Pseudomonas aeruginosa Strains Isolated from Different Ecological Niches" Antibiotics 12, no. 5: 866. https://doi.org/10.3390/antibiotics12050866
APA StyleGómez-Martínez, J., Rocha-Gracia, R. d. C., Bello-López, E., Cevallos, M. A., Castañeda-Lucio, M., Sáenz, Y., Jiménez-Flores, G., Cortés-Cortés, G., López-García, A., & Lozano-Zarain, P. (2023). Comparative Genomics of Pseudomonas aeruginosa Strains Isolated from Different Ecological Niches. Antibiotics, 12(5), 866. https://doi.org/10.3390/antibiotics12050866