
Computational Development of Inhibitors of Plasmid-Borne Bacterial Dihydrofolate Reductase


Abstract
:1. Introduction
2. Results and Discussion
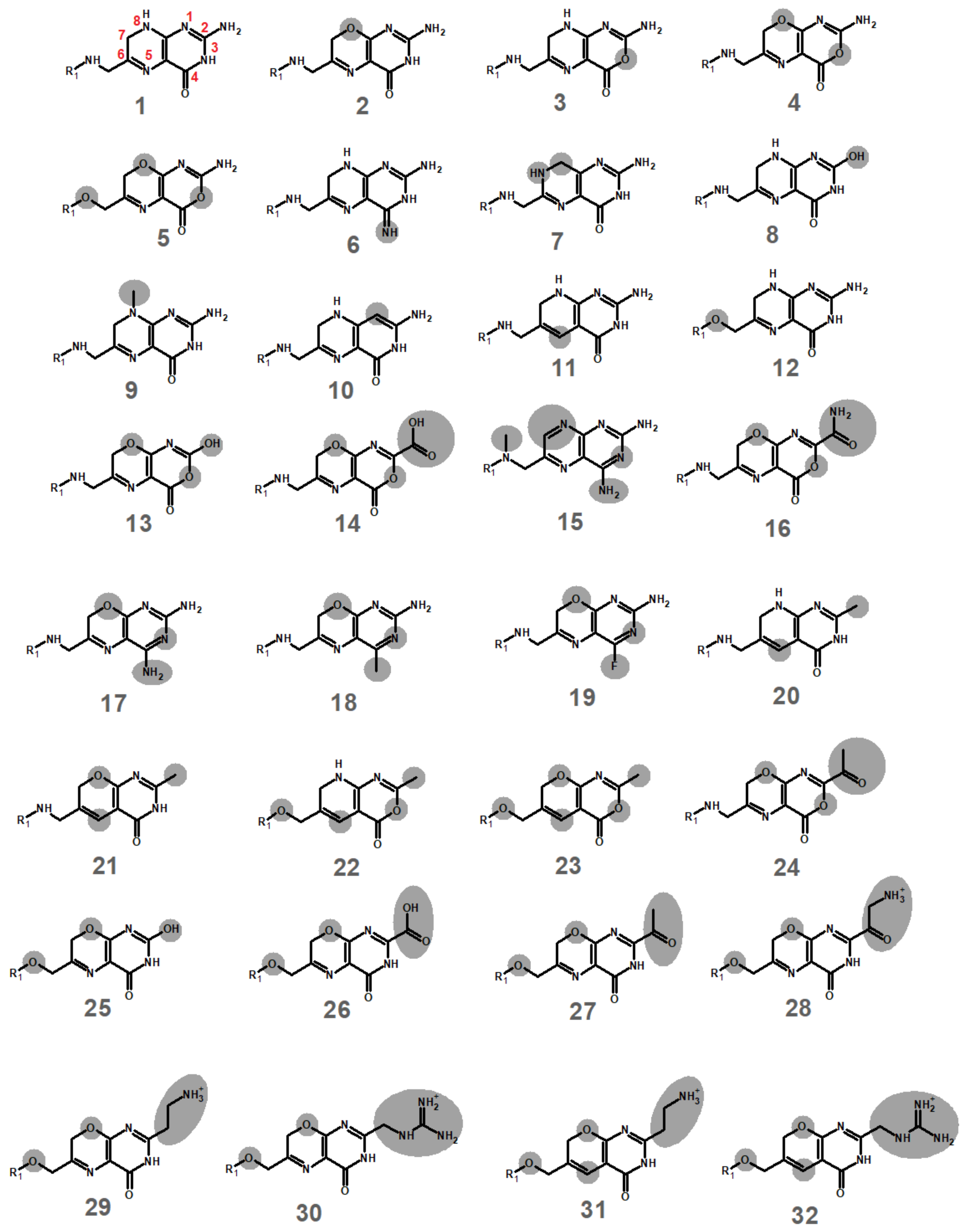
2.1. Computational Development of Unreactive Analogues of Folate
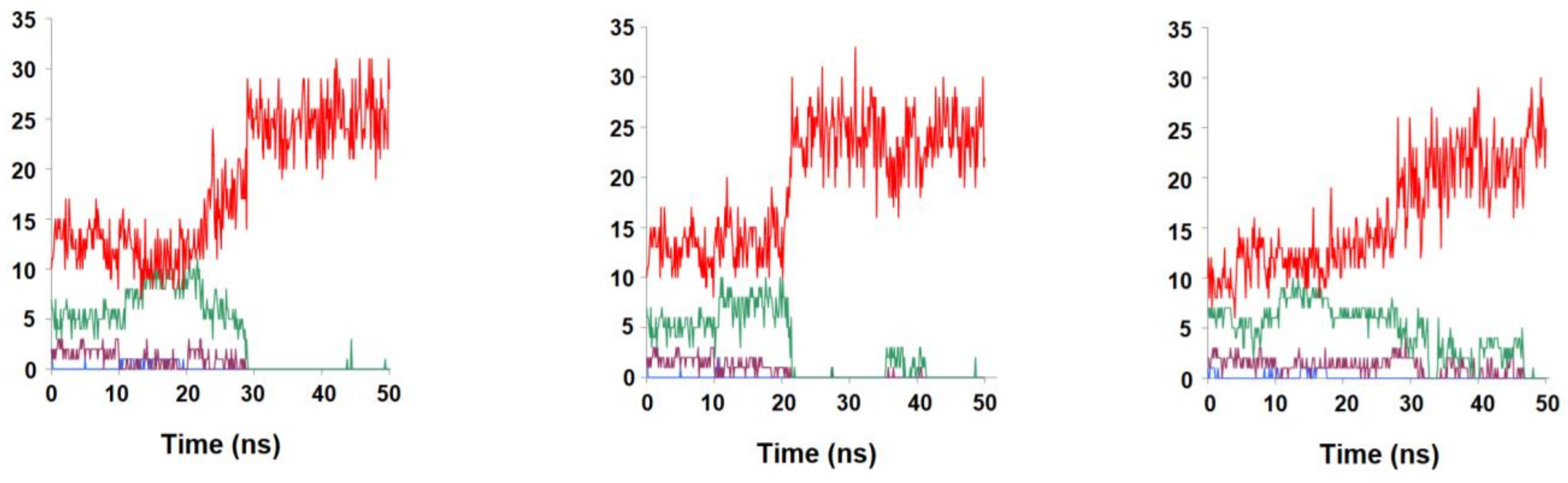
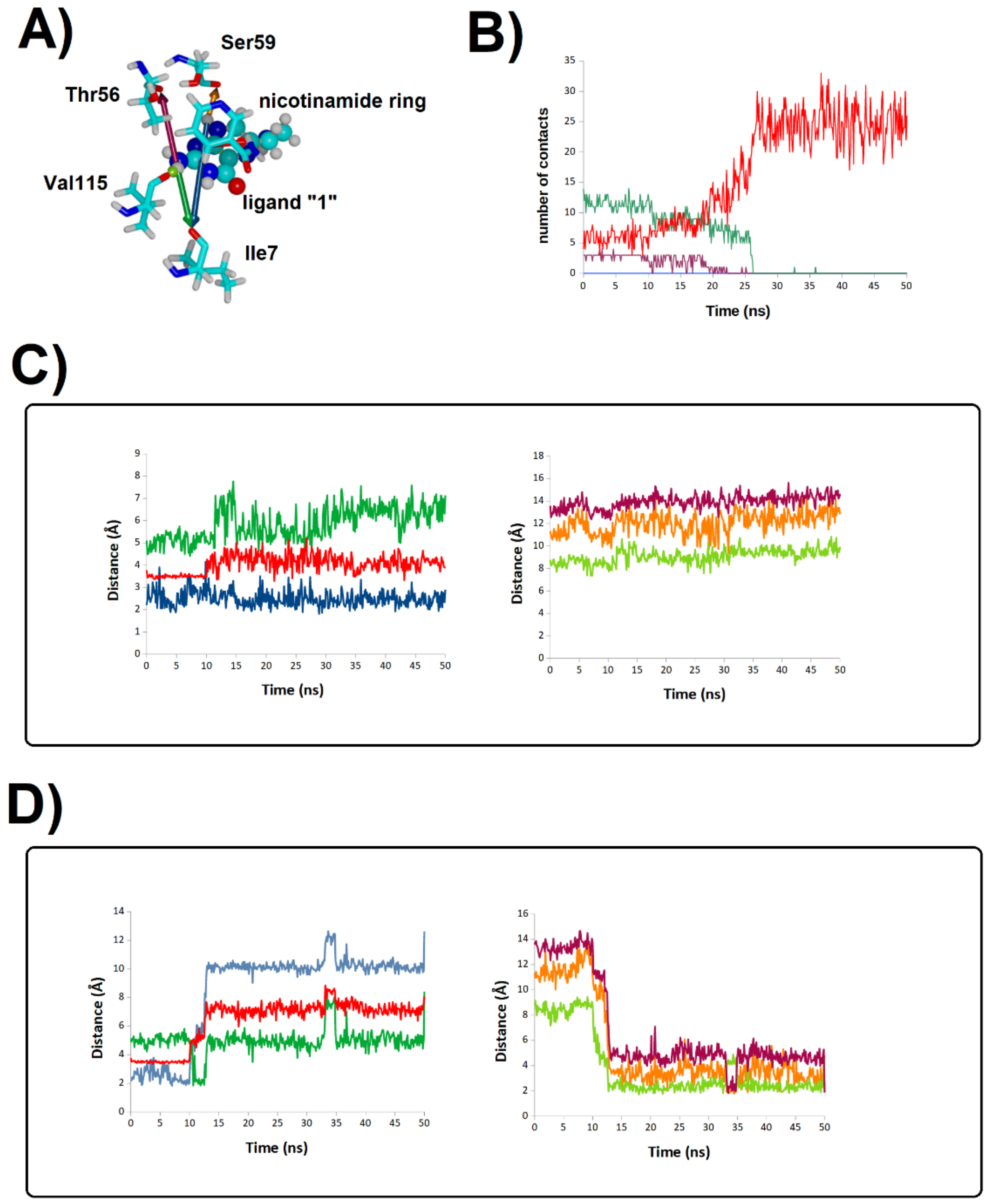
2.2. Binding Stability of the Unmodified Dihydropterin Core (Molecule 1)
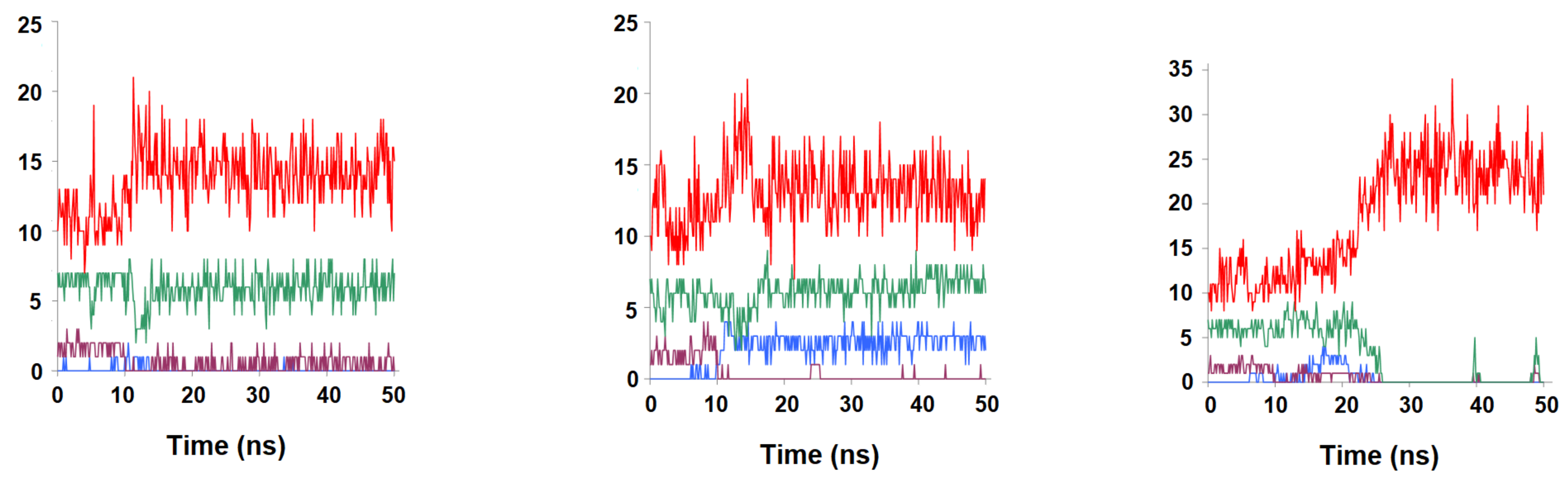
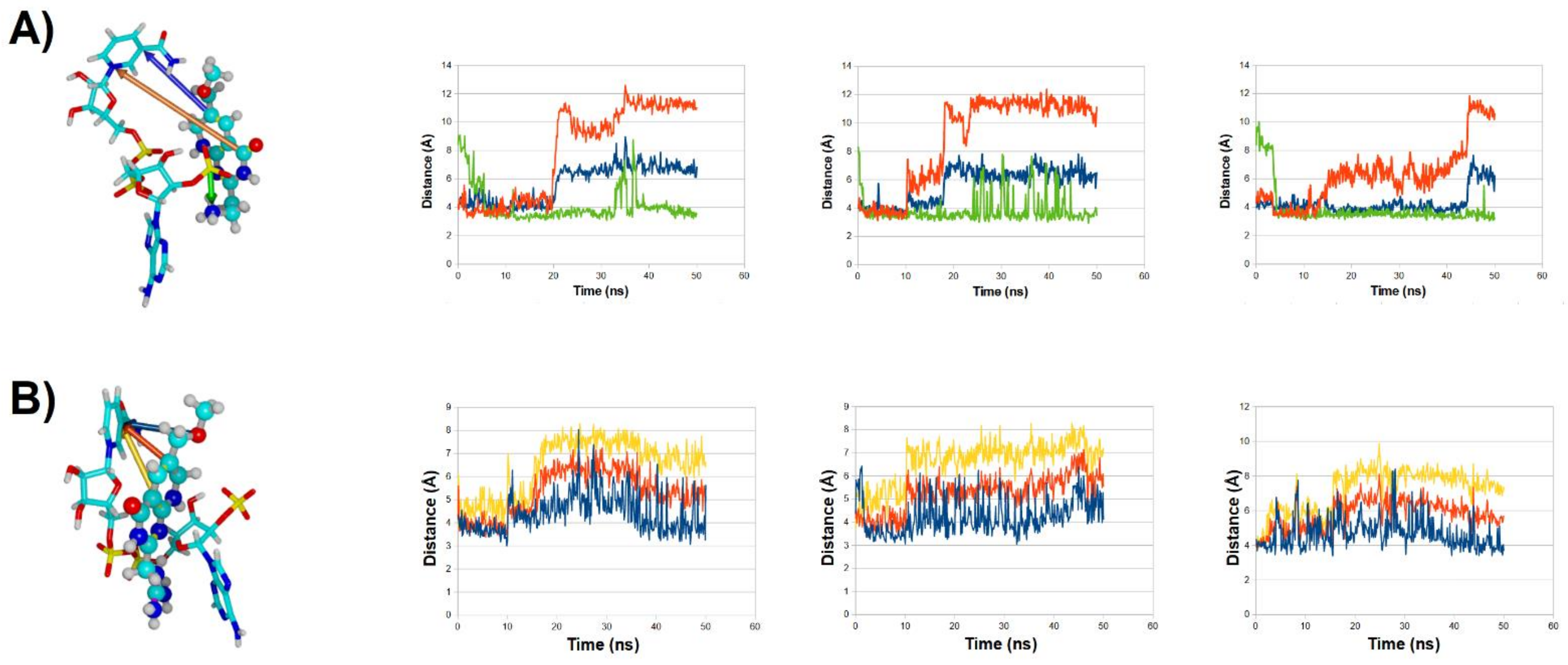
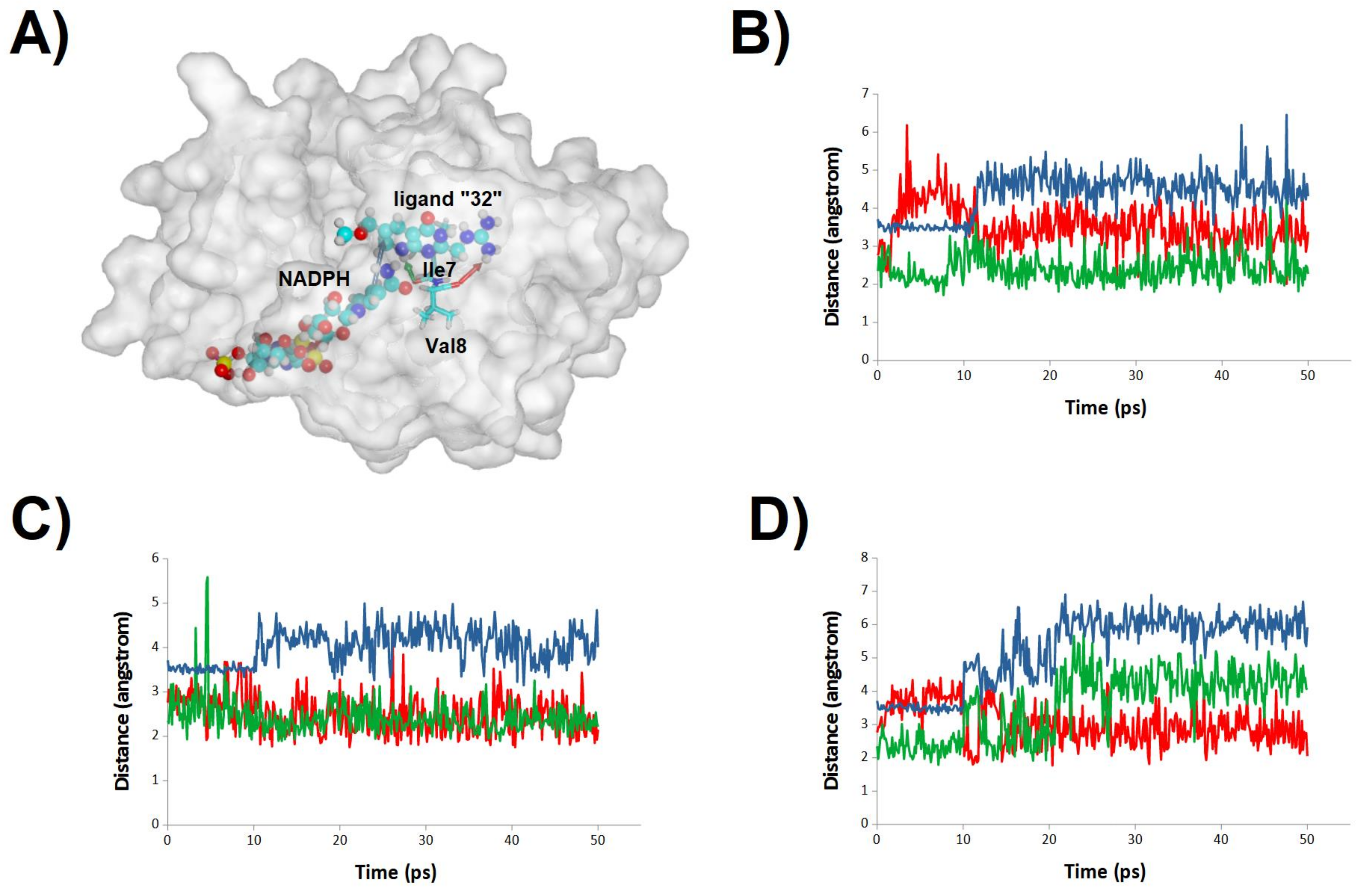
2.3. Binding Dynamics of Folate in the R67 DHFR Channel
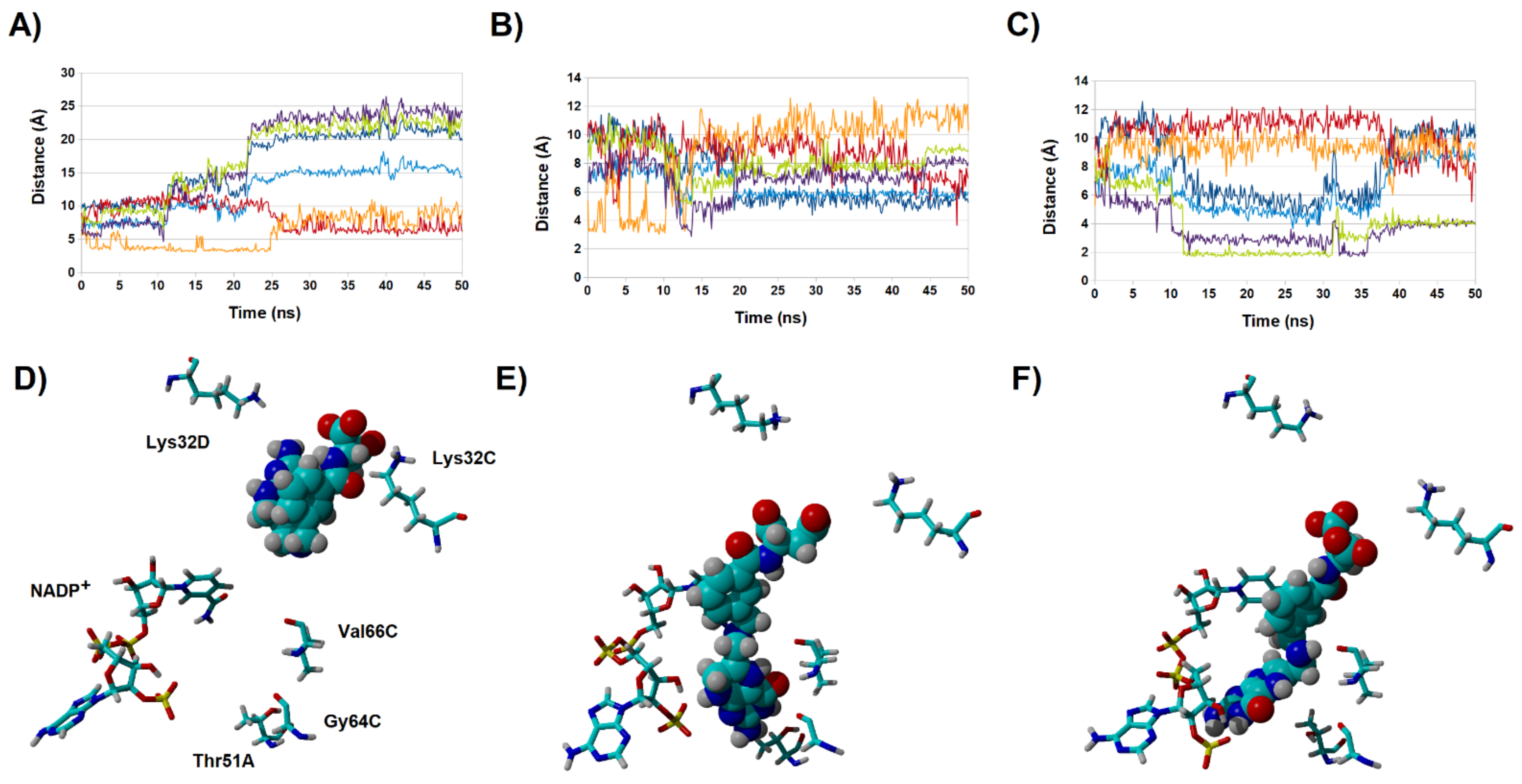
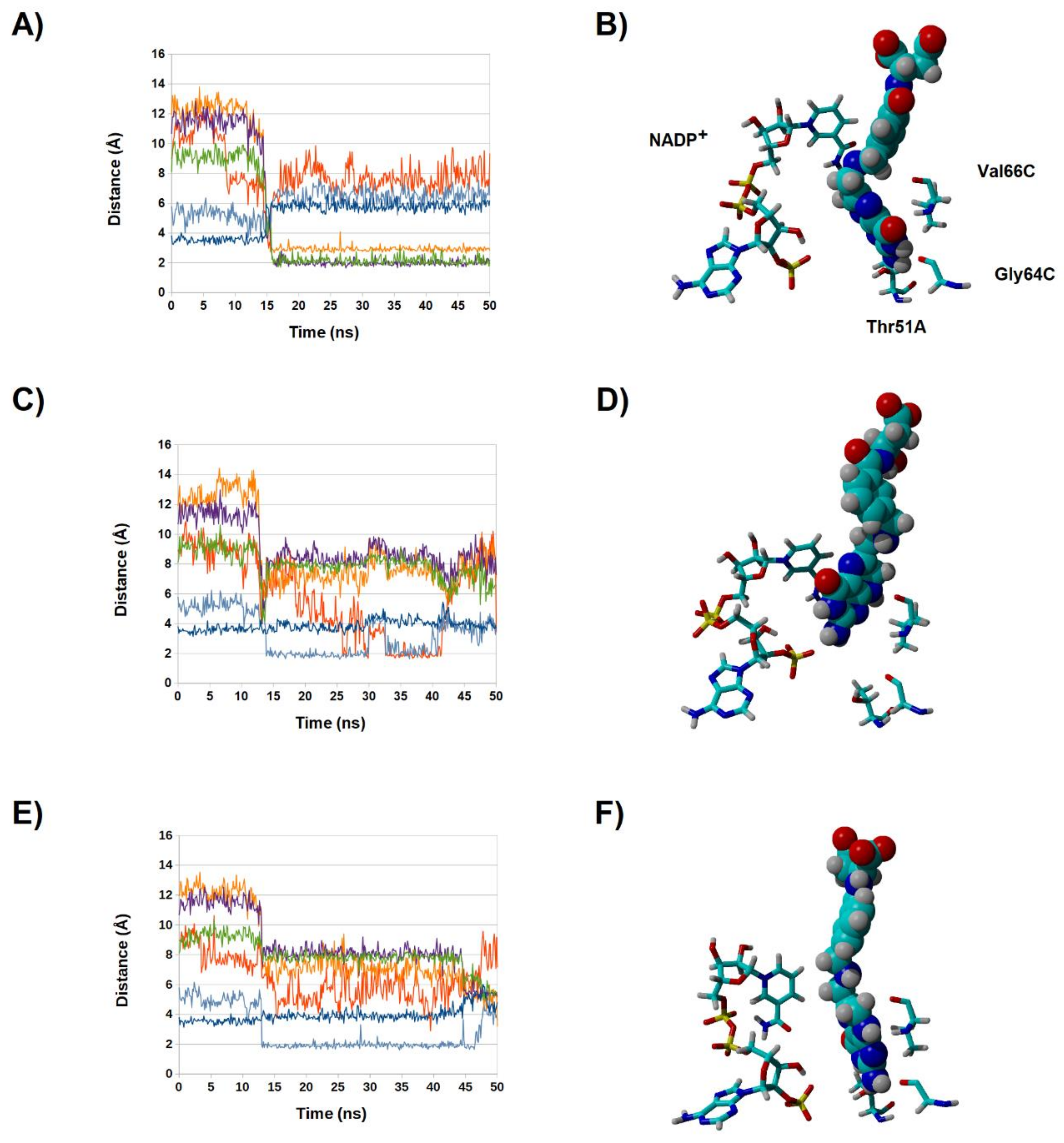
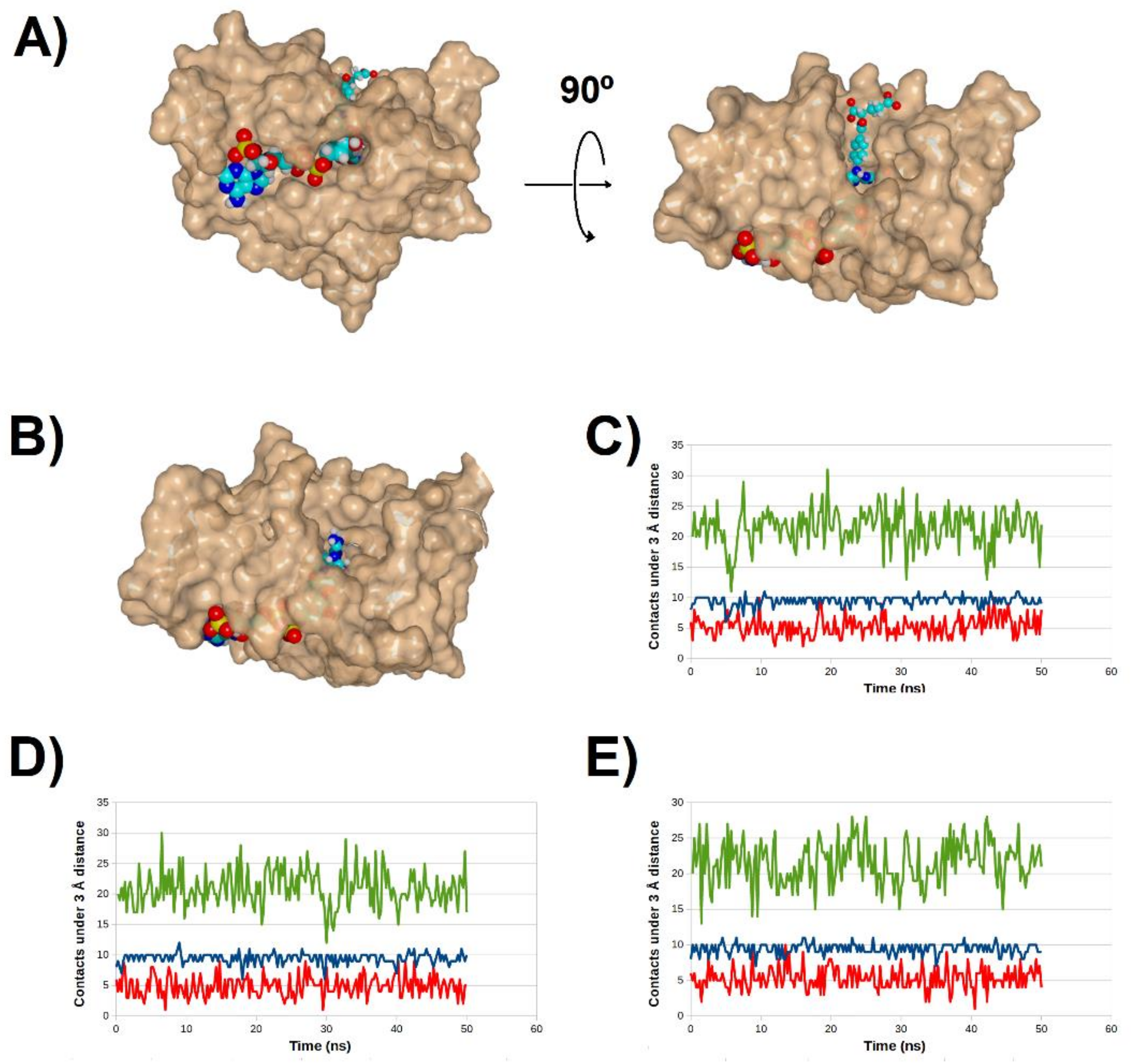
2.4. Binding Dynamics of Candidates 31 and 32 in the R67 DHFR Channel
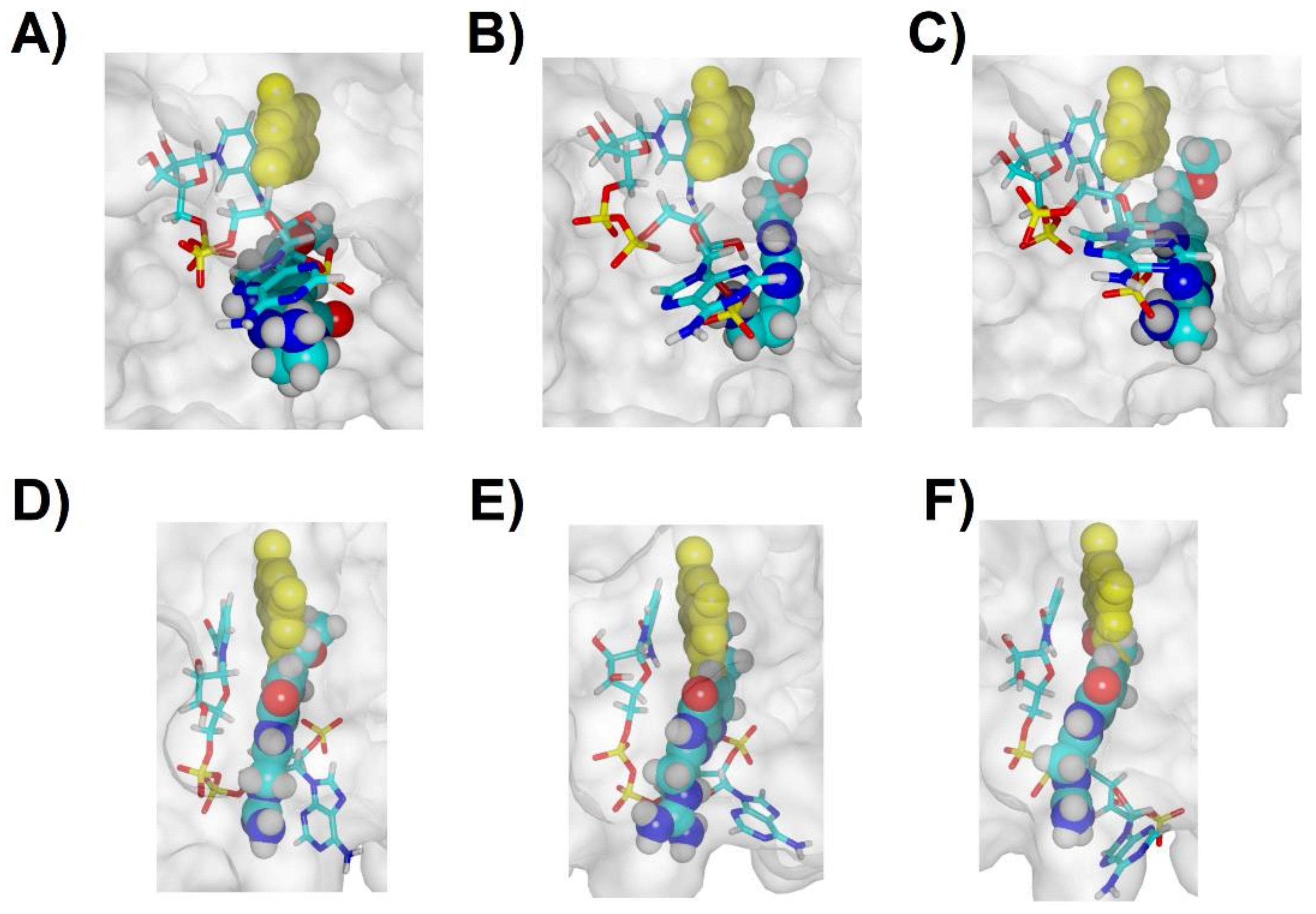
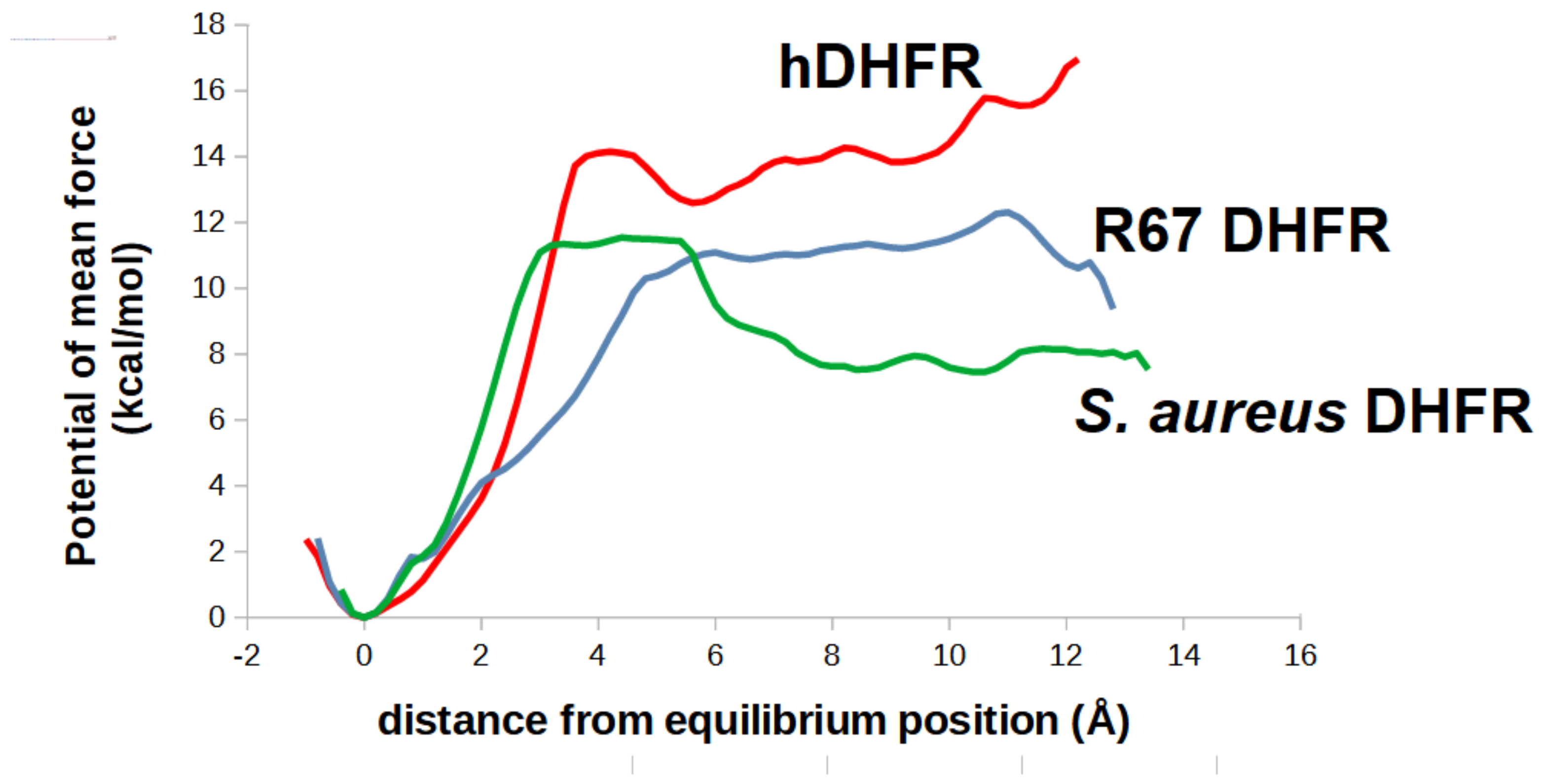
2.5. Analysis of the Binding of Molecule 32 Able to Other Dihydrofolate Reductases
3. Materials and Methods
3.1. Quantum Chemical Computations
3.2. Molecular Docking and Molecular Dynamics
3.3. Computation of Potentail of Mean Force Using Umbrella Sampling/Wheighted Histogram Analysis Method
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Appleman, J.R.; Prendergast, N.; Delcamp, T.J.; Freisheim, J.H.; Blakley, R.L. Kinetics of the formation and isomerization of methotrexate complexes of recombinant human dihydrofolate reductase. J. Biol. Chem. 1988, 263, 10304–10313. [Google Scholar] [CrossRef]
- Sköld, O.; Widh, A. A new dihydrofolate reductase with low trimethoprim sensitivity induced by an R factor mediating high resistance to trimethoprim. J. Biol. Chem. 1974, 249, 4324–4325. [Google Scholar] [CrossRef]
- Pattishall, K.H.; Acar, J.; Burchall, J.J.; Goldstein, F.W.; Harvey, R.J. Two distinct types of trimethoprim-resistant dihydrofolate reductase specified by R-plasmids of different compatibility groups. J. Biol. Chem. 1977, 252, 2319–2323. [Google Scholar] [CrossRef]
- Smith, S.L.; Stone, D.; Novak, P.; Baccanari, D.P.; Burchall, J.J. R plasmid dihydrofolate reductase with subunit structure. J. Biol. Chem. 1979, 254, 6222–6225. [Google Scholar] [CrossRef]
- Narayana, N.; Matthews, D.A.; Howell, E.; Xuong, N.-H. A plasmid-encoded dihydrofolate reductase from trimethoprim-resistant bacteria has a novel D2-symmetric active site. Nat. Struct. Mol. Biol. 1995, 2, 1018–1025. [Google Scholar] [CrossRef]
- Howell, E.E. Searching sequence space: Two different approaches to dihydrofolate reductase catalysis. ChemBioChem 2005, 6, 590–600. [Google Scholar] [CrossRef]
- Martinez, M.A.; Pezo, V.; Marlière, P.; Wain-Hobson, S. Exploring the functional robustness of an enzyme by in vitro evolution. EMBO J. 1996, 15, 1203–1210. [Google Scholar] [CrossRef]
- Park, H.; Bradrick, T.D.; Howell, E.E. A glutamine 67--> histidine mutation in homotetrameric R67 dihydrofolate reductase results in four mutations per single active site pore and causes substantial substrate and cofactor inhibition. Protein Eng. 1997, 10, 1415–1424. [Google Scholar] [CrossRef]
- Strader, M.B.; Smiley, R.D.; Stinnett, L.G.; VerBerkmoes, N.C.; Howell, E.E. Role of S65, Q67, I68, and Y69 residues in homotetrameric R67 dihydrofolate reductase. Biochemistry 2001, 40, 11344–11352. [Google Scholar] [CrossRef]
- Schmitzer, A.R.; Lépine, F.; Pelletier, J.N. Combinatorial exploration of the catalytic site of a drug-resistant dihydrofolate reductase: Creating alternative functional configurations. Protein Eng. Des. Sel. 2004, 17, 809–819. [Google Scholar] [CrossRef]
- Reece, L.J.; Nichols, R.; Ogden, R.C.; Howell, E.E. Construction of a synthetic gene for an R-plasmid-encoded dihydrofolate reductase and studies on the role of the N-terminus in the protein. Biochemistry 1991, 30, 10895–10904. [Google Scholar] [CrossRef]
- Fierke, C.; Kuchta, R.; Johnson, K.; Benkovic, S. Implications for enzymic catalysis from free-energy reaction coordinate profiles. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 631–638. [Google Scholar] [CrossRef]
- Bastien, D.; Ebert, M.C.C.J.C.; Forge, D.; Toulouse, J.; Kadnikova, N.; Perron, F.; Mayence, A.; Huang, T.L.; Eynde, J.J.V.; Pelletier, J.N. Fragment-based design of symmetrical bis-benzimidazoles as selective inhibitors of the trimethoprim-resistant, type II R67 dihydrofolate reductase. J. Med. Chem. 2012, 55, 3182–3192. [Google Scholar] [CrossRef]
- Toulouse, J.L.; Yachnin, B.; Ruediger, E.H.; Deon, D.; Gagnon, M.; Saint-Jacques, K.; Ebert, M.C.C.J.C.; Forge, D.; Bastien, D.; Colin, D.Y.; et al. Structure-based design of dimeric bisbenzimidazole inhibitors to an emergent trimethoprim-resistant type II dihydrofolate reductase guides the design of monomeric analogues. ACS Omega 2019, 4, 10056–10069. [Google Scholar] [CrossRef]
- Garcia-Viloca, M.; Truhlar, D.G.; Gao, J. Reaction-path energetics and kinetics of the hydride transfer reaction catalyzed by dihydrofolate reductase. Biochemistry 2003, 42, 13558–13575. [Google Scholar] [CrossRef]
- Park, H.; Zhuang, P.; Nichols, R.; Howell, E. Mechanistic studies of R67 dihydrofolate reductase. Effects of pH and H62C mutation. J. Biol. Chem. 1997, 272, 2252–2258. [Google Scholar] [CrossRef]
- Chopra, S.; Lynch, R.; Kim, S.-H.; Jackson, M.; Howell, E.E. Effects of temperature and viscosity on R67 dihydrofolate reductase catalysis. Biochemistry 2006, 45, 6596–6605. [Google Scholar] [CrossRef]
- Kamath, G.; Howell, E.E.; Agarwal, P.K. The tail wagging the dog: Insights into catalysis in R67 dihydrofolate reductase. Biochemistry 2010, 49, 9078–9088. [Google Scholar] [CrossRef]
- Li, D.; Levy, L.A.; Gabel, S.A.; Lebetkin, M.S.; DeRose, E.F.; Wall, M.J.; Howell, E.E.; London, R.E. Interligand overhauser effects in type II dihydrofolate reductase. Biochemistry 2001, 40, 4242–4252. [Google Scholar] [CrossRef]
- Krahn, J.M.; Jackson, M.R.; DeRose, E.F.; Howell, E.; London, R.E. Crystal structure of a type II dihydrofolate reductase catalytic ternary complex. Biochemistry 2007, 46, 14878–14888. [Google Scholar] [CrossRef]
- Bhabha, G.; Ekiert, D.C.; Jennewein, M.; Zmasek, C.M.; Tuttle, L.M.; Kroon, G.; Dyson, H.J.; Godzik, A.; Wilson, I.A.; Wright, P.E. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly 8.0.0. 2013. Available online: https://classic.chem.msu.su/gran/gamess/index.html (accessed on 1 April 2014).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Baker, J.; Kessi, A.; Delley, B. The generation and use of delocalized internal coordinates in geometry optimization. J. Chem. Phys. 1996, 105, 192–212. [Google Scholar] [CrossRef]
- Silva, P.J.; Ramos, M.J. Successes and failures of DFT functionals in acid/base and redox reactions of organic and biochemical interest. Comput. Theor. Chem. 2011, 966, 120–126. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Cossi, M.; Mennucci, B.; Pitarch, J.; Tomasi, J. Correction of cavity-induced errors in polarization charges of continuum solvation models. J. Comput. Chem. 1998, 19, 833–846. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Grossfield, A. WHAM: The Weighted Histogram Analysis Method. 2018. Available online: http://membrane.urmc.rochester.edu/?page_id=126 (accessed on 1 March 2022).
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Protonation Energy (kcal/mol) (vs. Dihydrofolate Core) | Reduction by NADH (kcal/mol) | Molecule | Protonation Energy (kcal/mol) (vs. Dihydrofolate Core) | Reduction by NADH (kcal/mol) |
|---|---|---|---|---|---|
| 1 | 0.0 | 35.2 | 17 | 10.6 | 28.0 |
| 2 | 4.0 | 28.8 | 18 | 8.2 | 26.2 |
| 3 | 2.8 | 33.5 | 19 | 10.5 | 22.9 |
| 4 | 7.0 | 26.3 | 20 | 20.1 | 46.0 |
| 5 | 9.4 | 22.0 | 21 | 26.0 | 37.2 |
| 6 | −2.9 | 38.4 | 22 | 25.1 | 40.2 |
| 7 | −9.1 | 39.6 | 23 | 33.8 | 32.2 |
| 8 | 1.8 | 33.2 | 24 | 7.9 | 4.5 |
| 9 | −0.4 | 36.3 | 25 | 8.2 | 20.3 |
| 10 | −2.4 | 37.8 | 26 | 11.4 | 2.3 |
| 11 | 13.9 | 53.8 | 27 | 11.7 | 3.6 |
| 12 | 2.2 | 30.5 | 28 | 16.1 | 2.2 |
| 13 | 9.8 | 23.8 | 29 | 8.6 | 16.1 |
| 14 | 9.0 | 5.8 | 30 | 11.7 | 16.2 |
| 15 | 16.8 | 40.7 | 31 | 20.7 | 42.8 |
| 16 | 8.7 | 10.4 | 32 | 24.1 | 39.7 |
| Molecules | R67 DHFR (PDB: 2RK1) | Human DHFR (PDB: 4M6K) | S. aureus DHFR (PDB: 3FRE) |
|---|---|---|---|
| 1 | 5.85 | 6.58 | 5.57 |
| 11 | 5.81 | 6.61 | 5.03 |
| 15 | 5.00 | 4.39 | 4.55 |
| 20 | 6.29 | 6.93 | 5.81 |
| 21 | 6.76 | 7.42 | 5.97 |
| 22 | 5.91 | 6.40 | 5.76 |
| 23 | 6.01 | 6.76 | 6.09 |
| 31 | 7.94 | 8.40 | 7.89 |
| 32 | 6.94 | 7.78 | 6.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, P.J. Computational Development of Inhibitors of Plasmid-Borne Bacterial Dihydrofolate Reductase. Antibiotics 2022, 11, 779. https://doi.org/10.3390/antibiotics11060779
Silva PJ. Computational Development of Inhibitors of Plasmid-Borne Bacterial Dihydrofolate Reductase. Antibiotics. 2022; 11(6):779. https://doi.org/10.3390/antibiotics11060779
Chicago/Turabian StyleSilva, Pedro J. 2022. "Computational Development of Inhibitors of Plasmid-Borne Bacterial Dihydrofolate Reductase" Antibiotics 11, no. 6: 779. https://doi.org/10.3390/antibiotics11060779
APA StyleSilva, P. J. (2022). Computational Development of Inhibitors of Plasmid-Borne Bacterial Dihydrofolate Reductase. Antibiotics, 11(6), 779. https://doi.org/10.3390/antibiotics11060779