
Antitubercular, Cytotoxicity, and Computational Target Validation of Dihydroquinazolinone Derivatives

 ,
,  , ,
, ,  , , , ,
, , , ,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. Antitubercular Activity
2.2. Cytotoxicity Assay
2.3. Computational Studies
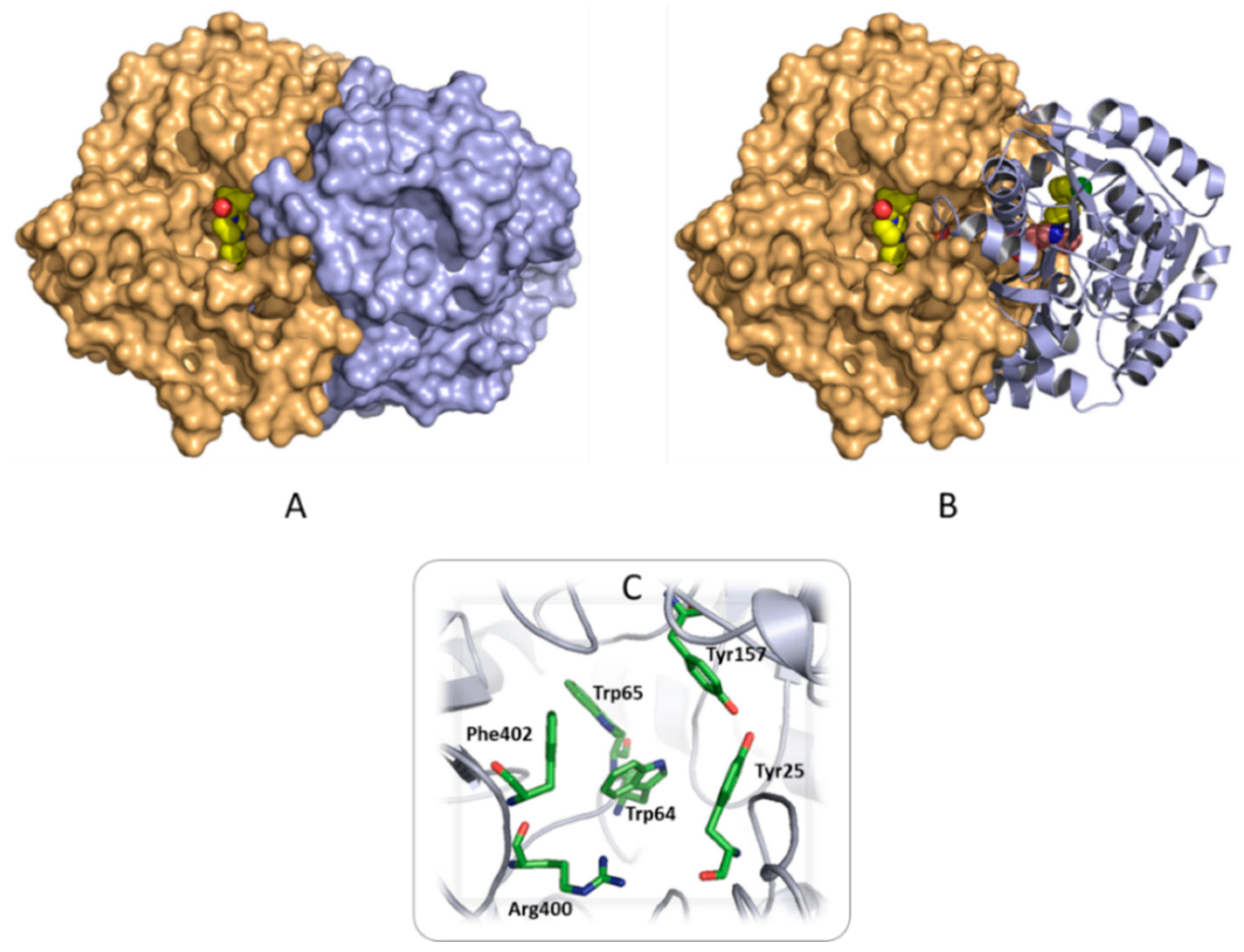
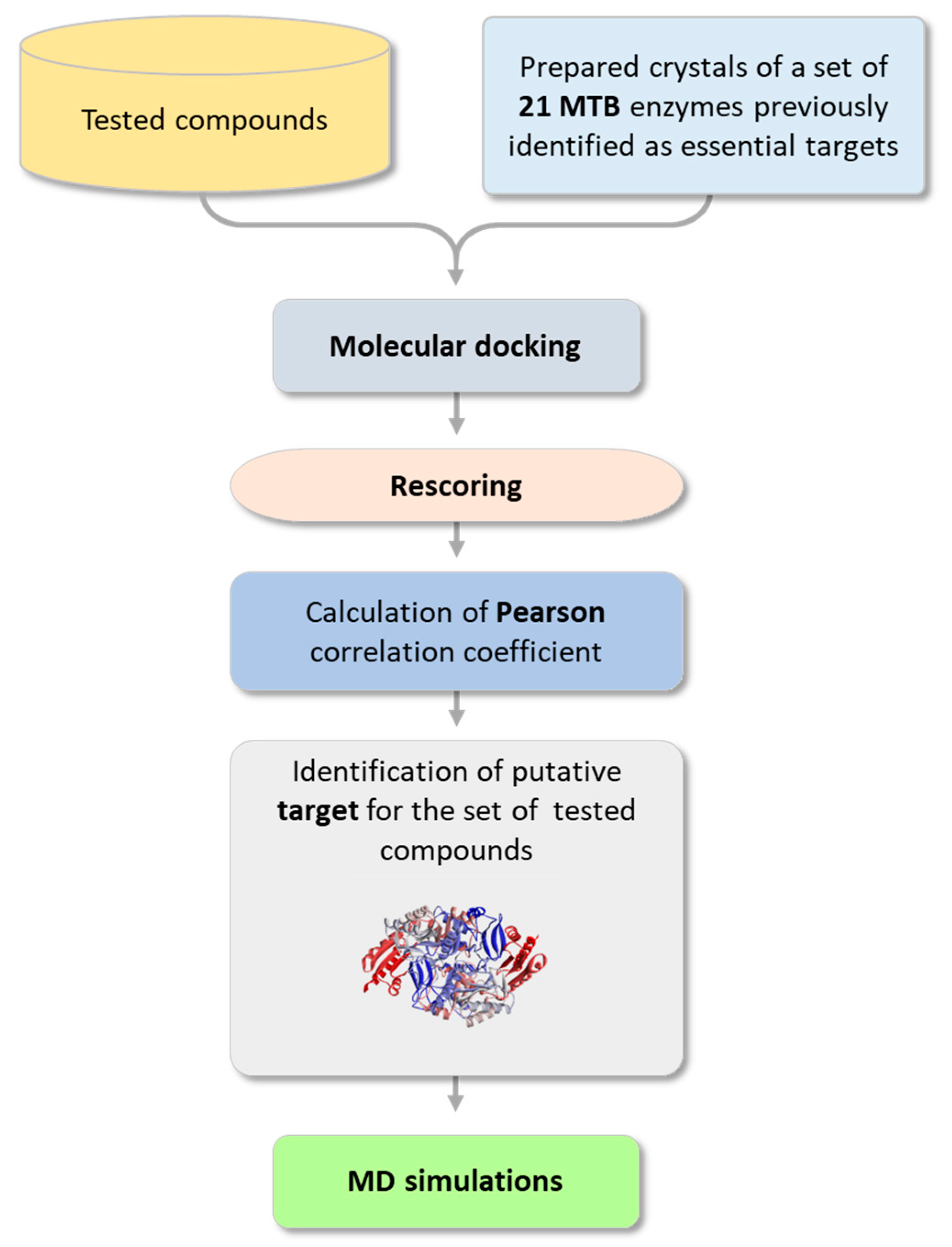
2.3.1. Searching for a Putative Drug Target
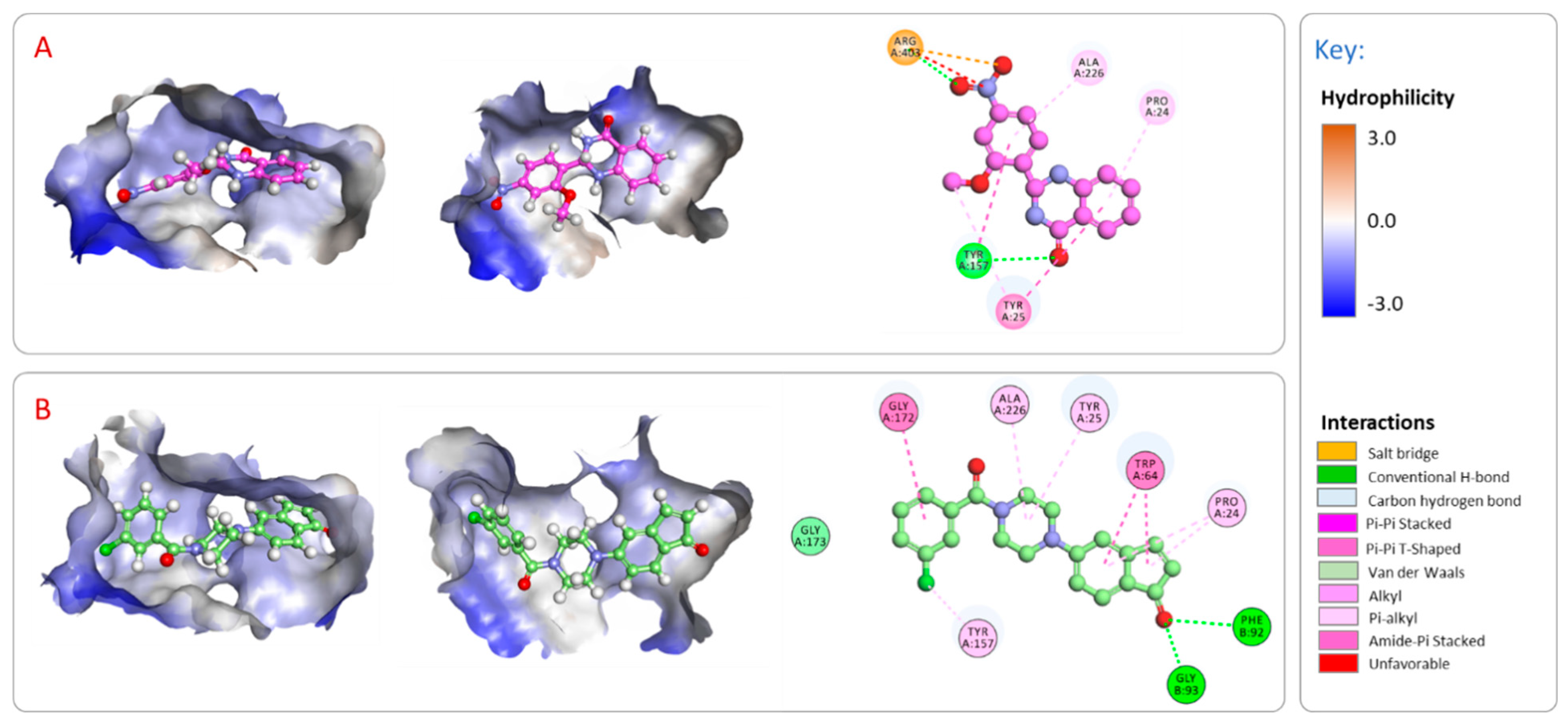
2.3.2. Analysis of the Binding Interactions with the Putative Target (BioA)
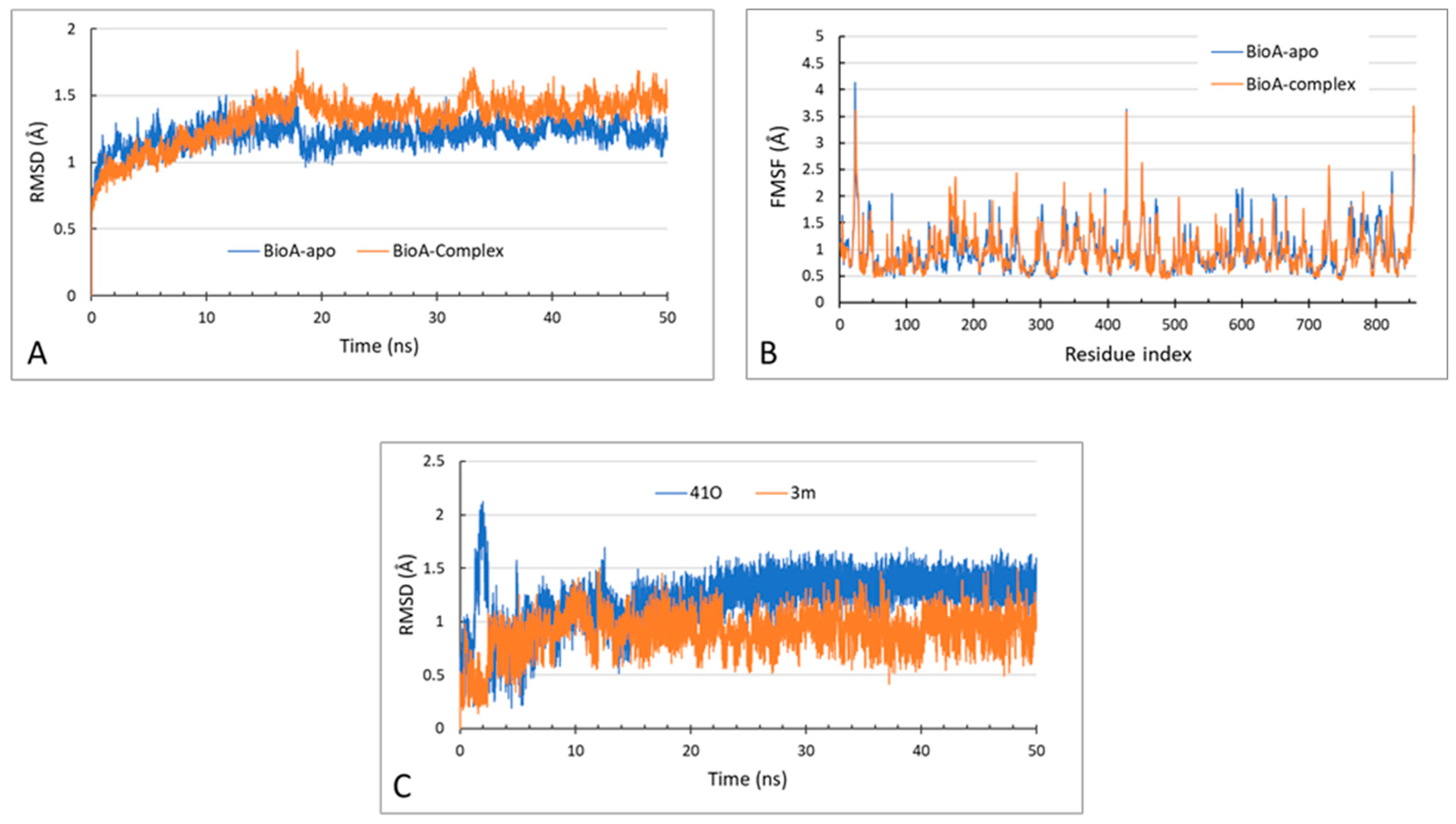
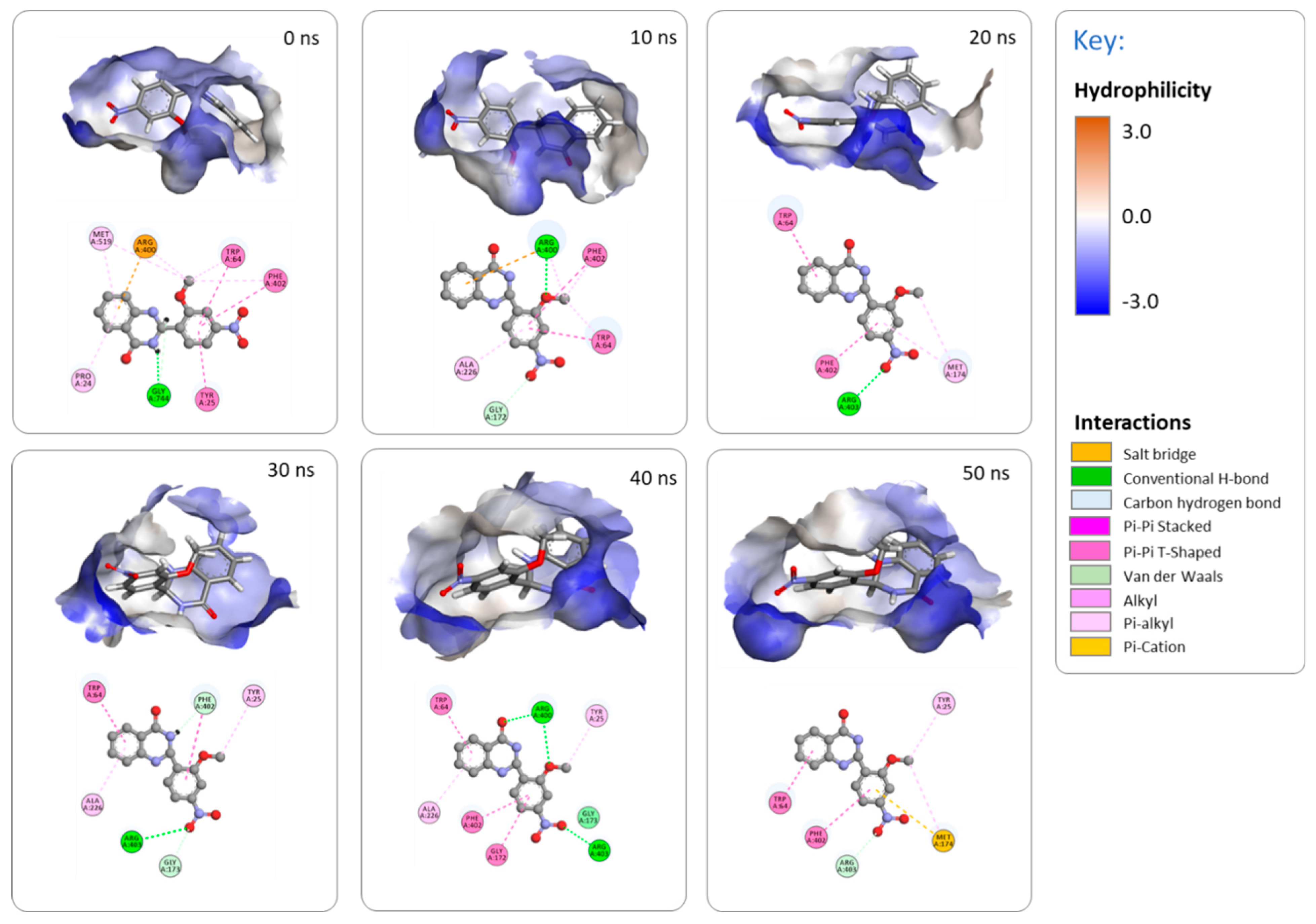
2.3.3. MD Simulations
2.3.4. ADME and Toxicity Predictions
3. Materials and Methods
3.1. General
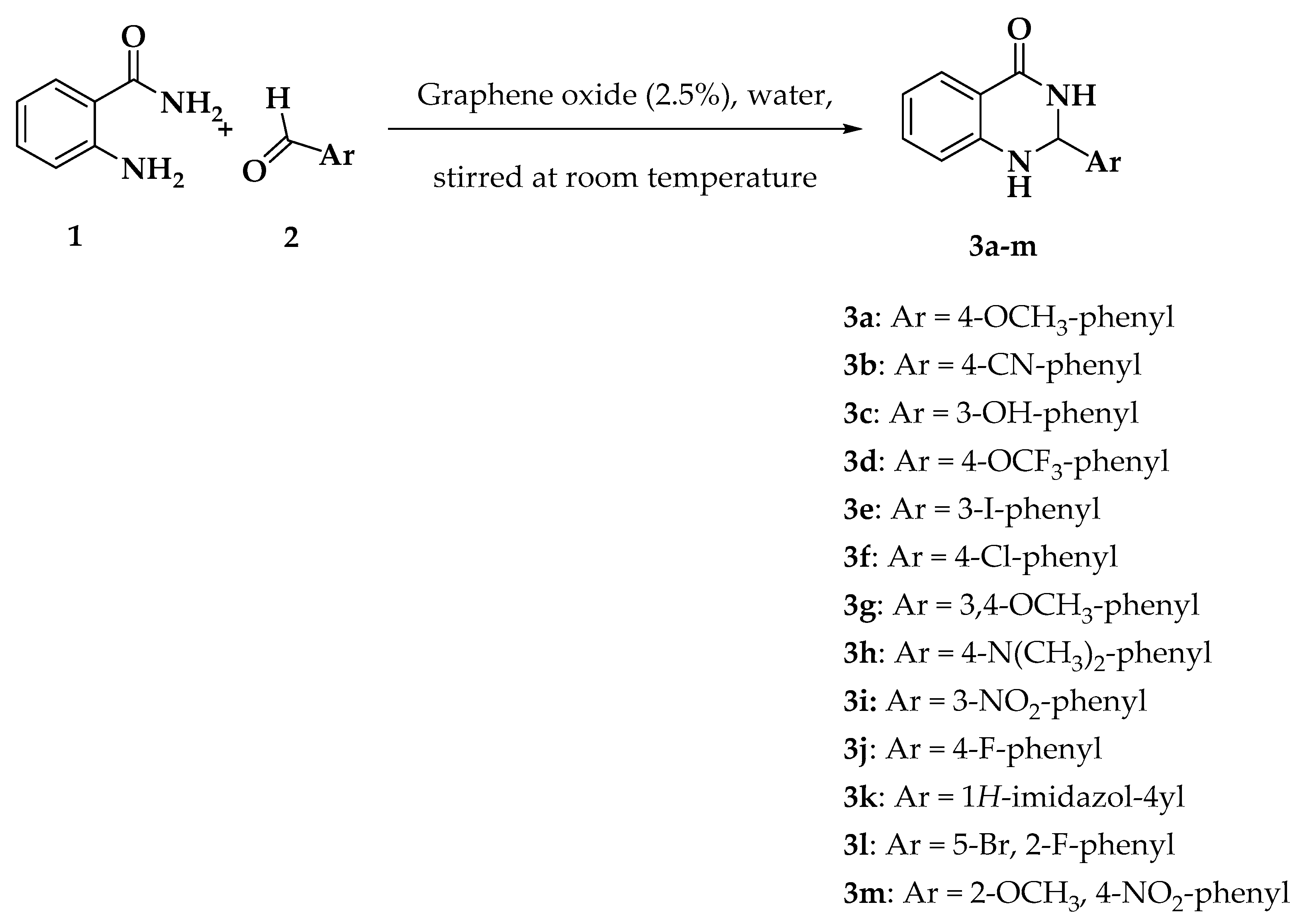
3.2. Chemistry
3.3. Antitubercular Activity
3.4. Cell Line
3.5. MTT Cytotoxicity Assay
3.6. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019 (Executive Summary). Available online: https://www.who.int/tb/publications/global_report/tb19_Exec_Sum_12Nov2019.pdf?ua=1 (accessed on 9 September 2020).
- Furin, J.; Cox, H.; Pai, M. Tuberculosis. Lancet 2019, 393, 1642–1656. [Google Scholar] [CrossRef]
- Lee, J.Y. Diagnosis and Treatment of Extrapulmonary Tuberculosis. Tuberc. Respir. Dis. 2015, 78, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Tetali, S.R.; Kunapaeddi, E.; Mailavaram, R.P.; Singh, V.; Borah, P.; Deb, P.K.; Venugopala, K.N.; Hourani, W.; Tekade, R.K. Current advances in the clinical development of anti-tubercular agents. Tuberculosis 2020, 125, 101989. [Google Scholar] [CrossRef] [PubMed]
- Udwadia, Z.F. MDR, XDR, TDR tuberculosis: Ominous progression. Thorax 2012, 67, 286–288. [Google Scholar] [CrossRef]
- Dheda, K.; Shean, K.; Zumla, A.; Badri, M.; Streicher, E.M.; Page-Shipp, L.; Willcox, P.; John, M.A.; Reubenson, G.; Govindasamy, D.; et al. Early treatment outcomes and HIV status of patients with extensively drug-resistant tuberculosis in South Africa: A retrospective cohort study. Lancet 2010, 375, 1798–1807. [Google Scholar] [CrossRef]
- Quan, D.; Nagalingam, G.; Payne, R.; Triccas, J.A. New tuberculosis drug leads from naturally occurring compounds. Int. J. Infect. Dis. 2017, 56, 212–220. [Google Scholar] [CrossRef]
- Yang, D.; Taylor, Z.E.; Handy, S.; Li, S.J.; Liu, J.W.; Stabenow, J.; Zalduondo, L.; Jonsson, C.B.; Altman, E.; Kong, Y. Identification of Anti-tuberculosis Compounds From Aurone Analogs. Front. Microbiol. 2020, 11, 1004. [Google Scholar] [CrossRef]
- Dasyam, N.; Munkacsi, A.B.; Fadzilah, N.H.; Senanayake, D.S.; O’Toole, R.F.; Keyzers, R.A. Identification and Bioactivity of 3-epi-Xestoaminol C Isolated from the New Zealand Brown Alga Xiphophora chondrophylla. J. Nat. Prod. 2014, 77, 1519–1523. [Google Scholar] [CrossRef]
- Tang, S.J.; Yao, L.; Hao, X.H.; Liu, Y.D.; Zeng, L.H.; Liu, G.; Li, M.W.; Li, F.J.; Wu, M.Y.; Zhu, Y.S.; et al. Clofazimine for the Treatment of Multidrug-Resistant Tuberculosis: Prospective, Multicenter, Randomized Controlled Study in China. Clin. Infect. Dis. 2015, 60, 1361–1367. [Google Scholar] [CrossRef]
- Lee, M.; Lee, J.; Carroll, M.W.; Choi, H.; Min, S.; Song, T.; Via, L.E.; Goldfeder, L.C.; Kang, E.; Jin, B.; et al. Linezolid for Treatment of Chronic Extensively Drug-Resistant Tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518. [Google Scholar] [CrossRef]
- Nguyen, T.V.A.; Anthony, R.; Bañuls, A.-L.; Vu, D.H.; Alffenaar, J. Bedaquiline resistance: Its emergence, mechanism and prevention. Clin. Infect. Dis. 2018, 66, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Kawasaki, M.; Hariguchi, N.; Liu, Y.; Matsumoto, M. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis 2018, 108, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Cox, H.; Ford, N. Linezolid for the treatment of complicated drug-resistant tuberculosis: A systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2012, 16, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Dalcolmo, M.; Gayoso, R.; Sotgiu, G.; D’Ambrosio, L.; Rocha, J.L.; Borga, L.; Fandinho, F.; Braga, J.U.; Galesi, V.M.N.; Barreira, D.; et al. Effectiveness and safety of clofazimine in multidrug-resistant tuberculosis: A nationwide report from Brazil. Eur. Respir. J. 2017, 49, 1602445. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.L.; Cha, H.C.; Jahng, Y. Recent advances in the studies on luotonins. Molecules 2011, 16, 4861–4883. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Z.; Luo, J.; Yang, F.; Liu, T.; Liu, M.; Qiu, W.-W.; Tang, J. Synthesis and Biological Evaluation of Heterocyclic Ring-Fused Betulinic Acid Derivatives as Novel Inhibitors of Osteoclast Differentiation and Bone Resorption. J. Med. Chem. 2012, 55, 3122–3134. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.-C.; Baek, H.Y.; Lee, K.-D.; Kim, Y.-C.; Oh, H. Anti-neuroinflammatory effects of tryptanthrin from Polygonum tinctorium Lour. in lipopolysaccharide-stimulated BV2 microglial cells. Arch. Pharmacal Res. 2018, 41, 419–430. [Google Scholar] [CrossRef]
- Tian, K.-M.; Li, J.-J.; Xu, S.-W. Rutaecarpine: A promising cardiovascular protective alkaloid from Evodia rutaecarpa (Wu Zhu Yu). Pharmacol. Res. 2019, 141, 541–550. [Google Scholar] [CrossRef]
- Estlin, E.J.; Pinkerton, C.R.; Lewis, I.J.; Lashford, L.; McDowell, H.; Morland, B.; Kohler, J.; Newell, D.R.; Boddy, A.V.; Taylor, G.A.; et al. A phase I study of nolatrexed dihydrochloride in children with advanced cancer. A United Kingdom Children’s Cancer Study Group Investigation. Br. J. Cancer 2001, 84, 11–18. [Google Scholar] [CrossRef][Green Version]
- Gudimella, K.K.; Bonige, K.B.; Gundla, R.; Katari, N.K.; Yamajala, B.; Battula, V.R. 2,4-Diphenyl-1,2-dihydroquinazoline Derivatives: Synthesis, Anticancer Activity and Docking Studies. ChemistrySelect 2019, 4, 12528–12533. [Google Scholar] [CrossRef]
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.N.C.V.L.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Madasu, C.; Xu, Y.-M.; Wijeratne, E.M.K.; Liu, M.X.; Molnár, I.; Gunatilaka, A.A.L. Semi-synthesis and cytotoxicity evaluation of pyrimidine, thiazole, and indole analogues of argentatins A–C from guayule (Parthenium argentatum) resin. Med. Chem. Res. 2022. [Google Scholar] [CrossRef]
- Mallavadhani, U.V.; Chandrashekhar, M.; Nayak, V.L.; Ramakrishna, S. Synthesis and anticancer activity of novel fused pyrimidine hybrids of myrrhanone C, a bicyclic triterpene of Commiphora mukul gum resin. Mol. Divers. 2015, 19, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Iyer, K.A.; Alix, K.; Eltit, J.M.; Solis, E., Jr.; Pan, X.; Argade, M.D.; Khatri, S.; De Felice, L.J.; Sweet, D.H.; Schulte, M.K.; et al. Multi-modal antidepressant-like action of 6- and 7-chloro-2-aminodihydroquinazolines in the mouse tail suspension test. Psychopharmacology 2019, 236, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Barmak, A.; Niknam, K.; Mohebbi, G. Synthesis, Structural Studies, and α-Glucosidase Inhibitory, Antidiabetic, and Antioxidant Activities of 2,3-Dihydroquinazolin-4(1H)-ones Derived from Pyrazol-4-carbaldehyde and Anilines. ACS Omega 2019, 4, 18087–18099. [Google Scholar] [CrossRef] [PubMed]
- Guillon, R.; Pagniez, F.; Picot, C.; Hédou, D.; Tonnerre, A.; Chosson, E.; Duflos, M.; Besson, T.; Logé, C.; Le Pape, P. Discovery of a novel broad-spectrum antifungal agent derived from albaconazole. ACS Med. Chem. Lett. 2013, 4, 288–292. [Google Scholar] [CrossRef]
- Mujeeb Ur, R.; Rathore, A.; Siddiqui, A.A.; Parveen, G.; Yar, M.S. Synthesis and characterization of quinazoline derivatives: Search for hybrid molecule as diuretic and antihypertensive agents. J. Enzym. Inhib. Med. Chem. 2014, 29, 733–743. [Google Scholar] [CrossRef]
- Obase, H.; Takai, H.; Teranishi, M.; Nakamizo, N. Synthesis of (1-substituted piperidin-4-yl)-1H-benzimidazoles and (1-substituted piperidin-4-yl)-3,4-dihydroquinazolines as possible antihypertensive agents. J. Heterocycl. Chem. 1983, 20, 565–573. [Google Scholar] [CrossRef]
- El-Sabbagh, O.I.; Ibrahim, S.M.; Baraka, M.M.; Kothayer, H. Synthesis of new 2,3-dihydroquinazolin-4(1H)-one derivatives for analgesic and anti-inflammatory evaluation. Arch. Pharm. 2010, 343, 274–281. [Google Scholar] [CrossRef]
- Clissold, S.P.; Beresford, R. Proquazone. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in rheumatic diseases and pain states. Drugs 1987, 33, 478–502. [Google Scholar] [CrossRef]
- Kamal, A.; Babu, K.S.; Poornachandra, Y.; Nagaraju, B.; Ali Hussaini, S.M.; Shaik, S.P.; Ganesh Kumar, C.; Alarifi, A. Efficient and green sulfamic acid catalyzed synthesis of new 1,2-dihydroquinazoline derivatives with antibacterial potential. Arab. J. Chem. 2015, 12, 3546–3554. [Google Scholar] [CrossRef]
- Salehi, P.; Ayyari, M.; Bararjanian, M.; Ebrahimi, S.N.; Aliahmadi, A. Synthesis, antibacterial and antioxidant activity of novel 2,3-dihydroquinazolin-4(1H)-one derivatives of dehydroabietylamine diterpene. J. Iran. Chem. Soc. 2014, 11, 607–613. [Google Scholar] [CrossRef]
- Chung, C.K.; Liu, Z.; Lexa, K.W.; Andreani, T.; Xu, Y.; Ji, Y.; DiRocco, D.A.; Humphrey, G.R.; Ruck, R.T. Asymmetric Hydrogen Bonding Catalysis for the Synthesis of Dihydroquinazoline-Containing Antiviral, Letermovir. J. Am. Chem. Soc. 2017, 139, 10637–10640. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.R.; Naravane, J.S.; Desai, R.M. Vasicinone. A Bronchodilator Principle from Adhatoda Vasica Nees (N. O. Acanthaceae). J. Org. Chem. 1963, 28, 445–448. [Google Scholar] [CrossRef]
- Yamamura, M.; Ochiai, T.; Ishida, R. Effects of afloqualone, a new centrally acting muscle relaxant, on DRL response and CER in rats (author’s transl). Nihon Yakurigaku Zasshi 1981, 78, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, C.; Foy, J.M.; Pratt, C.N.F.W.; Purvis, J.R. On the pharmacological actions of a diuretic, fenquizone, with particular reference to its site of action. J. Pharm. Pharmacol. 1981, 33, 219–222. [Google Scholar] [CrossRef] [PubMed]
- van Zyl, E.F. A survey of reported synthesis of methaqualone and some positional and structural isomers. Forensic Sci. Int. 2001, 122, 142–149. [Google Scholar] [CrossRef]
- Jafari, E.; Khajouei, M.R.; Hassanzadeh, F.; Hakimelahi, G.H.; Khodarahmi, G.A. Quinazolinone and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 2016, 11, 1–14. [Google Scholar]
- Li, W.J.; Li, Q.; Liu, D.L.; Ding, M.W. Synthesis, fungicidal activity, and sterol 14alpha-demethylase binding interaction of 2-azolyl-3,4-dihydroquinazolines on Penicillium digitatum. J. Agric. Food Chem. 2013, 61, 1419–1426. [Google Scholar] [CrossRef]
- Zhou, Y.; Feng, Q.; Di, F.; Liu, Q.; Wang, D.; Chen, Y.; Xiong, L.; Song, H.; Li, Y.; Li, Z. Synthesis and insecticidal activities of 2,3-dihydroquinazolin-4(1H)-one derivatives targeting calcium channel. Bioorganic Med. Chem. 2013, 21, 4968–4975. [Google Scholar] [CrossRef]
- Alveera, S.; Venugopala, K.N.; Khedr, M.A.; Pillay, M.; Nwaeze, K.U.; Coovadia, Y.; Shode, F.; Odhav, B. Antimycobacterial, docking and molecular dynamic studies of pentacyclic triterpenes from Buddleja saligna leaves. J. Biomol. Struct. Dyn. 2017, 35, 2654–2664. [Google Scholar] [CrossRef]
- Venugopala, K.N. Posological review of dose extrapolation methodologies in animal studies, early-phase clinical trials and special populations. Int. J. Res. Pharm. Sci. 2020, 11, 6079–6084. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Albericio, F.; Coovadia, Y.M.; Kruger, H.G.; Maguire, G.E.M.; Pillay, M.; Govender, T. Total synthesis of a depsidomycin analogue by convergent solid-phase peptide synthesis and macrolactonization strategy for antitubercular activity. J. Pept. Sci. 2011, 17, 683–689. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Nayak, S.K.; Pillay, M.; Prasanna, R.; Coovadia, Y.M.; Odhav, B. Synthesis and antitubercular activity of 2-(substituted phenyl/benzyl-amino)-6-(4-chlorophenyl)-5-(methoxycarbonyl)-4-methyl-3,6-dihydropyrimidin-1-ium chlorides. Chem. Biol. Drug Des. 2013, 81, 219–227. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Dharma Rao, G.B.; Bhandary, S.; Pillay, M.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; Alwassil, O.I.; Harsha, S.; Mlisana, K. Design, synthesis, and characterization of (1-(4-aryl)- 1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates against Mycobacterium tuberculosis. Drug Des. Dev. Ther. 2016, 10, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Chandrashekharappa, S.; Pillay, M.; Bhandary, S.; Kandeel, M.; Mahomoodally, F.M.; Morsy, M.A.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; et al. Synthesis and structural elucidation of novel benzothiazole derivatives as anti-tubercular agents: In-silico screening for possible target identification. Med. Chem. 2019, 15, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Chandrashekharappa, S.; Pillay, M.; Abdallah, H.H.; Mahomoodally, F.M.; Bhandary, S.; Chopra, D.; Attimarad, M.; Aldhubiab, B.E.; Nair, A.B. Computational, crystallographic studies, cytotoxicity and anti-tubercular activity of substituted 7-methoxy-indolizine analogues. PLoS ONE 2019, 14, e0217270. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Tratrat, C.; Pillay, M.; Mahomoodally, F.M.; Bhandary, S.; Chopra, D.; Morsy, M.A.; Haroun, M.; Aldhubiab, B.E.; Attimarad, M.; et al. Anti-tubercular activity of substituted 7-methyl and 7-formylindolizines and in silico study for prospective molecular target identification. Antibiotics 2019, 8, 247. [Google Scholar] [CrossRef]
- Khedr, M.A.; Pillay, M.; Chandrashekharappa, S.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; Alwassil, O.I.; Mlisana, K.; Odhav, B.; Venugopala, K.N. Molecular modeling studies and anti-TB activity of trisubstituted indolizine analogues; molecular docking and dynamic inputs. J. Biomol. Struct. Dyn. 2018, 36, 2163–2178. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Chandrashekharappa, S.; Deb, P.K.; Tratrat, C.; Pillay, M.; Chopra, D.; Al-Shar’i, N.A.; Hourani, W.; Dahabiyeh, L.A.; Borah, P.; et al. Anti-tubercular activity and molecular docking studies of indolizine derivatives targeting mycobacterial InhA enzyme. J. Enzym. Inhib. Med. Chem. 2021, 36, 1472–1487. [Google Scholar] [CrossRef]
- Venugopala, K.; Tratrat, C.; Chandrashekharappa, S.; Attimarad, M.; SreeHarsha, N.; Nair, A.; Pottathil, S.; Venugopala, R.; Al-Attraqchi, O.; Morsy, M.; et al. Anti-tubercular Potency and Computationallyassessed Drug-likeness and Toxicology of Diversely Substituted Indolizines. Indian J. Pharm. Educ. 2019, 53, 545–552. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Chandrashekharappa, S.; Tratrat, C.; Deb, P.K.; Nagdeve, R.D.; Nayak, S.K.; Morsy, M.A.; Borah, P.; Mahomoodally, F.M.; Mailavaram, R.P. Crystallography, Molecular Modeling, and COX-2 Inhibition Studies on Indolizine Derivatives. Molecules 2021, 26, 3550. [Google Scholar] [CrossRef] [PubMed]
- Uppar, V.; Chandrashekharappa, S.; Shivamallu, C.; Kollur, S.P.; Ortega-Castro, J.; Frau, J.; Flores-Holguín, N.; Basarikatti, A.I.; Chougala, M.; Mohan, M.M.; et al. Investigation of antifungal properties of synthetic dimethyl-4-bromo-1-(substituted benzoyl) pyrrolo[1,2-a] quinoline-2,3-dicarboxylates analogues: Molecular docking studies and conceptual DFT-based chemical reactivity descriptors and pharmacokinetics evaluation. Molecules 2021, 26, 2722. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Al-Attraqchi, O.H.; Tratrat, C.; Nayak, S.K.; Morsy, M.A.; Aldhubiab, B.E.; Attimarad, M.; Nair, A.B.; Sreeharsha, N.; Venugopala, R. Novel series of methyl 3-(substituted benzoyl)-7-substituted-2-phenylindolizine-1-carboxylates as promising anti-inflammatory agents: Molecular modeling studies. Biomolecules 2019, 9, 661. [Google Scholar] [CrossRef] [PubMed]
- Nefisath, P.; Dasappa, J.P.; Haripriya, B.; Chopra, D.; Venugopala, K.N.; Deb, P.K.; Gleiser, R.M.; Mohanlall, V.; Maharaj, R.; Shashiprabha; et al. Synthesis, structural elucidation and larvicidal activity of novel arylhydrazones. J. Mol. Struct. 2021, 1236, 130305. [Google Scholar] [CrossRef]
- Nefisath, P.; Dasappa, J.P.; Haripriya, B.; Chopra, D.; Venugopala, K.N.; Deb, P.K.; Gleiser, R.M.; Mohanlall, V.; Maharaj, R.; Shashiprabha, S.; et al. Synthesis, characterization and larvicidal activity of novel benzylidene derivatives of fenobam and its thio analogues with crystal insight. J. Mol. Struct. 2021, 1226, 129386. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Habeebuddin, M.; Aldhubiab, B.E.; Asif, A.H. Design, Synthesis, and In Vitro Evaluation of Novel Indolyl DiHydropyrazole Derivatives as Potential Anticancer Agents. Molecules 2021, 26, 5235. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Ćirić, A.; Soković, M.; Nagaraja, S.; Venugopala, K.N.; Balachandran Nair, A. Exploration of the antimicrobial effects of benzothiazolylthiazolidin-4-one and in silico mechanistic investigation. Molecules 2021, 26, 4061. [Google Scholar] [CrossRef]
- Tratrat, C.; Haroun, M.; Paparisva, A.; Kamoutsis, C.; Petrou, A.; Gavalas, A.; Eleftheriou, P.; Geronikaki, A.; Venugopala, K.N.; Kochkar, H. New Substituted 5-Benzylideno-2-Adamantylthiazol [3, 2-b][1, 2, 4] Triazol-6 (5H) ones as Possible Anti-Inflammatory Agents. Molecules 2021, 26, 659. [Google Scholar] [CrossRef]
- Morsy, M.A.; Ali, E.M.; Kandeel, M.; Venugopala, K.N.; Nair, A.B.; Greish, K.; El-Daly, M. Screening and molecular docking of novel benzothiazole derivatives as potential antimicrobial agents. Antibiotics 2020, 9, 221. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Khedr, M.A.; Girish, Y.R.; Bhandary, S.; Chopra, D.; Morsy, M.A.; Aldhubiab, B.E.; Deb, P.K.; Attimarad, M.; Nair, A.B. Crystallography, in silico studies, and In vitro antifungal studies of 2,4,5 trisubstituted 1,2,3-triazole analogues. Antibiotics 2020, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Haroun, M.; Tratrat, C.; Kolokotroni, A.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Kostic, M.; Sokovic, M.; Carazo, A.; Mladěnka, P. 5-Benzyliden-2-(5-methylthiazol-2-ylimino) thiazolidin-4-ones as Antimicrobial Agents. Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Antibiotics 2021, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Al-Shar’i, N.A.; Al-Balas, Q.A.; Al-Waqfi, R.A.; Hassan, M.A.; Alkhalifa, A.E.; Ayoub, N.M. Discovery of a nanomolar inhibitor of the human glyoxalase-I enzyme using structure-based poly-pharmacophore modelling and molecular docking. J. Comput.-Aided Mol. Des. 2019, 33, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Al-Shar’i, N.A.; Al-Balas, Q.A.; Hassan, M.A.; El-Elimat, T.M.; Aljabal, G.A.; Almaaytah, A.M. Ellagic acid: A potent glyoxalase-I inhibitor with a unique scaffold. Acta Pharm. 2021, 71, 115–130. [Google Scholar] [CrossRef]
- Deb, P.K.; Al-Shar’i, N.A.; Venugopala, K.N.; Pillay, M.; Borah, P. In vitro anti-TB properties, in silico target validation, molecular docking and dynamics studies of substituted 1,2,4-oxadiazole analogues against Mycobacterium tuberculosis. J. Enzym. Inhib. Med. Chem. 2021, 36, 869–884. [Google Scholar] [CrossRef]
- Al-Shar’i, N.A.; Musleh, S.S. Identification of CHK1 Kinase Inhibitors Using Structure Based Pharmacophore Modelling and Molecular Docking. Indian J. Pharm. Sci. 2020, 82, 472–482. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Al-Balas, Q.A.; Hassan, M.A.; Al-Shar’i, N.A.; El-Elimat, T.; Almaaytah, A.M. Computational and experimental exploration of the structure–activity relationships of flavonoids as potent glyoxalase-I inhibitors. Drug Dev. Res. 2018, 79, 58–69. [Google Scholar] [CrossRef]
- Liu, F.; Dawadi, S.; Maize, K.M.; Dai, R.; Park, S.W.; Schnappinger, D.; Finzel, B.C.; Aldrich, C.C. Structure-Based Optimization of Pyridoxal 5′-Phosphate-Dependent Transaminase Enzyme (BioA) Inhibitors that Target Biotin Biosynthesis in Mycobacterium tuberculosis. J. Med. Chem. 2017, 60, 5507–5520. [Google Scholar] [CrossRef]
- Dey, S.; Lane, J.M.; Lee, R.E.; Rubin, E.J.; Sacchettini, J.C. Structural Characterization of the Mycobacterium tuberculosis Biotin Biosynthesis Enzymes 7,8-Diaminopelargonic Acid Synthase and Dethiobiotin Synthetase. Biochemistry 2010, 49, 6746–6760. [Google Scholar] [CrossRef]
- Singh, S.; Khare, G.; Bahal, R.K.; Ghosh, P.C.; Tyagi, A.K. Identification of Mycobacterium tuberculosis BioA inhibitors by using structure-based virtual screening. Drug Des. Dev. Ther. 2018, 12, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Geders, T.W.; Liu, F.; Park, S.W.; Schnappinger, D.; Aldrich, C.C.; Finzel, B.C. Fragment-based exploration of binding site flexibility in Mycobacterium tuberculosis BioA. J. Med. Chem. 2015, 58, 5208–5217. [Google Scholar] [CrossRef] [PubMed]
- Al-Shar’i, N.A.; Alnabulsi, S.M. Explaining the autoinhibition of the SMYD enzyme family: A theoretical study. J. Mol. Graph. Model. 2016, 68, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Al-Shar’i, N.A.; Al-Balas, Q.A. Molecular Dynamics Simulations of Adenosine Receptors: Advances, Applications and Trends. Curr. Pharm. Des. 2019, 25, 783–816. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Zhang, W.; Xu, X. ADME Evaluation in Drug Discovery. 6. Can Oral Bioavailability in Humans Be Effectively Predicted by Simple Molecular Property-Based Rules? J. Chem. Inf. Model. 2007, 47, 460–463. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Ramachandra, P.; Tratrat, C.; Gleiser, R.M.; Bhandary, S.; Chopra, D.; Morsy, M.A.; Aldhubiab, B.E.; Attimarad, M.; Nair, A.B.; et al. Larvicidal activities of 2-aryl-2,3-dihydroquinazolin -4-ones against malaria vector Anopheles arabiensis, In silico ADMET prediction and molecular target investigation. Molecules 2020, 25, 1316. [Google Scholar] [CrossRef]
- Bairagi, K.M.; Venugopala, K.N.; Alwassil, O.I.; Mohanlall, V.; Chandrashekharappa, S.; Nayak, S.K. Synthesis, crystal structure and Hirshfeld surface analysis of 2-(4-fluorophenyl)-2,3–dihydroquinazolin-4(1H)-one. Chem. Data Collect. 2020, 26, 100355. [Google Scholar] [CrossRef]
- Martin, A.; Morcillo, N.; Lemus, D.; Montoro, E.; Telles, M.A.; Simboli, N.; Pontino, M.; Porras, T.; Leon, C.; Velasco, M.; et al. Multicenter study of MTT and resazurin assays for testing susceptibility to first-line anti-tuberculosis drugs. Int. J. Tuberc. Lung Dis. 2005, 9, 901–906. [Google Scholar]
- Middlebrook, G.; Reggiards, Z.; Tigertt, W.D. Automable radiometric detection of growth of Mycobacterium tuberculosis in selective media. Am. Rev. Respir. Dis. 1977, 115, 1067–1069. [Google Scholar]
- Deb, P.K.; Mailavaram, R.; Chandrasekaran, B.; Kaki, V.R.; Kaur, R.; Kachler, S.; Klotz, K.N.; Akkinepally, R.R. Synthesis, adenosine receptor binding and molecular modelling studies of novel thieno[2,3-d]pyrimidine derivatives. Chem. Biol. Drug Des. 2018, 91, 962–969. [Google Scholar] [CrossRef]
- Dhingra, M.S.; Deb, P.K.; Chadha, R.; Singh, T.; Karan, M. Synthesis, evaluation, and molecular docking studies of cycloalkyl/aryl-3,4,5-trimethylgallates as potent non-ulcerogenic and gastroprotective anti-inflammatory agents. Med. Chem. Res. 2014, 23, 87–106. [Google Scholar] [CrossRef]
- Deb, P.K.; Sharma, A.; Piplani, P.; Akkinepally, R.R. Molecular docking and receptor-specific 3D-QSAR studies of acetylcholinesterase inhibitors. Mol. Divers. 2012, 16, 803–823. [Google Scholar] [CrossRef]
- Deb, P.K.; Junaid, A.; El-Rabie, D.; Hon, T.; Nasr, E.; Pichika, M. Molecular Docking Studies and Comparative Binding Mode Analysis of FDA Approved HIV Protease Inhibitors. Asian J. Chem. 2014, 26, 6227–6232. [Google Scholar] [CrossRef]
- Al-Jaidi, B.A.; Telfah, S.T.; Bardaweel, S.K.; Deb, P.K.; Borah, P.; Venugopala, K.N.; Bataineh, Y.A.; Al Khames Aga, Q.A. Anticancer Activity and in Silico ADMET Properties of 2,4,5-Trisubstitutedthiazole Derivatives. Curr. Drug Metab. 2021, 22, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E2510–E2517. [Google Scholar] [CrossRef] [PubMed]
- Torfs, E.; Piller, T.; Cos, P.; Cappoen, D. Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy. Int. J. Mol. Sci. 2019, 20, 2868. [Google Scholar] [CrossRef]
- Piton, J.; Foo, C.S.-Y.; Cole, S.T. Structural studies of Mycobacterium tuberculosis DprE1 interacting with its inhibitors. Drug Discov. Today 2017, 22, 526–533. [Google Scholar] [CrossRef]
- Kamsri, P.; Hanwarinroj, C.; Phusi, N.; Pornprom, T.; Chayajarus, K.; Punkvang, A.; Suttipanta, N.; Srimanote, P.; Suttisintong, K.; Songsiriritthigul, C.; et al. Discovery of New and Potent InhA Inhibitors as Antituberculosis Agents: Structure-Based Virtual Screening Validated by Biological Assays and X-ray Crystallography. J. Chem. Inf. Model. 2019, 60, 226–234. [Google Scholar] [CrossRef]
- Huang, C.-C.; Smith, C.V.; Glickman, M.S.; Jacobs, W.R.; Sacchettini, J.C. Crystal Structures of Mycolic Acid Cyclopropane Synthases from Mycobacterium tuberculosis. J. Biol. Chem. 2002, 277, 11559–11569. [Google Scholar] [CrossRef]
- LeMagueres, P.; Im, H.; Ebalunode, J.; Strych, U.; Benedik, M.J.; Briggs, J.M.; Kohn, A.H.; Krause, K.L. The 1.9 Å Crystal Structure of Alanine Racemase from Mycobacterium tuberculosis Contains a Conserved Entryway into the Active Site. Biochemistry 2005, 44, 1471–1481. [Google Scholar] [CrossRef]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Gonsaud, M.; Ducasse, S.; Hoh, F.; Zerbib, D.; Labesse, G.; Quemard, A. Crystal Structure of MabA from Mycobacterium tuberculosis, a Reductase involved in Long-chain Fatty Acid Biosynthesis. J. Mol. Biol. 2002, 320, 249–261. [Google Scholar] [CrossRef]
- Gurcha, S.S.; Usha, V.; Cox, J.; Fütterer, K.; Abrahams, K.; Bhatt, A.; Alderwick, L.; Reynolds, R.C.; Loman, N.; Nataraj, V.; et al. Biochemical and Structural Characterization of Mycobacterial Aspartyl-tRNA Synthetase AspS, a Promising TB Drug Target. PLoS ONE 2014, 9, e113568. [Google Scholar] [CrossRef]
- Yuan, T.; Sampson, N.S. Hit Generation in TB Drug Discovery: From Genome to Granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Han, X.; Yan, Q.; Wang, C.; Jia, L.; Taj, A.; Zhao, L.; Ma, Y. The Inhibitory Effect of GlmU Acetyltransferase Inhibitor TPSA on Mycobacterium tuberculosis May Be Affected Due to Its Methylation by Methyltransferase Rv0560c. Front. Cell. Infect. Microbiol. 2019, 9, 251. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, G.; Orena, B.S.; Esposito, M.; Lane, T.; Ribeiro, A.L.D.J.L.; Degiacomi, G.; Zemanová, J.; Szádocka, S.; Huszar, S.; et al. A multitarget approach to drug discovery inhibiting Mycobacterium tuberculosis PyrG and PanK. Sci. Rep. 2018, 8, 3187. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Teachout, N.; Beczkiewicz, J.; Procknow, R.; Schaenzer, A.J.; Satyshur, K.; Pavelka, M.; Zuercher, W.; Drewry, D.; Sauer, J.-D.; et al. In Silico Screen and Structural Analysis Identifies Bacterial Kinase Inhibitors which Act with β-Lactams To Inhibit Mycobacterial Growth. Mol. Pharm. 2018, 15, 5410–5426. [Google Scholar] [CrossRef]
- Kang, C.-M.; Abbott, D.W.; Park, S.T.; Dascher, C.C.; Cantley, L.C.; Husson, R.N. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: Substrate identification and regulation of cell shape. Genes Dev. 2005, 19, 1692–1704. [Google Scholar] [CrossRef]
- Ioerger, T.R.; O’Malley, T.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Mohaideen, N.; Murphy, K.C.; Boshoff, H.I.M.; Mizrahi, V.; Rubin, E.J.; et al. Identification of New Drug Targets and Resistance Mechanisms in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e75245. [Google Scholar] [CrossRef]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259. [Google Scholar] [CrossRef]
- Jansen, R.S.; Mandyoli, L.; Hughes, R.; Wakabayashi, S.; Pinkham, J.T.; Selbach, B.; Guinn, K.M.; Rubin, E.J.; Sacchettini, J.C.; Rhee, K.Y. Aspartate aminotransferase Rv3722c governs aspartate-dependent nitrogen metabolism in Mycobacterium tuberculosis. Nat. Commun. 2020, 11, 1960. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Abrahams, K.; Alemparte, C.; Ghidelli-Disse, S.; Rullas, J.; Angulo-Barturen, I.; Singh, A.; Gurcha, S.S.; Nataraj, V.; Bethell, S.; et al. THPP target assignment reveals EchA6 as an essential fatty acid shuttle in mycobacteria. Nat. Microbiol. 2016, 1, 15006. [Google Scholar] [CrossRef] [PubMed]
- Willand, N.; Dirié, B.; Carette, X.; Bifani, P.; Singhal, A.; Desroses, M.; Leroux, F.; Willery, E.; Mathys, V.; Déprez-Poulain, R.; et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 2009, 15, 537–544. [Google Scholar] [CrossRef]
- Munshi, T.; Gupta, A.; Evangelopoulos, D.; Guzman, J.D.; Gibbons, S.; Keep, N.H.; Bhakta, S. Characterisation of ATP-Dependent Mur Ligases Involved in the Biogenesis of Cell Wall Peptidoglycan in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e60143. [Google Scholar] [CrossRef] [PubMed]
- VanderVen, B.C.; Fahey, R.J.; Lee, W.; Liu, Y.; Abramovitch, R.B.; Memmott, C.; Crowe, A.M.; Eltis, L.D.; Perola, E.; Deininger, D.D.; et al. Novel Inhibitors of Cholesterol Degradation in Mycobacterium tuberculosis Reveal How the Bacterium’s Metabolism Is Constrained by the Intracellular Environment. PLoS Pathog. 2015, 11, e1004679. [Google Scholar] [CrossRef]
- Mori, G.; Chiarelli, L.R.; Esposito, M.; Makarov, V.; Bellinzoni, M.; Hartkoorn, R.C.; Degiacomi, G.; Boldrin, F.; Ekins, S.; Ribeiro, A.L.D.J.L.; et al. Thiophenecarboxamide Derivatives Activated by EthA Kill Mycobacterium tuberculosis by Inhibiting the CTP Synthetase PyrG. Chem. Biol. 2015, 22, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Baugh, L.; Phan, I.; Begley, D.W.; Clifton, M.C.; Armour, B.; Dranow, D.M.; Taylor, B.M.; Muruthi, M.M.; Abendroth, J.; Fairman, J.W.; et al. Increasing the structural coverage of tuberculosis drug targets. Tuberculosis 2014, 95, 142–148. [Google Scholar] [CrossRef]
- Connor, S.E.; Capodagli, G.C.; Deaton, M.K.; Pegan, S.D. Structural and functional characterization ofMycobacterium tuberculosistriosephosphate isomerase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Bhakuni, V. Unusual Structural, Functional, and Stability Properties of Serine Hydroxymethyltransferase from Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 40793–40805. [Google Scholar] [CrossRef]
- Fivian-Hughes, A.S.; Houghton, J.; Davis, E.O. Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology 2012, 158, 308–318. [Google Scholar] [CrossRef]
- Singh, V.; Mizrahi, V. Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov. Today 2017, 22, 503–509. [Google Scholar] [CrossRef]
- Fu, G.; Wu, J.; Liu, W.; Zhu, D.; Hu, Y.; Deng, J.; Zhang, X.-E.; Bi, L.; Wang, D.-C. Crystal structure of DNA gyrase B′ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 2009, 37, 5908–5916. [Google Scholar] [CrossRef] [PubMed]
- Krieger, I.V.; Freundlich, J.S.; Gawandi, V.B.; Roberts, J.P.; Gawandi, V.B.; Sun, Q.; Owen, J.L.; Fraile, M.T.; Huss, S.I.; Lavandera, J.-L.; et al. Structure-Guided Discovery of Phenyl-diketo Acids as Potent Inhibitors of M. tuberculosis Malate Synthase. Chem. Biol. 2012, 19, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.B.; Murillo, A.C.; Chacon, O.; Barletta, R.G.; Sacchettini, J.C. Structure of the Mycobacterium tuberculosis d -Alanine: D -Alanine Ligase, a Target of the Antituberculosis Drug d -Cycloserine. Antimicrob. Agents Chemother. 2011, 55, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Sundaramurthi, J.C.; Hanna, L.E.; Selvaraju, S.; Brindha, S.; Gnanadoss, J.J.; Vincent, S.; Singh, H.; Swaminathan, S. TBDRUGS—Database of drugs for tuberculosis. Tuberculosis 2016, 100, 69–71. [Google Scholar] [CrossRef]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The Critical Role of embC in Mycobacterium tuberculosis. J. Bacteriol. 2008, 190, 4335–4341. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huszár, S.; Singh, V.; Polčicová, A.; Baráth, P.; Barrio, M.B.; Lagrange, S.; Leblanc, V.; Nacy, C.A.; Mizrahi, V.; Mikušová, K. N -Acetylglucosamine-1-Phosphate Transferase, WecA, as a Validated Drug Target in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e01310-17. [Google Scholar] [CrossRef]
- Venugopala, K.; Kandeel, M.; Pillay, M.; Deb, P.; Abdallah, H.; Mahomoodally, M.; Chopra, D. Anti-Tubercular Properties of 4-Amino-5-(4-Fluoro-3- Phenoxyphenyl)-4H-1,2,4-Triazole-3-Thiol and Its Schiff Bases: Computational Input and Molecular Dynamics. Antibiotics 2020, 9, 559. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | Compound Structure | Level of Activity (µg/mL) | Cytotoxicity (%) at 100 µM * | |
|---|---|---|---|---|
| Susceptible (H37Rv) | MDR | |||
| 3a |  | 64 | >128 | 28.8 ± 0.6 |
| 3b |  | >64 | No activity | 19.5 ± 5.0 |
| 3c |  | 8 | >32 | 58.5 ± 1.1 |
| 3d |  | >64 | No activity | 47.6 ± 1.6 |
| 3e |  | 128 | No activity | 72.0 ± 8.3 |
| 3f |  | >128 | No activity | 57.6 ± 3.7 |
| 3g |  | 64 | No activity | 32.5 ± 11.0 |
| 3h |  | >128 | No activity | 54.6 ± 2.8 |
| 3i |  | >64 | No activity | 24.1 ± 3.9 |
| 3j |  | >64 | No activity | 44.1 ± 11.5 |
| 3k |  | 4 | 16 | 19.2 ± 4.9 |
| 3l |  | 2 | >64 | 36.3 ± 4.2 |
| 3m |  | 2 | >64 | 30.9 ± 8.8 |
| Index | Name | LS1 | LS2 | PLP1 | PLP2 | Jain | PMF | PMF4 | CDE | CDIE | Ludi-1 | Ludi-2 | Ludi-3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Alanine racemase | −0.71 | 0.29 | −0.16 | −0.12 | 0 | −0.06 | −0.51 | 0.38 | 0.31 | −0.15 | −0.19 | −0.35 |
| 2 | CamA | −0.57 | −0.39 | −0.12 | −0.05 | −0.09 | −0.07 | −0.45 | −0.3 | −0.38 | 0.44 | 0.19 | −0.06 |
| 3 | DprE1 | −0.3 | 0.62 | 0.1 | −0.01 | −0.12 | 0.28 | −0.82 | 0.51 | 0.38 | 0.33 | 0.2 | 0.3 |
| 4 | FabH | −0.67 | 0.26 | −0.42 | −0.44 | −0.02 | −0.24 | −0.75 | 0.35 | 0.32 | −0.24 | −0.3 | −0.31 |
| 5 | InhA | −0.59 | −0.5 | 0.12 | 0.32 | 0.32 | 0.12 | −0.29 | 0.02 | −0.08 | 0.4 | 0.49 | −0.05 |
| 6 | MabA | −0.4 | 0.35 | −0.3 | −0.23 | −0.51 | −0.46 | −0.66 | 0.28 | 0.16 | −0.13 | −0.15 | −0.34 |
| 7 | AspS | −0.68 | −0.32 | −0.14 | −0.05 | 0.05 | 0.08 | −0.51 | 0.06 | −0.16 | 0.06 | 0.01 | −0.03 |
| 8 | LeuRS | −0.82 | 0.54 | 0.3 | −0.06 | −0.15 | −0.27 | −0.62 | 0.41 | 0.08 | 0.58 | 0.54 | 0.13 |
| 9 | GlmU | −0.3 | 0.52 | −0.32 | 0.11 | −0.36 | 0.05 | −0.81 | 0.43 | 0.2 | 0.17 | 0.17 | 0.33 |
| 10 | PanK | −0.02 | 0.75 | −0.14 | −0.12 | −0.08 | −0.15 | −0.31 | 0.25 | 0.25 | 0.56 | 0.38 | 0.08 |
| 11 | PknB | −0.36 | −0.58 | −0.56 | −0.3 | 0.03 | −0.03 | −0.67 | 0.03 | −0.2 | 0.1 | 0.12 | −0.12 |
| 12 | PknA | −0.42 | 0.4 | −0.07 | 0.11 | −0.76 | 0.13 | −0.52 | 0.17 | 0.01 | 0.25 | 0.05 | 0.03 |
| 13 | KasA | −0.28 | −0.08 | −0.62 | −0.48 | −0.38 | −0.24 | −0.38 | −0.04 | −0.26 | −0.27 | −0.25 | −0.43 |
| 14 | KasA (BS2) * | −0.27 | 0.38 | 0.17 | 0.06 | 0.32 | 0.43 | −0.44 | 0.14 | 0.43 | −0.02 | 0.06 | 0.16 |
| 15 | Pks13 | −0.1 | 0.32 | 0.3 | 0.39 | 0.66 | −0.12 | −0.66 | 0.59 | 0.29 | 0.1 | 0.22 | 0.25 |
| 16 | BioA # | −0.63 | 0.1 | 0.14 | 0.28 | 0.65 | −0.42 | −0.86 | 0.74 | 0.55 | 0.52 | 0.58 | 0.65 |
| 17 | EchA6 | −0.33 | 0.17 | −0.04 | 0.3 | 0.31 | −0.17 | −0.69 | 0.38 | 0.31 | 0.46 | 0.05 | 0.18 |
| 18 | MmpL3 | −0.77 | −0.38 | 0.25 | 0.31 | 0.14 | 0.32 | −0.39 | 0.04 | −0.07 | 0.1 | 0.1 | 0.42 |
| 19 | AspAT | −0.41 | 0.67 | −0.24 | 0.24 | 0.16 | 0.01 | −0.62 | 0.23 | 0.14 | 0.15 | 0.41 | 0.44 |
| 20 | MurE | −0.83 | −0.05 | 0.25 | 0.35 | 0.6 | −0.31 | −0.38 | 0.31 | 0.15 | 0.33 | 0.29 | 0.2 |
| 21 | EthR | −0.39 | 0.17 | 0.06 | 0.19 | −0.41 | −0.24 | −0.78 | 0.48 | 0.13 | 0.13 | 0.22 | 0.12 |
| 22 | PrpC | −0.4 | 0.29 | −0.44 | −0.46 | 0.08 | 0.04 | −0.56 | 0.37 | 0.3 | 0.08 | 0.16 | 0.05 |
| Compounds | MIC (µg/mL) | ADME Descriptors * | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Index | Code | H37Rv | MDR-MTB | AS | BBB | CYP2D6 Inhibition | Hepatotoxicity | HIA a | PPB b | AlogP | PSA c | |
| 1 | 3a | 64 | >128 | 3 | 2 | FALSE | TRUE | 0 | TRUE | 2.432 | 51.851 | |
| 2 | 3b | >64 | No activity | 3 | 2 | FALSE | TRUE | 0 | TRUE | 2.327 | 65.856 | |
| 3 | 3c | 8 | >32 | 3 | 2 | FALSE | TRUE | 0 | TRUE | 2.206 | 63.736 | |
| 4 | 3d | >64 | No activity | 1 | 1 | FALSE | TRUE | 0 | TRUE | 4.568 | 51.851 | |
| 5 | 3e | 128 | No activity | 2 | 1 | FALSE | FALSE | 0 | TRUE | 3.026 | 42.921 | |
| 6 | 3f | >128 | No activity | 2 | 1 | FALSE | TRUE | 0 | TRUE | 3.113 | 42.921 | |
| 7 | 3g | 64 | No activity | 3 | 2 | FALSE | TRUE | 0 | TRUE | 2.415 | 60.781 | |
| 8 | 3h | >128 | No activity | 3 | 2 | FALSE | TRUE | 0 | TRUE | 2.61 | 46.273 | |
| 9 | 3i | >64 | No activity | 3 | 3 | FALSE | TRUE | 0 | TRUE | 2.343 | 85.744 | |
| 10 | 3j | >64 | No activity | 3 | 2 | FALSE | TRUE | 0 | TRUE | 2.654 | 42.921 | |
| 11 | 3k | 4 | 16 | 4 | 3 | FALSE | TRUE | 0 | FALSE | 0.666 | 73.031 | |
| 12 | 3l | 2 | >64 | 2 | 1 | FALSE | TRUE | 0 | TRUE | 3.402 | 42.921 | |
| 13 | 3m | 2 | >64 | 2 | 3 | FALSE | TRUE | 0 | TRUE | 2.326 | 94.674 | |
| Aqueous Solubility (AS) | BBB Penetration | Human Intestinal Absorption (HIA) | ||||||||||
| Level | Drug-likeness | Level | Description | Level | Description | |||||||
| 0 | Extremely low | 0 | Very High | 0 | Good absorption | |||||||
| 1 | No, very low, but possible | 1 | High | 1 | Moderate absorption | |||||||
| 2 | Yes, low | 2 | Medium | 2 | Low absorption | |||||||
| 3 | Yes, good | 3 | Low | 3 | Very low absorption | |||||||
| 4 | Yes, optimal | 4 | Undefined | |||||||||
| 5 | No, too soluble | |||||||||||
| Compounds | MIC (µg/mL) | Toxicity Parameters * | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Index | Code | H37Rv | MDR-MTB | AM a | SI b | OI c | AB d | DTP e | CMR f | CFR | CLM | CFM |
| 1 | 3a | 64 | >128 | 0.997 | 0.016 | 0 | 0 | 0.026 | 0 | 0 | 0.988 | 1 |
| 2 | 3b | >64 | No activity | 1 | 0 | 0 | 0.001 | 0 | 0 | 0 | 0 | 1 |
| 3 | 3c | 8 | >32 | 1 | 0 | 0 | 0 | 0.002 | 0 | 0 | 0.007 | 1 |
| 4 | 3d | >64 | No activity | 1 | 0.219 | 0 | 0 | 0.997 | 0 | 0 | 1 | 1 |
| 5 | 3e | 128 | No activity | 1 | 0 | 0 | 0.003 | 0.001 | 0 | 0 | 0.27 | 1 |
| 6 | 3f | >128 | No activity | 0.998 | 0 | 0 | 0 | 0.004 | 0 | 0 | 0.978 | 1 |
| 7 | 3g | 64 | No activity | 1 | 0.061 | 0 | 0 | 0.275 | 0 | 0 | 0.956 | 1 |
| 8 | 3h | >128 | No activity | 1 | 0 | 0 | 0.969 | 0 | 0 | 0 | 0 | 1 |
| 9 | 3i | >64 | No activity | 1 | 0 | 0 | 0 | 0.002 | 0 | 0 | 1 | 1 |
| 10 | 3j | >64 | No activity | 0.078 | 0 | 0 | 0 | 0.101 | 0 | 0 | 1 | 1 |
| 11 | 3k | 4 | 16 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 |
| 12 | 3l | 2 | >64 | 0.956 | 0 | 0 | 0 | 0.154 | 0 | 0 | 1 | 1 |
| 13 | 3m | 2 | >64 | 1 | 0 | 0 | 0 | 0.056 | 0 | 0 | 1 | 1 |
| Probability Values | Probability Level | Description | ||||||||||
| 0.0 to 0.30 | Low probability | Such a chemical is not likely to produce a positive response in an experimental assay | ||||||||||
| >0.30 but <0.70 | Intermediate probability | |||||||||||
| >0.70 | High probability | Likely to produce a positive response in an experimental assay | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venugopala, K.N.; Al-Shar’i, N.A.; Dahabiyeh, L.A.; Hourani, W.; Deb, P.K.; Pillay, M.; Abu-Irmaileh, B.; Bustanji, Y.; Chandrashekharappa, S.; Tratrat, C.; et al. Antitubercular, Cytotoxicity, and Computational Target Validation of Dihydroquinazolinone Derivatives. Antibiotics 2022, 11, 831. https://doi.org/10.3390/antibiotics11070831
Venugopala KN, Al-Shar’i NA, Dahabiyeh LA, Hourani W, Deb PK, Pillay M, Abu-Irmaileh B, Bustanji Y, Chandrashekharappa S, Tratrat C, et al. Antitubercular, Cytotoxicity, and Computational Target Validation of Dihydroquinazolinone Derivatives. Antibiotics. 2022; 11(7):831. https://doi.org/10.3390/antibiotics11070831
Chicago/Turabian StyleVenugopala, Katharigatta N., Nizar A. Al-Shar’i, Lina A. Dahabiyeh, Wafa Hourani, Pran Kishore Deb, Melendhran Pillay, Bashaer Abu-Irmaileh, Yasser Bustanji, Sandeep Chandrashekharappa, Christophe Tratrat, and et al. 2022. "Antitubercular, Cytotoxicity, and Computational Target Validation of Dihydroquinazolinone Derivatives" Antibiotics 11, no. 7: 831. https://doi.org/10.3390/antibiotics11070831
APA StyleVenugopala, K. N., Al-Shar’i, N. A., Dahabiyeh, L. A., Hourani, W., Deb, P. K., Pillay, M., Abu-Irmaileh, B., Bustanji, Y., Chandrashekharappa, S., Tratrat, C., Attimarad, M., Nair, A. B., Sreeharsha, N., Shinu, P., Haroun, M., Kandeel, M., Balgoname, A. A., Venugopala, R., & Morsy, M. A. (2022). Antitubercular, Cytotoxicity, and Computational Target Validation of Dihydroquinazolinone Derivatives. Antibiotics, 11(7), 831. https://doi.org/10.3390/antibiotics11070831