
Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents

 ,
,  , , , , , ,
, , , , , ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis of CAM-Cn-BER
2.2. Synthesis of CH3-Cn-BER
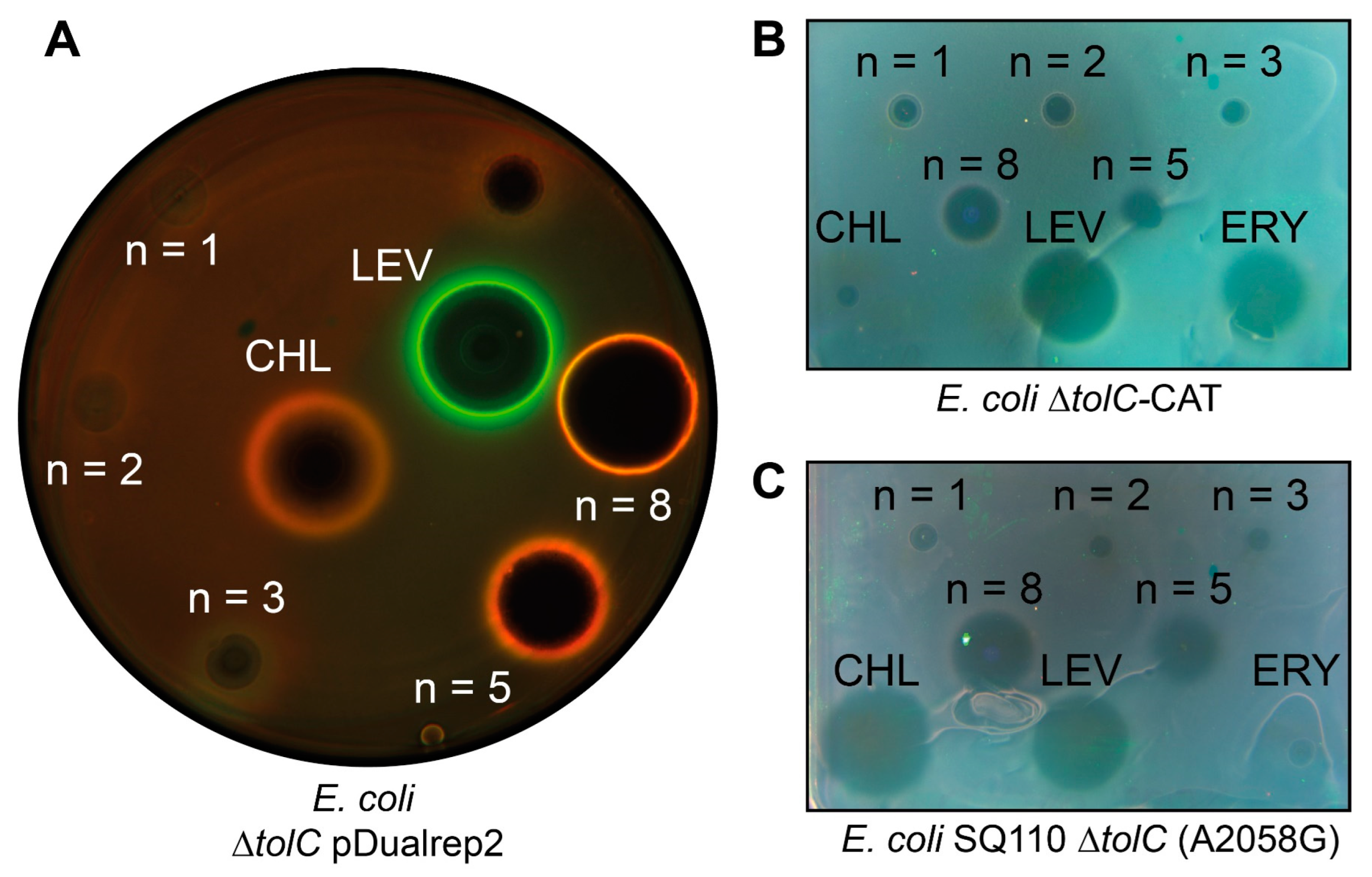
2.3. Preliminary Assessment of the Antibacterial Activity and Mechanism of Action of CAM-Cn-BER in the Double Reporter System pDualrep2
2.4. CAM-Cn-BER and CH3-Cn-BER Exhibit Antibacterial Activity against Various Strains, including Resistant
2.5. CAM-Cn-BER Selectively Inhibit Prokaryotic Translation, Allowing the Formation of Short Peptides, and Bind to the Bacterial Ribosome
2.6. CAM-Cn-BER (n = 5, 8) Act according to the Context Specificity of Chloramphenicol
2.7. Molecular Docking of CAM-Cn-BER Suggests Possible Explanation of Toeprinting Results
2.8. CAM-C8-BER Causes a Decrease in the Membrane Potential of B. subtilis
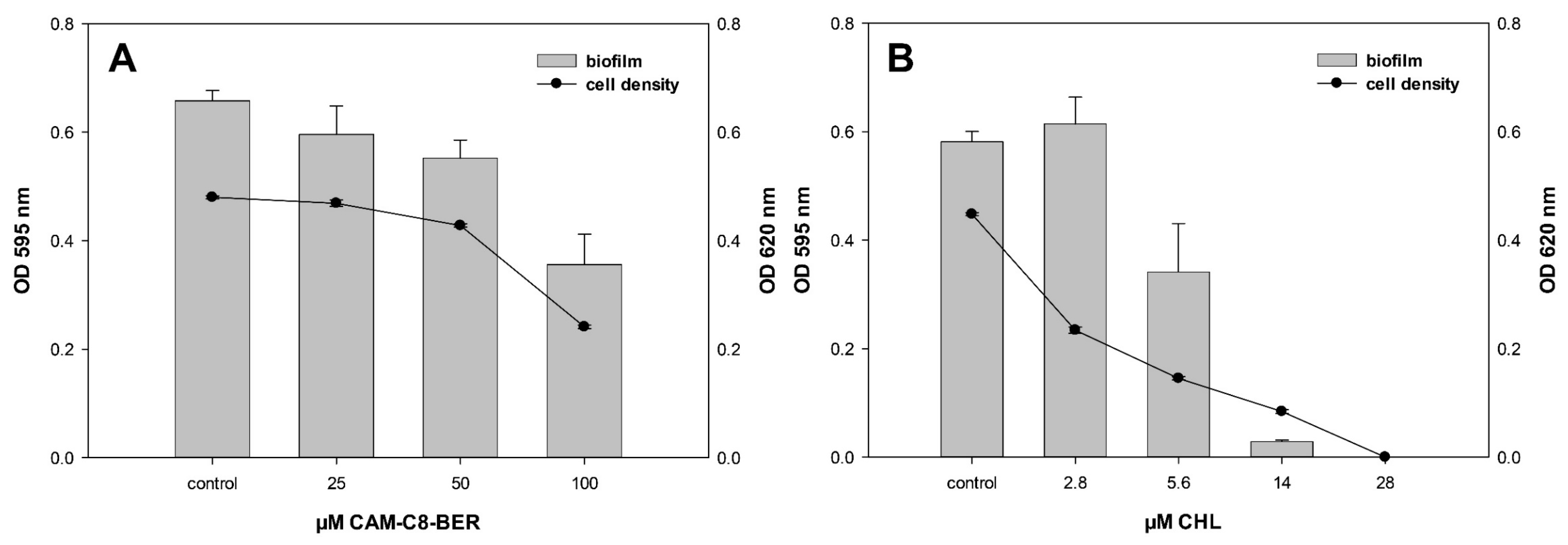
2.9. CAM-C8-BER Does Not Have CHL Ability to Induce Biofilms
2.10. CAM-C8-BER and CAM-C5-BER Are Non-Toxic for Mammalian Cells
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Chromatography
3.3. Liquid Chromatography-Mass Spectrometry
3.4. 1H and 13C NMR
3.5. Synthetic Procedures
3.6. Bacteria Inhibition Assays
3.6.1. Bacterial Strains
3.6.2. Detection of Translation Inhibitors Using pDualrep2 Reporter Strain
3.6.3. Testing the Antibacterial Activity of Substances on Plates with LB and Agar
3.6.4. MIC Determination
3.6.5. Assessment of Antibiotic Activity of Substances in Wells
3.7. In Vitro Translation Inhibition Assay
3.8. In Vitro Binding Assay
3.9. Toeprinting Analysis
3.10. Molecular Docking
3.11. Measurement of B. subtilis Membrane Potential
3.12. Biofilm Development
3.13. In Vitro Survival Assay (MTT Assay)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wencewicz, T.A. Crossroads of Antibiotic Resistance and Biosynthesis. J. Mol. Biol. 2019, 431, 3370–3399. [Google Scholar] [CrossRef] [PubMed]
- Schaenzer, A.J.; Wright, G.D. Antibiotic Resistance by Enzymatic Modification of Antibiotic Targets. Trends Mol. Med. 2020, 26, 768–782. [Google Scholar] [CrossRef]
- Tevyashova, A.N.; Olsufyeva, E.N.; Preobrazhenskaya, M.N. Design of Dual Action Antibiotics as an Approach to Search for New Promising Drugs. Russ. Chem. Rev. 2015, 84, 61–97. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-Targeting Antibiotics and Mechanisms of Bacterial Resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Barbachyn, M.R. Recent Advances in the Discovery of Hybrid Antibacterial Agents. Annu. Rep. Med. Chem. 2008, 43, 281–290. [Google Scholar] [CrossRef]
- Bremner, J.; Ambrus, J.; Samosorn, S. Dual Action-Based Approaches to Antibacterial Agents. Curr. Med. Chem. 2007, 14, 1459–1477. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef]
- Contreras, A.; Vazquez, D. Cooperative and Antagonistic Interactions of Peptidyl-TRNA and Antibiotics with Bacterial Ribosomes. Eur. J. Biochem. 1977, 74, 539–547. [Google Scholar] [CrossRef]
- Levin, B.R.; McCall, I.C.; Perrot, V.; Weiss, H.; Ovesepian, A.; Baquero, F. A Numbers Game: Ribosome Densities, Bacterial Growth, and Antibiotic-Mediated Stasis and Death. mBio 2017, 8, e02253-16. [Google Scholar] [CrossRef]
- Kaplan, J.B. Antibiotic-Induced Biofilm Formation. Int. J. Artif. Organs 2011, 34, 737–751. [Google Scholar] [CrossRef]
- Boehm, A.; Steiner, S.; Zaehringer, F.; Casanova, A.; Hamburger, F.; Ritz, D.; Keck, W.; Ackermann, M.; Schirmer, T.; Jenal, U. Second Messenger Signalling Governs Escherichia Coli Biofilm Induction upon Ribosomal Stress. Mol. Microbiol. 2009, 72, 1500–1516. [Google Scholar] [CrossRef] [PubMed]
- Tevyashova, A.N. Recent Trends in Synthesis of Chloramphenicol New Derivatives. Antibiotics 2021, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.; Athanassopoulos, C.; Missiri, D.; Giannopoulou, P.; Vlachogiannis, I.; Papadopoulos, G.; Papaioannou, D.; Kalpaxis, D. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.; Kannan, K.; Roncase, E.J.; Klepacki, D.; Kefi, A.; Orelle, C.; Vázquez-Laslop, N.; Mankin, A.S. Context-Specific Inhibition of Translation by Ribosomal Antibiotics Targeting the Peptidyl Transferase Center. Proc. Natl. Acad. Sci. USA 2016, 113, 12150–12155. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Marks, J.; Zhang, J.; Chen, D.-H.; Wang, J.; Vázquez-Laslop, N.; Mankin, A.S.; Puglisi, J.D. Dynamics of the Context-Specific Translation Arrest by Chloramphenicol and Linezolid. Nat. Chem. Biol. 2020, 16, 310–317. [Google Scholar] [CrossRef]
- Syroegin, E.A.; Flemmich, L.; Klepacki, D.; Vazquez-Laslop, N.; Micura, R.; Polikanov, Y.S. Structural Basis for the Context-Specific Action of Classic Peptidyl Transferase Inhibitor Chloramphenicol. Nat. Struct. Mol. Biol. 2022, 29, 152–161. [Google Scholar] [CrossRef]
- Rebstock, M.C.; Crooks, H.M.; Controulis, J.; Bartz, Q.R. Chloramphenicol (Chloromycetin). 1 IV. 1a Chemical Studies. J. Am. Chem. Soc. 1949, 71, 2458–2462. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Shishkina, A.V.; Tashlitsky, V.N.; Korshunova, G.A.; Bogdanov, A.A.; Sumbatyan, N.V. Interaction of Chloramphenicol Tripeptide Analogs with Ribosomes. Biochemistry 2016, 81, 392–400. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with Polyamines Enhances the Antibacterial and Anticancer Activity of Chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef]
- Giannopoulou, P.; Missiri, D.; Kournoutou, G.; Sazakli, E.; Papadopoulos, G.; Papaioannou, D.; Dinos, G.; Athanassopoulos, C.; Kalpaxis, D. New Chloramphenicol Derivatives from the Viewpoint of Anticancer and Antimicrobial Activity. Antibiotics 2019, 8, 9. [Google Scholar] [CrossRef]
- Tsirogianni, A.; Kournoutou, G.G.; Bougas, A.; Poulou-Sidiropoulou, E.; Dinos, G.; Athanassopoulos, C.M. New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents. Antibiotics 2021, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, J.A.; Khairullina, Z.Z.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Volynkina, I.A.; Skvortsov, D.A.; Makarov, G.I.; Abad, E.; Murayama, S.Y.; et al. Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents. Antibiotics 2021, 10, 489. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Pavlova, J.A.; Lukianov, D.A.; Tereshchenkov, A.G.; Makarov, G.I.; Khairullina, Z.Z.; Tashlitsky, V.N.; Paleskava, A.; Konevega, A.L.; Bogdanov, A.A.; et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics 2021, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Severina, I.I.; Muntyan, M.S.; Lewis, K.; Skulachev, V.P. Transfer of Cationic Antibacterial Agents Berberine, Palmatine, and Benzalkonium Through Bimolecular Planar Phospholipid Film and Staphylococcus Aureus Membrane. IUBMB Life 2001, 52, 321–324. [Google Scholar] [CrossRef]
- Davidson, M.W.; Lopp, I.; Alexander, S.; Wilson, W.D. The Interaction of Plant Alkaloids with DNA. II. Berberinium Chloride. Nucl Acids Res. 1977, 4, 2697–2712. [Google Scholar] [CrossRef][Green Version]
- Bhowmik, D.; Suresh Kumar, G. Recent Advances in Nucleic Acid Binding Aspects of Berberine Analogs and Implications for Drug Design. MRMC 2015, 16, 104–109. [Google Scholar] [CrossRef]
- Jin, J.; Hua, G.; Meng, Z.; Gao, P. Antibacterial Mechanisms of Berberine and Reasons for Little Resistance of Bacteria. Chin. Herb. Med. 2010, 3, 27–35. [Google Scholar] [CrossRef]
- Jang, M.H.; Kim, H.Y.; Kang, K.S.; Yokozawa, T.; Park, J.H. Hydroxyl Radical Scavenging Activities of Isoquinoline Alkaloids Isolated from Coptis Chinensis. Arch. Pharm. Res. 2009, 32, 341–345. [Google Scholar] [CrossRef]
- Li, Z.; Geng, Y.-N.; Jiang, J.-D.; Kong, W.-J. Antioxidant and Anti-Inflammatory Activities of Berberine in the Treatment of Diabetes Mellitus. Evid.-Based Complement. Altern. Med. 2014, 2014, 289264. [Google Scholar] [CrossRef]
- Bashir, S.; Gilani, A.H. Antiurolithic Effect of Berberine Is Mediated through Multiple Pathways. Eur. J. Pharmacol. 2011, 651, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gaba, S.; Saini, A.; Singh, G.; Monga, V. An Insight into the Medicinal Attributes of Berberine Derivatives: A Review. Bioorg. Med. Chem. 2021, 38, 116143. [Google Scholar] [CrossRef] [PubMed]
- Guamán Ortiz, L.; Lombardi, P.; Tillhon, M.; Scovassi, A. Berberine, an Epiphany Against Cancer. Molecules 2014, 19, 12349–12367. [Google Scholar] [CrossRef]
- Zou, K.; Li, Z.; Zhang, Y.; Zhang, H.; Li, B.; Zhu, W.; Shi, J.; Jia, Q.; Li, Y. Advances in the Study of Berberine and Its Derivatives: A Focus on Anti-Inflammatory and Anti-Tumor Effects in the Digestive System. Acta Pharmacol. Sin. 2017, 38, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sheng, J.; Li, G.; Zhao, L.; Wang, Y.; Yang, W.; Yao, X.; Sun, L.; Zhang, Z.; Cui, R. Effects of Berberine and Its Derivatives on Cancer: A Systems Pharmacology Review. Front. Pharmacol. 2020, 10, 1461. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Salam, M.; Mekky, H.; El-Naggar, E.M.B.; Ghareeb, D.; El-Demellawy, M.; El-Fiky, F. Hepatoprotective Properties and Biotransformation of Berberine and Berberrubine by Cell Suspension Cultures of Dodonaea Viscosa and Ocimum Basilicum. S. Afr. J. Bot. 2015, 97, 191–195. [Google Scholar] [CrossRef]
- Caliceti, C.; Franco, P.; Spinozzi, S.; Roda, A.; FG Cicero, A. Berberine: New Insights from Pharmacological Aspects to Clinical Evidences in the Management of Metabolic Disorders. Curr. Med. Chem. 2016, 23, 1460–1476. [Google Scholar] [CrossRef]
- Pang, B.; Zhao, L.-H.; Zhou, Q.; Zhao, T.-Y.; Wang, H.; Gu, C.-J.; Tong, X.-L. Application of Berberine on Treating Type 2 Diabetes Mellitus. Int. J. Endocrinol. 2015, 2015, 905749. [Google Scholar] [CrossRef]
- Warowicka, A.; Nawrot, R.; Goździcka-Józefiak, A. Antiviral Activity of Berberine. Arch. Virol. 2020, 165, 1935–1945. [Google Scholar] [CrossRef]
- Amin, A.H.; Subbaiah, T.V.; Abbasi, K.M. Berberine Sulfate: Antimicrobial Activity, Bioassay, and Mode of Action. Can. J. Microbiol. 1969, 15, 1067–1076. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Patra, P.H.; Mahanti, A.; Mondal, D.K.; Dandapat, P.; Bandyopadhyay, S.; Samanta, I.; Lodh, C.; Bera, A.K.; Bhattacharyya, D.; et al. Potential Antibacterial Activity of Berberine against Multi Drug Resistant Enterovirulent Escherichia Coli Isolated from Yaks (Poephagus Grunniens) with Haemorrhagic Diarrhoea. Asian Pac. J. Trop. Med. 2013, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.-B.; Yan, Y.; Liang, Y.; Bao, Z. Evaluation of the Antimicrobial Mode of Berberine by LC/ESI-MS Combined with Principal Component Analysis. J. Pharm. Biomed. Anal. 2007, 44, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Azimi, G.; Hakakian, A.; Ghanadian, M.; Joumaa, A.; Alamian, S. Bioassay-Directed Isolation of Quaternary Benzylisoquinolines from Berberis Integerrima with Bactericidal Activity against Brucella Abortus. Res. Pharma Sci. 2018, 13, 149. [Google Scholar] [CrossRef]
- Neag, M.A.; Mocan, A.; Echeverría, J.; Pop, R.M.; Bocsan, C.I.; Crişan, G.; Buzoianu, A.D. Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders. Front. Pharmacol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Porras, G.; Chassagne, F.; Lyles, J.T.; Marquez, L.; Dettweiler, M.; Salam, A.M.; Samarakoon, T.; Shabih, S.; Farrokhi, D.R.; Quave, C.L. Ethnobotany and the Role of Plant Natural Products in Antibiotic Drug Discovery. Chem. Rev. 2021, 121, 3495–3560. [Google Scholar] [CrossRef]
- Kim, S.A.; Kwon, Y.; Kim, J.H.; Muller, M.T.; Chung, I.K. Induction of Topoisomerase II-Mediated DNA Cleavage by a Protoberberine Alkaloid, Berberrubine. Biochemistry 1998, 37, 16316–16324. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Chan, F.-Y.; Lu, Y.-J.; Neves, M.A.C.; Lui, H.-K.; Wang, Y.; Chow, K.-Y.; Chan, K.-F.; Yan, S.-C.; Leung, Y.-C.; et al. Rational Design of Berberine-Based FtsZ Inhibitors with Broad-Spectrum Antibacterial Activity. PLoS ONE 2014, 9, e97514. [Google Scholar] [CrossRef]
- Olleik, H.; Yacoub, T.; Hoffer, L.; Gnansounou, S.M.; Benhaiem-Henry, K.; Nicoletti, C.; Mekhalfi, M.; Pique, V.; Perrier, J.; Hijazi, A.; et al. Synthesis and Evaluation of the Antibacterial Activities of 13-Substituted Berberine Derivatives. Antibiotics 2020, 9, E381. [Google Scholar] [CrossRef]
- Ball, A.R.; Casadei, G.; Samosorn, S.; Bremner, J.B.; Ausubel, F.M.; Moy, T.I.; Lewis, K. Conjugating Berberine to a Multidrug Resistance Pump Inhibitor Creates an Effective Antimicrobial. ACS Chem. Biol. 2006, 1, 594–600. [Google Scholar] [CrossRef]
- Nazarov, P.A. MDR Pumps as Crossroads of Resistance: Antibiotics and Bacteriophages. Antibiotics 2022, 11, 734. [Google Scholar] [CrossRef]
- Stermitz, F.R.; Lorenz, P.; Tawara, J.N.; Zenewicz, L.A.; Lewis, K. Synergy in a Medicinal Plant: Antimicrobial Action of Berberine Potentiated by 5′-Methoxyhydnocarpin, a Multidrug Pump Inhibitor. Proc. Natl. Acad. Sci. USA 2000, 97, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Ettefagh, K.; Burns, J.; Junio, H.; Kaatz, G.; Cech, N. Goldenseal (Hydrastis Canadensis L.) Extracts Synergistically Enhance the Antibacterial Activity of Berberine via Efflux Pump Inhibition. Planta Med. 2011, 77, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wu, L.-L.; Li, Q.; Hu, Q.-M.; Zhang, S.-Y.; Liu, K.; Jiang, J.-Q. Novel Berberine Derivatives: Design, Synthesis, Antimicrobial Effects, and Molecular Docking Studies. Chin. J. Nat. Med. 2018, 16, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, T.; Chen, H.; Xu, Y.-N.; Yu, L.-F.; Liu, T.; Tang, J.; Yi, Z.; Yang, C.-G.; Xue, W.; et al. The Synthesis and Antistaphylococcal Activity of 9, 13-Disubstituted Berberine Derivatives. Eur. J. Med. Chem. 2017, 127, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Demekhin, O.D.; Zagrebaev, A.D.; Burov, O.N.; Kletskii, M.E.; Pavlovich, N.V.; Bereznyak, E.A.; Tsimbalistova, M.V.; Kurbatov, S.V. The First 13-Vinyl Derivatives of Berberine: Synthesis and Antimicrobial Activity. Chem. Heterocycl. Comp. 2019, 55, 1128–1130. [Google Scholar] [CrossRef]
- Zhang, S.-L.; Chang, J.-J.; Damu, G.L.V.; Fang, B.; Zhou, X.-D.; Geng, R.-X.; Zhou, C.-H. Novel Berberine Triazoles: Synthesis, Antimicrobial Evaluation and Competitive Interactions with Metal Ions to Human Serum Albumin. Bioorg. Med. Chem. Lett. 2013, 23, 1008–1012. [Google Scholar] [CrossRef]
- Zhang, L.; Chang, J.-J.; Zhang, S.-L.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Synthesis and Bioactive Evaluation of Novel Hybrids of Metronidazole and Berberine as New Type of Antimicrobial Agents and Their Transportation Behavior by Human Serum Albumin. Bioorg. Med. Chem. 2013, 21, 4158–4169. [Google Scholar] [CrossRef]
- Jeyakkumar, P.; Zhang, L.; Avula, S.R.; Zhou, C.-H. Design, Synthesis and Biological Evaluation of Berberine-Benzimidazole Hybrids as New Type of Potentially DNA-Targeting Antimicrobial Agents. Eur. J. Med. Chem. 2016, 122, 205–215. [Google Scholar] [CrossRef]
- Fan, T.; Wang, Y.; Tang, S.; Hu, X.; Zen, Q.; Pang, J.; Yang, Y.; You, X.; Song, D. Synthesis and Antibacterial Evaluation of 13-Substituted Cycloberberine Derivatives as a Novel Class of Anti-MRSA Agents. Eur. J. Med. Chem. 2018, 157, 877–886. [Google Scholar] [CrossRef]
- Yang, Y.-S.; Wei, W.; Hu, X.-X.; Tang, S.; Pang, J.; You, X.-F.; Fan, T.-Y.; Wang, Y.-X.; Song, D.-Q. Evolution and Antibacterial Evaluation of 8-Hydroxy-Cycloberberine Derivatives as a Novel Family of Antibacterial Agents Against MRSA. Molecules 2019, 24, 984. [Google Scholar] [CrossRef]
- Yang, Y.-S.; Lu, X.; Zeng, Q.-X.; Pang, J.; Fan, T.-Y.; You, X.-F.; Tang, S.; Wang, Y.-X.; Song, D.-Q. Synthesis and Biological Evaluation of 7-Substituted Cycloberberine Derivatives as Potent Antibacterial Agents against MRSA. Eur. J. Med. Chem. 2019, 168, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Samosorn, S.; Tanwirat, B.; Muhamad, N.; Casadei, G.; Tomkiewicz, D.; Lewis, K.; Suksamrarn, A.; Prammananan, T.; Gornall, K.C.; Beck, J.L.; et al. Antibacterial Activity of Berberine-NorA Pump Inhibitor Hybrids with a Methylene Ether Linking Group. Bioorg. Med. Chem. 2009, 17, 3866–3872. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Pustovidko, A.V.; Simonyan, R.A.; Rokitskaya, T.I.; Domnina, L.V.; Ivanova, O.Y.; Severina, I.I.; Sumbatyan, N.V.; Korshunova, G.A.; Tashlitsky, V.N.; et al. Novel Mitochondria-Targeted Antioxidants: Plastoquinone Conjugated with Cationic Plant Alkaloids Berberine and Palmatine. Pharm. Res. 2011, 28, 2883–2895. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jhong, T.N.; Paik, Y.K.; Park, J.S.; Kim, E.D.; Lee, Y.S.; Kim, S.U.; Hanwha Corp. Antifungal Formulation Comprising Protoberberine Derivatives and Salts Thereof. U.S. Patent 6,030,978, 29 February 2000. [Google Scholar]
- Lombardi, P.; Buzzetti, F.; Guido Arcamone, A. Benzooquinolizinium Salt Derivatives as Anticancer Agents. WO2011009714A3, 27 January 2011. [Google Scholar]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting Out Antibiotics’ Mechanisms of Action: A Double Fluorescent Protein Reporter for High-Throughput Screening of Ribosome and DNA Biosynthesis Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef]
- Wright, G. Bacterial Resistance to Antibiotics: Enzymatic Degradation and Modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Plessa, E.; Chen, C.-W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-Resolution Crystal Structures of Ribosome-Bound Chloramphenicol and Erythromycin Provide the Ultimate Basis for Their Competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Syroegin, E.A.; Aleksandrova, E.V.; Atkinson, G.C.; Gregory, S.T.; Mankin, A.S.; Polikanov, Y.S. Structure of Erm-Modified 70S Ribosome Reveals the Mechanism of Macrolide Resistance. Nat. Chem. Biol. 2021, 17, 412–420. [Google Scholar] [CrossRef]
- Sulavik, M.C.; Houseweart, C.; Cramer, C.; Jiwani, N.; Murgolo, N.; Greene, J.; DiDomenico, B.; Shaw, K.J.; Miller, G.H.; Hare, R.; et al. Antibiotic Susceptibility Profiles of Escherichia Coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob. Agents Chemother. 2001, 45, 1126–1136. [Google Scholar] [CrossRef]
- Franco-Cendejas, R.; Colín-Castro, C.A.; Hernández-Durán, M.; López-Jácome, L.E.; Ortega-Peña, S.; Cerón-González, G.; Vanegas-Rodríguez, S.; Mondragón-Eguiluz, J.A.; Acosta-Rodríguez, E. Leuconostoc Mesenteroides Periprosthetic Knee Infection, an Unusual Fastidious Gram-Positive Bacteria: A Case Report. BMC Infect. Dis. 2017, 227, 5. [Google Scholar] [CrossRef]
- Menegueti, M.G.; Gaspar, G.G.; Laus, A.M.; Basile-Filho, A.; Bellissimo-Rodrigues, F.; Auxiliadora-Martins, M. Bacteremia by Leuconostoc Mesenteroides in an Immunocompetent Patient with Chronic Chagas Disease: A Case Report. BMC Infect. Dis. 2018, 18, 547. [Google Scholar] [CrossRef]
- Yan, K.; Hunt, E.; Berge, J.; May, E.; Copeland, R.A.; Gontarek, R.R. Fluorescence Polarization Method To Characterize Macrolide-Ribosome Interactions. Antimicrob. Agents Chemother. 2005, 49, 3367–3372. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenkov, A.G.; Shishkina, A.V.; Karpenko, V.V.; Chertkov, V.A.; Konevega, A.L.; Kasatsky, P.S.; Bogdanov, A.A.; Sumbatyan, N.V. New Fluorescent Macrolide Derivatives for Studying Interactions of Antibiotics and Their Analogs with the Ribosomal Exit Tunnel. Biochemistry 2016, 81, 1163–1172. [Google Scholar] [CrossRef]
- Syroegin, E.A.; Aleksandrova, E.V.; Polikanov, Y.S. Structural Basis for the Inability of Chloramphenicol to Inhibit Peptide Bond Formation in the Presence of A-Site Glycine. Nucleic Acids Res. 2022, 50, 7669–7679. [Google Scholar] [CrossRef] [PubMed]
- Syroegin, E.A.; Aleksandrova, E.V.; Polikanov, Y.S. Insights into the Ribosome Function from the Structures of Non-Arrested Ribosome–Nascent Chain Complexes. Nat. Chem. 2022, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia Coli Ribosome with Antibiotics Bound near the Peptidyl Transferase Center Explain Spectra of Drug Action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed]
- Maravic, G. Macrolide Resistance Based on the Erm-Mediated RRNA Methylation. Curr. Drug Targets Infect. Disord. 2004, 4, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Pustovidko, A.V.; Rokitskaya, T.I.; Severina, I.I.; Simonyan, R.A.; Trendeleva, T.A.; Lyamzaev, K.G.; Antonenko, Y.N.; Rogov, A.G.; Zvyagilskaya, R.A.; Skulachev, V.P.; et al. Derivatives of the Cationic Plant Alkaloids Berberine and Palmatine Amplify Protonophorous Activity of Fatty Acids in Model Membranes and Mitochondria. Mitochondrion 2013, 13, 520–525. [Google Scholar] [CrossRef]
- Maeda, T.; Fukushima, Y.; Yoshida, H.; Goto, M.; Fujita, T.; Tsuyuki, Y.; Takahashi, T. Biofilm Production Ability and Associated Characteristics of Streptococcus Agalactiae Isolates from Companion Animals and Humans. J. Infect. Chemother. 2021, 27, 1571–1577. [Google Scholar] [CrossRef]
- Xu, C.; Wang, F.; Huang, F.; Yang, M.; He, D.; Deng, L. Targeting Effect of Berberine on Type I Fimbriae of Salmonella Typhimurium and Its Effective Inhibition of Biofilm. Appl. Microbiol. Biotechnol. 2021, 105, 1563–1573. [Google Scholar] [CrossRef]
- Tseng, C.-Y.; Sun, M.-F.; Li, T.-C.; Lin, C.-T. Effect of Coptis Chinensis on Biofilm Formation and Antibiotic Susceptibility in Mycobacterium Abscessus. Evid.-Based Complement. Altern. Med. 2020, 2020, 9754357. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, X.; Zhou, P. In Vitro Antifungal Effects of Berberine Against Candida Spp. In Planktonic and Biofilm Conditions. Drug Des. Devel. Ther. 2020, 14, 87–101. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.R.; de Andrade Neto, J.B.; da Silva, C.R.; de Campos, R.S.; Costa Silva, R.A.; Freitas, D.D.; do Nascimento, F.B.S.A.; de Andrade, L.N.D.; Sampaio, L.S.; Grangeiro, T.B.; et al. Berberine Antifungal Activity in Fluconazole-Resistant Pathogenic Yeasts: Action Mechanism Evaluated by Flow Cytometry and Biofilm Growth Inhibition in Candida spp. Antimicrob. Agents Chemother. 2016, 60, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Aswathanarayan, J.B.; Vittal, R.R. Inhibition of Biofilm Formation and Quorum Sensing Mediated Phenotypes by Berberine in Pseudomonas Aeruginosa and Salmonella Typhimurium. RSC Adv. 2018, 8, 36133–36141. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, X.-D.; Hong, J.; Liu, C.; Zhang, X.-L.; Zheng, J.-P.; Xu, Y.-J.; Ou, Z.-Y.; Zheng, J.-L.; Yu, D.-J. Inhibitory Effect of Two Traditional Chinese Medicine Monomers, Berberine and Matrine, on the Quorum Sensing System of Antimicrobial-Resistant Escherichia Coli. Front. Microbiol. 2019, 10, 2584. [Google Scholar] [CrossRef]
- Jhanji, R.; Bhati, V.; Singh, A.; Kumar, A. Phytomolecules against Bacterial Biofilm and Efflux Pump: An in Silico and in Vitro Study. J. Biomol. Struct. Dyn. 2020, 38, 5500–5512. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Dai, K.; Nomoto, K.; Ueno, S.; Tomono, K.; Miyamura, K. Odd–Even Effect and Unusual Behavior of Dodecyl-Substituted Analogue Observed in the Crystal Structure of Alkyltrimethylammonium–[Ni(Dmit)2]− Salts. BCSJ 2011, 84, 312–319. [Google Scholar] [CrossRef]
- Li, J.; Kim, I.H.; Roche, E.D.; Beeman, D.; Lynch, A.S.; Ding, C.Z.; Ma, Z. Design, Synthesis, and Biological Evaluation of BODIPY®–Erythromycin Probes for Bacterial Ribosomes. Bioorg. Med. Chem. Lett. 2006, 16, 794–797. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Samosorn, S. Development of Berberine-Based Derivatives as Novel Antimicrobial Agents. Ph.D. Thesis, University of Wollongong, Wollongong, NSW, Australia, 2005. [Google Scholar]
- Zakalyukina, Y.V.; Birykov, M.V.; Lukianov, D.A.; Shiriaev, D.I.; Komarova, E.S.; Skvortsov, D.A.; Kostyukevich, Y.; Tashlitsky, V.N.; Polshakov, V.I.; Nikolaev, E.; et al. Nybomycin-Producing Streptomyces Isolated from Carpenter Ant Camponotus Vagus. Biochimie 2019, 160, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lukianov, D.A.; Buev, V.S.; Ivanenkov, Y.A.; Kartsev, V.G.; Skvortsov, D.A.; Osterman, I.A.; Sergiev, P.V. Imidazole Derivative As a Novel Translation Inhibitor. Acta Nat. 2022, 14, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X. An Exact Mathematical Expression for Describing Competitive Binding of Two Different Ligands to a Protein Molecule. FEBS Lett. 1995, 360, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Denisov, S.S.; Khailova, L.S.; Nazarov, P.A.; Rokitskaya, T.; Tashlitsky, V.N.; Firsov, A.M.; Korshunova, G.A.; Kotova, E.A. Alkyl-Substituted Phenylamino Derivatives of 7-Nitrobenz-2-Oxa-1,3-Diazole as Uncouplers of Oxidative Phosphorylation and Antibacterial Agents: Involvement of Membrane Proteins in the Uncoupling Action. Biochim. Biophys. Acta Biomembr. 2017, 1859, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-C.; Guo, W.-Q.; Zheng, H.-S.; Wu, Q.-L.; Luo, H.-C.; Ren, N.-Q. Effect of Metabolic Uncoupler, 3,3′,4′,5-Tetrachlorosalicylanilide (TCS) on Bacillus Subtilis: Biofilm Formation, Flocculability and Surface Characteristics. RSC Adv. 2018, 8, 16178–16186. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, P.A.; Osterman, I.A.; Tokarchuk, A.V.; Karakozova, M.V.; Korshunova, G.A.; Lyamzaev, K.G.; Skulachev, M.V.; Kotova, E.A.; Skulachev, V.P.; Antonenko, Y.N. Mitochondria-Targeted Antioxidants as Highly Effective Antibiotics. Sci. Rep. 2017, 7, 1394. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli K12 | E. coli JW5503 ΔtolC | E. coli JW5503 ΔtolC-CAT | B. subtilis 168 | B. subtilis-CAT 168 | |
|---|---|---|---|---|---|
| CAM-C8-BER | >100 | 12.5 | 50 | 100 | 50 |
| CH3-C8-BER | >100 | 12.5 | 12.5 | 12.5 | 12.5 |
| CHL | 6 | 2.5 | >100 | 12.5 | >100 |
| B. subtilis ATCC 6633 | L. mesenteroidesVKPM B-4177 | St. aureus INA 00761 | Myc. smegmatisVKPM Ac 1339 | Myc. smegmatis mc2155 | E. coli ATCC 25922 | E. coli K-12 | A. niger INA 00760 | S. cerevisiae RIA 259 | |
|---|---|---|---|---|---|---|---|---|---|
| CAM-C8-BER | NO 2 | 11(13) | NO | 13 | 15 | NO | NO | NO | NO |
| CH3-C8-BER | 15 | 18 | 20 | 14 | 22 | NO | NO | 16 | 12 |
| BER | NO | NO | NO | 14 | 12 | NO | NO | NO | NO |
| CHL | 30 | 25 | 23 | 11 | 21 | 18 | 26 | NO | NO |
| HEK293T | MCF7 | A549 | VA13 | |
|---|---|---|---|---|
| CHL | >50 | >50 | >50 | >50 |
| CAM-C5-BER | >50 | >50 | >50 | >50 |
| CAM-C8-BER | 28 ± 3 | >50 | >50 | >50 |
| CH3-C5-BER | 11 ± 1 | >50 | 34 ± 6 | >50 |
| CH3-C8-BER | 1.39 ± 0.07 | 1.8 ± 0.2 | 1.7 ± 0.1 | 2.9 ± 0.3 |
| BER | 0.70 ± 0.08 | 25 ± 4 | 4.6 ± 0.5 | 17 ± 2 |
| Doxorubicin | 0.007 ± 0.001 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.18 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlova, J.A.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Skvortsov, D.A.; Polshakov, V.I.; Vasilieva, B.F.; Efremenkova, O.V.; Kaiumov, M.Y.; Paleskava, A.; et al. Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics 2023, 12, 15. https://doi.org/10.3390/antibiotics12010015
Pavlova JA, Tereshchenkov AG, Nazarov PA, Lukianov DA, Skvortsov DA, Polshakov VI, Vasilieva BF, Efremenkova OV, Kaiumov MY, Paleskava A, et al. Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics. 2023; 12(1):15. https://doi.org/10.3390/antibiotics12010015
Chicago/Turabian StylePavlova, Julia A., Andrey G. Tereshchenkov, Pavel A. Nazarov, Dmitrii A. Lukianov, Dmitry A. Skvortsov, Vladimir I. Polshakov, Byasilya F. Vasilieva, Olga V. Efremenkova, Mikhail Y. Kaiumov, Alena Paleskava, and et al. 2023. "Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents" Antibiotics 12, no. 1: 15. https://doi.org/10.3390/antibiotics12010015
APA StylePavlova, J. A., Tereshchenkov, A. G., Nazarov, P. A., Lukianov, D. A., Skvortsov, D. A., Polshakov, V. I., Vasilieva, B. F., Efremenkova, O. V., Kaiumov, M. Y., Paleskava, A., Konevega, A. L., Dontsova, O. A., Osterman, I. A., Bogdanov, A. A., & Sumbatyan, N. V. (2023). Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics, 12(1), 15. https://doi.org/10.3390/antibiotics12010015