
Predicting Antibiotic Effect of Vancomycin Using Pharmacokinetic/Pharmacodynamic Modeling and Simulation: Dense Sampling versus Sparse Sampling


Abstract
:1. Introduction
2. Materials and Methods
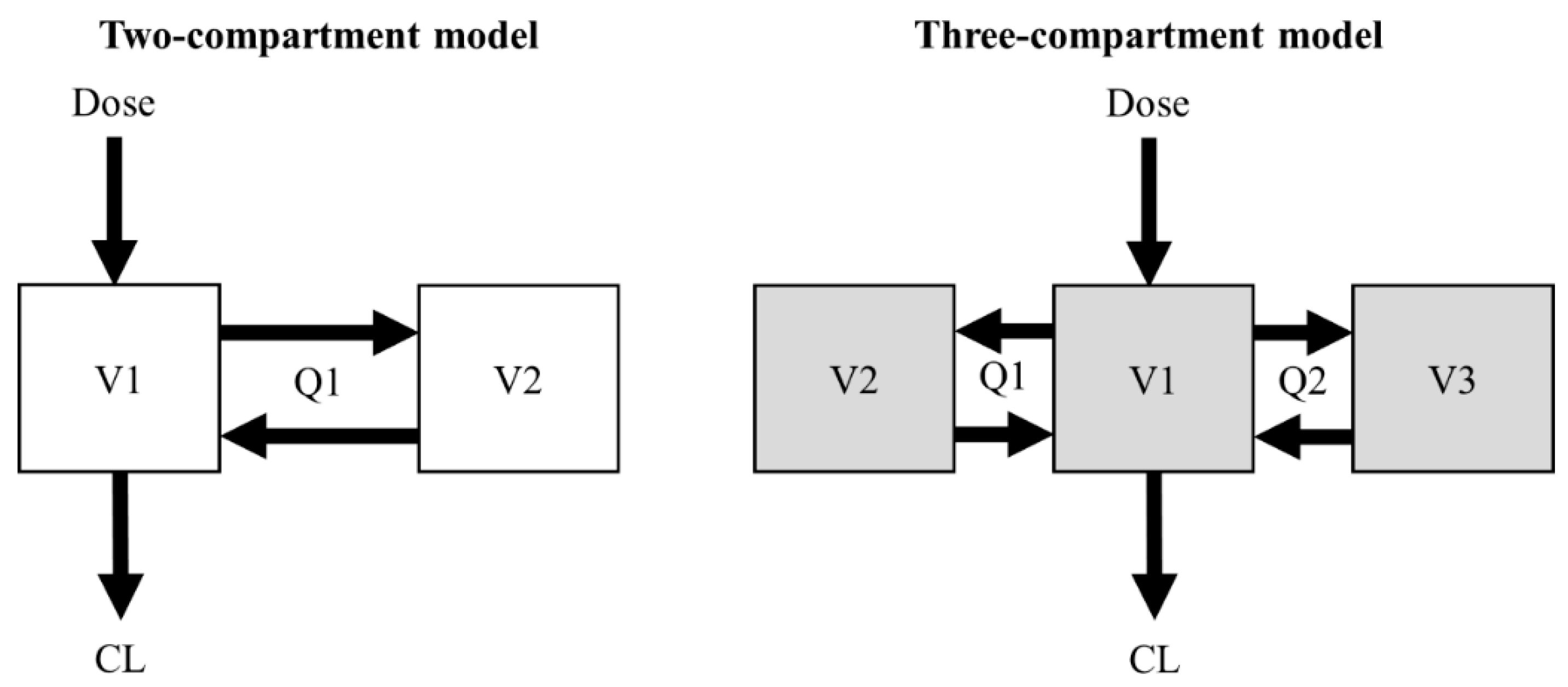
2.1. Simulation of the Concentration–Time Profiles
2.2. Estimation of PK Parameters
2.3. Evaluation of the PK/PD Index of Vancomycin
3. Results
3.1. Estimation of PK Parameters
3.2. Bias and Precision of PK Parameter Estimates
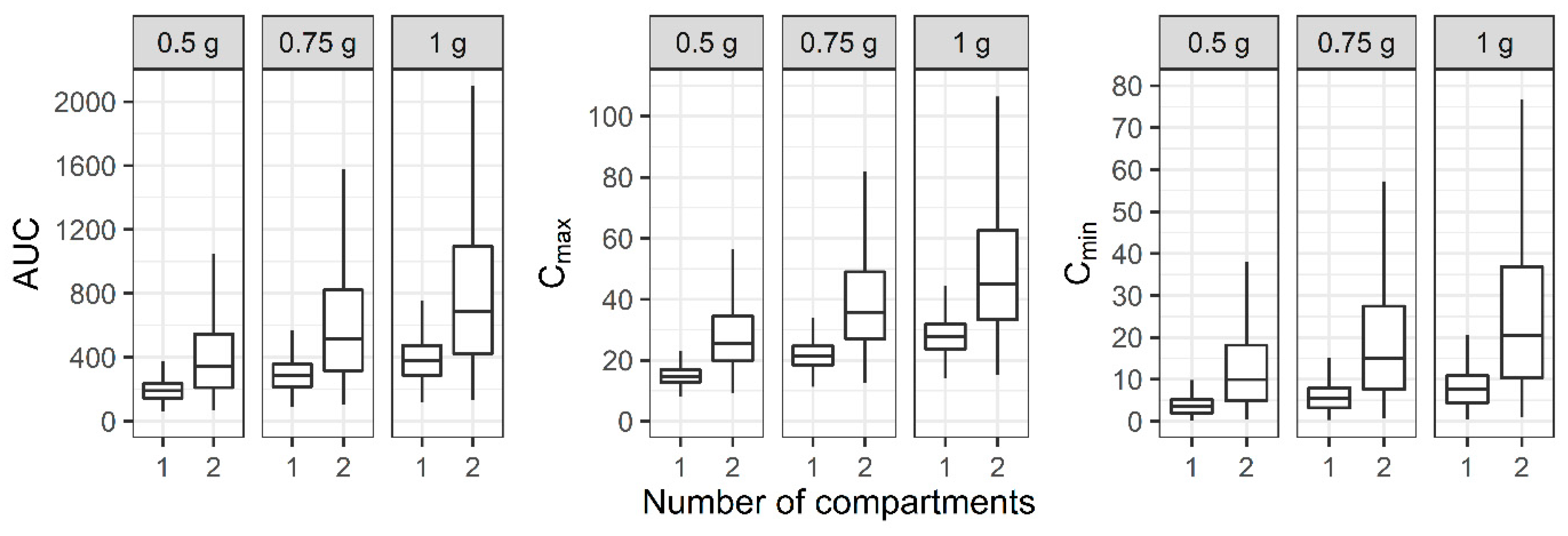
3.3. Evaluation of the PK/PD Index of Vancomycin
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yim, G.; Thaker, M.N.; Koteva, K.; Wright, G. Glycopeptide antibiotic biosynthesis. J. Antibiot. 2014, 67, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruniera, F.R.; Ferreira, F.M.; Saviolli, L.R.M.; Bacci, M.R.; Feder, D.; Pedreira, M.D.G.; Peterlini, M.A.S.; Azzalis, L.A.; Junqueira, V.B.C.; Fonseca, F.L.A. The use of vancomycin with its therapeutic and adverse effects: A review. Eur. Rev. Med. Pharmacol. 2015, 19, 694–700. [Google Scholar]
- van Hal, S.J.; Paterson, D.L.; Lodise, T.P. Systematic review and meta-analysis of vancomycin-induced nephrotoxicity associated with dosing schedules that maintain troughs between 15 and 20 milligrams per liter. Antimicrob. Agents Chemother. 2013, 57, 734–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyamlani, G.; Potukuchi, P.K.; Thomas, F.; Akbilgic, O.; Soohoo, M.; Streja, E.; Naseer, A.; Sumida, K.; Molnar, M.Z.; Kalantar-Zadeh, K.; et al. Vancomycin-associated acute kidney injury in a large veteran population. Am. J. Nephrol. 2019, 49, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Girgis, S.; Pai, S.M.; Girgis, I.G.; Batra, V.K. Pharmacodynamic parameter estimation: Population size versus number of samples. AAPS J. 2005, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C.; et al. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American society of health-system pharmacists, the infectious diseases society of america, the pediatric infectious diseases society, and the society of infectious diseases pharmacists. Am. J. Health Syst. Pharm. 2020, 77, 835–864. [Google Scholar] [CrossRef] [Green Version]
- Colin, P.J.; Allegaert, K.; Thomson, A.H.; Touw, D.J.; Dolton, M.; de Hoog, M.; Roberts, J.A.; Adane, E.D.; Yamamoto, M.; Santos-Buelga, D.; et al. Vancomycin pharmacokinetics throughout life: Results from a pooled population analysis and evaluation of current dosing recommendations. Clin. Pharmacokinet. 2019, 58, 767–780. [Google Scholar] [CrossRef] [Green Version]
- Aljutayli, A.; Marsot, A.; Nekka, F. An update on population pharmacokinetic analyses of vancomycin, part I: In adults. Clin. Pharmacokinet. 2020, 59, 671–698. [Google Scholar] [CrossRef]
- Box, G.E.P.; Draper, N.R. Empirical Model-Building and Response Surfaces; Wiley: New York, NY, USA, 1987; p. 669. [Google Scholar]
- Guo, T.; van Hest, R.M.; Roggeveen, L.F.; Fleuren, L.M.; Thoral, P.J.; Bosman, R.J.; van der Voort, P.H.J.; Girbes, A.R.J.; Mathot, R.A.A.; Elbers, P.W.G. External evaluation of population pharmacokinetic models of vancomycin in large cohorts of intensive care unit patients. Antimicrob. Agents Chemother. 2019, 63, e02543-18. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Kim, D.J.; Zang, D.Y.; Lee, D.H. Effect of pharmacokinetic model misspecification on antibiotic probability of target attainment predicted by Monte Carlo simulation. Int. J. Clin. Pharmacol. Ther. 2019, 57, 362–374. [Google Scholar] [CrossRef]
- Moore, J.N.; Healy, J.R.; Thoma, B.N.; Peahota, M.M.; Ahamadi, M.; Schmidt, L.; Cavarocchi, N.C.; Kraft, W.K. A population pharmacokinetic model for vancomycin in adult patients receiving extracorporeal membrane oxygenation therapy. CPT Pharmacometrics Syst. Pharmacol. 2016, 5, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Lee, D.H.; Kim, H.S. Prospective cohort study of population pharmacokinetics and pharmacodynamic target attainment of vancomycin in adults on extracorporeal membrane oxygenation. Antimicrob. Agents Chemother. 2021, 65, e02408-20. [Google Scholar] [CrossRef] [PubMed]
- Plan, E.L.; Maloney, A.; Mentre, F.; Karlsson, M.O.; Bertrand, J. Performance comparison of various maximum likelihood nonlinear mixed-effects estimation methods for dose-response models. AAPS J. 2012, 14, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, C.; Staatz, C.E.; Unwin, S.; Hennig, S. Assessing predictive performance of published population pharmacokinetic models of intravenous tobramycin in pediatric patients. Antimicrob. Agents Chemother. 2016, 60, 3407–3414. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Lipman, J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit. Care Med. 2009, 37, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, A.J.; Mohd Sazlly Lim, S.; Lipman, J.; Roberts, J.A. A personalised approach to antibiotic pharmacokinetics and pharmacodynamics in critically ill patients. Anaesth. Crit. Care Pain Med. 2021, 40, 100970. [Google Scholar] [CrossRef]
- Rybak, M.; Lomaestro, B.; Rotschafer, J.C.; Moellering, R., Jr.; Craig, W.; Billeter, M.; Dalovisio, J.R.; Levine, D.P. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American society of health-system pharmacists, the infectious diseases society of America, and the Society of infectious diseases pharmacists. Am. J. Health Syst. Pharm. 2009, 66, 82–98. [Google Scholar] [CrossRef]
- Darwich, A.S.; Ogungbenro, K.; Vinks, A.A.; Powell, J.R.; Reny, J.L.; Marsousi, N.; Daali, Y.; Fairman, D.; Cook, J.; Lesko, L.J.; et al. Why has model-informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin. Pharmacol. Ther. 2017, 101, 646–656. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Hodson, R. Precision medicine. Nature 2016, 537, S49. [Google Scholar] [CrossRef] [Green Version]
- Denny, J.C.; Collins, F.S. Precision medicine in 2030-seven ways to transform healthcare. Cell 2021, 184, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, F.; Michelet, R.; Mueller-Schoell, A.; Maier, C.; Klopp-Schulze, L.; van Dyk, M.; Mikus, G.; Huisinga, W.; Kloft, C. Perspectives on model-informed precision dosing in the digital health era: Challenges, opportunities, and recommendations. Clin. Pharmacol. Ther. 2021, 109, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kantasiripitak, W.; Van Daele, R.; Gijsen, M.; Ferrante, M.; Spriet, I.; Dreesen, E. Software tools for model-informed precision dosing: How well do they satisfy the needs? Front. Pharmacol. 2020, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Wicha, S.G.; Martson, A.G.; Nielsen, E.I.; Koch, B.C.P.; Friberg, L.E.; Alffenaar, J.W.; Minichmayr, I.K. From therapeutic drug monitoring to model-informed precision dosing for antibiotics. Clin. Pharmacol. Ther. 2021, 109, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Kim, D.J.; Zang, D.Y.; Lee, D.H. Impact of sampling period on population pharmacokinetic analysis of antibiotics: Why do you take blood samples following the fourth dose? Pharmaceuticals 2020, 13, 249. [Google Scholar] [CrossRef]
- Ogungbenro, K.; Aarons, L. How many subjects are necessary for population pharmacokinetic experiments? Confidence interval approach. Eur. J. Clin. Pharmacol. 2008, 64, 705–713. [Google Scholar] [CrossRef]
- Kang, D.; Schwartz, J.B.; Verotta, D. Sample size computations for PK/PD population models. J. Pharmacokinet. Pharmacodyn. 2005, 32, 685–701. [Google Scholar] [CrossRef]
- Ribbing, J.; Jonsson, E.N. Power, selection bias and predictive performance of the population pharmacokinetic covariate model. J. Pharmacokinet. Pharmacodyn. 2004, 31, 109–134. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Estimate | BSV |
|---|---|---|
| Two-compartment model [12] | ||
| θ1 (L/h) | 2.83 | 77% |
| θ2 | 0.0154 | |
| θ3 (L) | 24.2 | 34% |
| θ4 | 0.00638 | |
| Q1 (L/h) | 11.2 | |
| θ5 (L) | 32.3 | |
| θ6 | 0.0169 | |
| Residual proportional error | 8.19% | |
| Three-compartment model [13] | ||
| θ7 (L/h) | 4.01 | 33.9% |
| θ8 | 0.00752 | |
| V1 (L) | 8.01 | 27.9% |
| Q2 (L/h) | 4.95 | |
| V2 (L) | 15.4 | 34.3% |
| Q3 (L/h) | 9.09 | |
| V3 (L) | 6.21 | 56.9% |
| Residual proportional error | 6.64% |
| Number of Compartments | Subject Number | CL | VSS | |||
|---|---|---|---|---|---|---|
| Simulation | Estimation | RBias (%) | RRMSE (%) | RBias (%) | RRMSE (%) | |
| 2 | 1 | 12 | 90.1 | 92.2 | −28.5 | 28.7 |
| 25 | 92.0 | 93.2 | −28.1 | 28.2 | ||
| 50 | 92.6 | 93.1 | −27.7 | 27.8 | ||
| 100 | 91.4 | 91.7 | −27.9 | 27.9 | ||
| 2 | 12 | −2.30 | 35.1 | 1.87 | 14.9 | |
| 25 | 3.10 | 25.8 | 0.053 | 10.2 | ||
| 50 | 3.02 | 17.7 | 0.661 | 6.93 | ||
| 100 | 1.96 | 13.0 | 0.441 | 5.27 | ||
| 3 | 1 | 12 | 15.6 | 18.3 | −7.94 | 9.31 |
| 25 | 14.0 | 15.4 | −8.59 | 9.17 | ||
| 50 | 14.3 | 15.0 | −8.55 | 8.88 | ||
| 100 | 14.5 | 14.8 | −8.40 | 8.59 | ||
| 2 | 12 | 7.79 | 13.0 | 13.2 | 15.8 | |
| 25 | 6.35 | 9.62 | 13.5 | 14.7 | ||
| 50 | 6.98 | 8.61 | 13.1 | 13.7 | ||
| 100 | 7.27 | 8.03 | 13.3 | 13.6 | ||
| 3 | 12 | 1.81 | 9.66 | 1.44 | 7.75 | |
| 25 | 0.395 | 6.42 | 0.684 | 4.75 | ||
| 50 | 0.715 | 4.64 | 0.327 | 3.51 | ||
| 100 | 0.929 | 3.34 | 0.325 | 2.49 | ||
| Dose | PK/PD Index | 1 Compartment | 2 Compartments |
|---|---|---|---|
| 0.5 g | AUC<400 | 99.9 | 59.0 |
| AUC400–600 | 0.120 | 20.2 | |
| AUC>600 | 0.000 | 20.8 | |
| 0.75 g | AUC<400 | 86.3 | 36.3 |
| AUC400–600 | 13.5 | 22.6 | |
| AUC>600 | 0.120 | 41.1 | |
| 1 g | AUC<400 | 55.2 | 22.6 |
| AUC400–600 | 39.4 | 19.6 | |
| AUC>600 | 5.42 | 57.8 |
| Dose | PK/PD Index | 1 Compartment | 2 Compartments | 3 Compartments |
|---|---|---|---|---|
| 0.5 g | AUC<400 | 100 | 96.5 | 95.5 |
| AUC400–600 | 0.000 | 3.50 | 4.50 | |
| AUC>600 | 0.000 | 0.000 | 0.000 | |
| 0.75 g | AUC<400 | 83.0 | 65.0 | 59.1 |
| AUC400–600 | 17.0 | 31.5 | 36.4 | |
| AUC>600 | 0.000 | 3.51 | 4.54 | |
| 1 g | AUC<400 | 35.6 | 32.7 | 25.8 |
| AUC400–600 | 58.8 | 45.1 | 47.7 | |
| AUC>600 | 5.56 | 22.2 | 26.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.K.; Lee, J.H.; Jang, H.-J.; Zang, D.Y.; Lee, D.-H. Predicting Antibiotic Effect of Vancomycin Using Pharmacokinetic/Pharmacodynamic Modeling and Simulation: Dense Sampling versus Sparse Sampling. Antibiotics 2022, 11, 743. https://doi.org/10.3390/antibiotics11060743
Kim YK, Lee JH, Jang H-J, Zang DY, Lee D-H. Predicting Antibiotic Effect of Vancomycin Using Pharmacokinetic/Pharmacodynamic Modeling and Simulation: Dense Sampling versus Sparse Sampling. Antibiotics. 2022; 11(6):743. https://doi.org/10.3390/antibiotics11060743
Chicago/Turabian StyleKim, Yong Kyun, Jae Ha Lee, Hang-Jea Jang, Dae Young Zang, and Dong-Hwan Lee. 2022. "Predicting Antibiotic Effect of Vancomycin Using Pharmacokinetic/Pharmacodynamic Modeling and Simulation: Dense Sampling versus Sparse Sampling" Antibiotics 11, no. 6: 743. https://doi.org/10.3390/antibiotics11060743
APA StyleKim, Y. K., Lee, J. H., Jang, H.-J., Zang, D. Y., & Lee, D.-H. (2022). Predicting Antibiotic Effect of Vancomycin Using Pharmacokinetic/Pharmacodynamic Modeling and Simulation: Dense Sampling versus Sparse Sampling. Antibiotics, 11(6), 743. https://doi.org/10.3390/antibiotics11060743