
Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment


Abstract
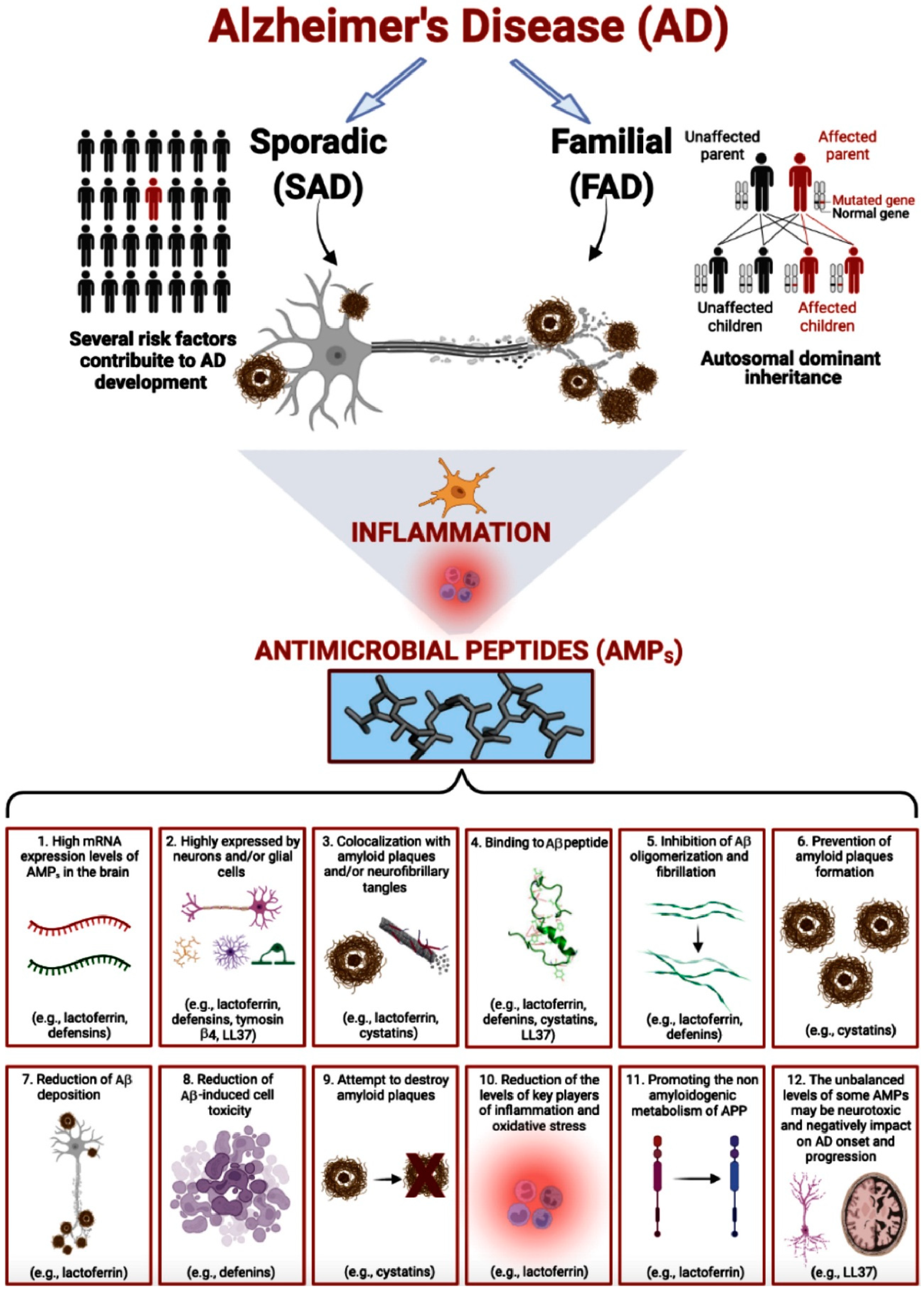
1. Introduction
2. The Infectious Hypothesis of AD
3. AMPs Involvement in AD
3.1. Aβ
3.2. Lactoferrin
3.3. Defensins
3.4. Cystatins
3.5. Thymosin β4
3.6. LL37
3.7. Histatin 1 and Statherin
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- García-Blanco, A.; Baquero, M.; Vento, M.; Gil, E.; Bataller, L.; Cháfer-Pericás, C. Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J. Neurol. Sci. 2017, 373, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Altomari, N.; Bruno, F.; Laganà, V.; Smirne, N.; Colao, R.; Curcio, S.; Bruni, A.C. A Comparison of Behavioral and Psychological Symptoms of Dementia (BPSD) and BPSD Sub-Syndromes in Early-Onset and Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Laganà, V.; Bruno, F.; Altomari, N.; Giulia, B.; Nicoletta, S.; Sabrina, C.; Maria, M.; Rosanna, C.; Gianfranco, P.; Francesca, F.; et al. Neuropsychiatric or Behavioral and Psychological Symptoms of Dementia (BPSD): Focus on prevalence and natural history in Alzheimer’s Disease and Frontotemporal Dementia. Front. Neurol. 2022; in press. [Google Scholar]
- Abondio, P.; Sarno, S.; Giuliani, C.; Laganà, V.; Maletta, R.; Bernardi, L.; Bruni, A. Amyloid Precursor Protein A713T Mutation in Calabrian Patients with Alzheimer’s Disease: A Population Genomics Approach to Estimate Inheritance from a Common Ancestor. Biomedicines 2021, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Mattsson, N. Understanding the cause of sporadic Alzheimer’s disease. Expert Rev. Neurother. 2014, 14, 621–630. [Google Scholar] [CrossRef]
- Thompson, P.M.; Vinters, H.V. Pathologic lesions in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 2012, 107, 1–40. [Google Scholar]
- Padurariu, M.; Ciobica, A.; Mavroudis, I.; Fotiou, D.; Baloyannis, S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr. Danub. 2012, 24, 152–158. [Google Scholar]
- Skaper, S.D. Alzheimer’s disease and amyloid: Culprit or coincidence? Int. Rev. Neurobiol. 2012, 102, 277–316. [Google Scholar]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Zilkova, M.; Koson, P.; Zilka, N. The hunt for dying neurons: Insight into the neuronal loss in Alzheimer’s disease. Bratisl. Lek. Listy 2006, 107, 366–373. [Google Scholar]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Gong, N.J.; Dibb, R.; Bulk, M.; van der Weerd, L.; Liu, C. Imaging beta amyloid aggregation and iron accumulation in Alzheimer’s disease using quantitative susceptibility mapping MRI. Neuroimage 2019, 191, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Hondius, D.C.; Koopmans, F.; Leistner, C.; Pita-Illobre, D.; Peferoen-Baert, R.M.; Marbus, F.; Paliukhovich, I.; Li, K.W.; Rozemuller, A.J.; Hoozemans, J.J. The proteome of granulovacuolar degeneration and neurofibrillary tangles in Alzheimer’s disease. Acta Neuropathol. 2021, 141, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Komaroff, A.L. Can infections cause Alzheimer disease? JAMA 2020, 324, 239–240. [Google Scholar] [CrossRef]
- Seaks, C.E.; Wilcock, D.M. Infectious hypothesis of Alzheimer disease. PLoS Pathog. 2020, 16, e1008596. [Google Scholar] [CrossRef]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. Correction: The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2022, 51, 792. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Boparai, J.K.; Sharma, P.K. Mini Review on Antimicrobial Peptides, Sources, Mechanism and Recent Applications. Protein Pept. Lett. 2020, 27, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Georgountzou, A.; Papadopoulos, N.G. Postnatal Innate Immune Development: From Birth to Adulthood. Front. Immunol. 2017, 8, 957. [Google Scholar] [CrossRef] [PubMed]
- Lupetti, A.; Welling, M.M.; Pauwels, E.K.; Nibbering, P.H. Radiolabelled antimicrobial peptides for infection detection. Lancet Infect. Dis. 2003, 3, 223–229. [Google Scholar] [CrossRef]
- Welling, M.M.; Nabuurs, R.J.; van der Weerd, L. Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 51–57. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Karimi, A.; Fallah, F.; Akhavan, M.M. Overview of ribosomal and non-ribosomal antimicrobial peptides produced by Gram positive bacteria. Cell. Mol. Biol. 2017, 63, 20–32. [Google Scholar] [CrossRef]
- Papagianni, M. Ribosomally synthesized peptides with antimicrobial properties: Biosynthesis, structure, function, and applications. Biotechnol. Adv. 2003, 21, 465–499. [Google Scholar] [CrossRef]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial peptides: Interaction with model and biological membranes and synergism with chemical antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Cudic, M.; Otvos, L., Jr. Intracellular targets of antibacterial peptides. Curr. Drug Targets 2002, 3, 101–106. [Google Scholar] [CrossRef]
- Krizsan, A.; Volke, D.; Weinert, S.; Sträter, N.; Knappe, D.; Hoffmann, R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew. Chem. Int. Ed. Engl. 2014, 53, 12236–12239. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Tomasinsig, L.; Skerlavaj, B.; Papo, N.; Giabbai, B.; Shai, Y.; Zanetti, M. Mechanistic and functional studies of the interaction of a proline-rich antimicrobial peptide with mammalian cells. J. Biol. Chem. 2006, 281, 383–391. [Google Scholar] [CrossRef]
- Blondelle, S.E.; Jerala, R.; Lamata, M.; Moriyon, I.; Brandenburg, K.; Andra, J.; Porro, M.; Lohner, K. Structure-function studies of antimicrobial and endotoxin neutralizing peptides. In Peptides-American Symposium; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; Volume 18. [Google Scholar]
- McPhee, J.B.; Scott, M.G.; Hancock, R.E. Design of host defence peptides for antimicrobial and immunity enhancing activities. Comb. Chem. High Throughput Screen. 2005, 8, 257–272. [Google Scholar] [CrossRef]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M. The role and potential application of antimicrobial peptides in autoimmune diseases. Front. Immunol. 2020, 11, 859. [Google Scholar] [CrossRef]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Carro, E.; Bartolomé, F.; Bermejo-Pareja, F.; Villarejo-Galende, A.; Molina, J.A.; Ortiz, P.; Orive, G. Early diagnosis of mild cognitive impairment and Alzheimer’s disease based on salivary lactoferrin. Alzheimer’sDement. Diagn. Assess. Dis. Monit. 2017, 8, 131–138. [Google Scholar] [CrossRef]
- Lee, E.Y.; Srinivasan, Y.; De Anda, J.; Nicastro, L.K.; Tükel, Ç.; Wong, G.C. Functional reciprocity of amyloids and antimicrobial peptides: Rethinking the role of supramolecular assembly in host defense, immune activation, and inflammation. Front Immun. 2020, 11, 1629. [Google Scholar] [CrossRef] [PubMed]
- Contini, C.; Olianas, A.; Serrao, S.; Deriu, C.; Iavarone, F.; Boroumand, M.; Bizzarro, A.; Lauria, A.; Faa, G.; Castagnola, M.; et al. Top-down proteomics of human saliva highlights anti-inflammatory, antioxidant, and antimicrobial defense responses in alzheimer disease. Front. Neurosci. 2021, 15, 478. [Google Scholar]
- Williams, W.M.; Torres, S.; Siedlak, S.L.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. Antimicrobial peptide β-defensin-1 expression is upregulated in Alzheimer’s brain. J. Neuroinflamm. 2013, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Tang, Y.; Zhang, D.; He, H.; Wu, J.; Zheng, J. Antimicrobial α-defensins as multi-target inhibitors against amyloid formation and microbial infection. Chem. Sci. 2021, 12, 9124–9139. [Google Scholar] [CrossRef]
- Allnutt, M.A.; Jacobson, S. Do herpesviruses play a role in Alzheimer’s disease pathogenesis? Drug Discov. Today Dis. Models 2020, 32, 21–26. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Wilcock, G.K.; Craske, J.; Itzhaki, R.F. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 1991, 33, 224–227. [Google Scholar] [CrossRef]
- Vigasova, D.; Nemergut, M.; Liskova, B.; Damborsky, J. Multi-pathogen infections and Alzheimer’s disease. Microb. Cell Factories 2021, 20, 25. [Google Scholar] [CrossRef]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Does antiherpetic antiviral therapy reduce the risk of dementia? Nat. Rev. Neurol. 2021, 18, 63–64. [Google Scholar] [CrossRef]
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Chiang, C.P.; Yeh, C.B.; Huang, S.Y.; Chien, W.C. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections—a nationwide, population-based cohort study in Taiwan. Neurother. 2018, 15, 417–429. [Google Scholar] [CrossRef]
- Tharp, W.G.; Sarkar, I.N. Origins of amyloid-beta. BMC Genom. 2013, 14, 290. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Fahrenholz, F. The non-amyloidogenic pathway: Structure and function of α-secretases. Alzheimer’s Dis. 2005, 38, 105–127. [Google Scholar]
- Palladino, G.; Nicolia, V.; Kovacs, G.G.; Canterini, S.; Ciraci, V.; Fuso, A.; Mangia, F.; Scarpa, S.; Fiorenza, M.T. Sexually dimorphic expression of reelin in the brain of a mouse model of Alzheimer disease. J. Mol. Neurosci. 2017, 61, 359–367. [Google Scholar] [CrossRef]
- Maler, J.M.; Klafki, H.W.; Paul, S.; Spitzer, P.; Groemer, T.W.; Henkel, A.W.; Wiltfang, J. Urea-based two-dimensional electrophoresis of beta-amyloid peptides in human plasma: Evidence for novel Abeta species. Proteomics 2007, 7, 3815–3820. [Google Scholar] [CrossRef]
- Guntert, A.; Dobeli, H.; Bohrmann, B. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neurosci. 2006, 143, 461–475. [Google Scholar] [CrossRef]
- Portelius, E.; Brinkmalm, G.; Tran, A.J.; Zetterberg, H.; Westman-Brinkmalm, A.; Blennow, K. Identification of novel APP/Abeta isoforms in human cerebrospinal fluid. Neurodegener. Dis. 2009, 6, 87–94. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Smirnov, A.; Otto, M.; Paul, S.; Kornhuber, J. Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1–37/38/39 in addition to 1–40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J. Neurochem. 2002, 81, 481–496. [Google Scholar] [CrossRef]
- Little, C.S.; Joyce, T.A.; Hammond, C.J.; Matta, H.; Cahn, D.; Appelt, D.M.; Balin, B.J. Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 2014, 6, 304. [Google Scholar] [CrossRef] [PubMed]
- Schluesener, H.J.; Su, Y.; Ebrahimi, A.; Pouladsaz, D. Antimicrobial peptides in the brain: Neuropeptides and amyloid. Front. Biosci. (Schol. Ed.) 2012, 4, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Kagan, B.L.; Jang, H.; Capone, R.; Arce, F.T.; Ramachandran, S.; Lal, R.; Nussinov, R. Antimicrobial properties of amyloid peptides. Mol. Pharm. 2012, 9, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Bourgade, K.; Dupuis, G.; Frost, E.H.; Fülöp, T., Jr. Anti-viral properties of amyloid-β peptides. J. Alzheimer’s Dis. 2016, 54, 859–878. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.R.; Bishop, G.M. Aβ as a bioflocculant: Implications for the amyloid hypothesis of Alzheimer’s disease. Neurobiol. Aging 2002, 23, 1051–1072. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R.; Liu, Q.; Perry, G.; Atwood, C.S.; Smith, M.A. Iron: A pathological mediator of Alzheimer disease? Dev. Neurosci. 2002, 24, 184–187. [Google Scholar] [CrossRef]
- Dominguez, D.; Tournoy, J.; Hartmann, D.; Huth, T.; Cryns, K.; Deforce, S.; Serneels, L.; Camacho, I.E.; Marjaux, E.; Craessaerts, K.; et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J. Biol. Chem. 2005, 280, 30797–30806. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Tarenflurbil Phase 3 Study Group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef]
- Spitzer, P.; Condic, M.; Herrmann, M.; Oberstein, T.J.; Scharin-Mehlmann, M.; Gilbert, D.F.; Maler, J.M. Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 2016, 6, 32228. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Zetterberg, H. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Toombs, J.; Zetterberg, H. In the blood: Biomarkers for amyloid pathology and neurodegeneration in Alzheimer’s disease. Brain Commun. 2020, 2, fcaa054. [Google Scholar] [CrossRef] [PubMed]
- Gleerup, H.S.; Jensen, C.S.; Høgh, P.; Hasselbalch, S.G.; Simonsen, A.H. Cerebrospinal fluid and saliva lactoferrin as a diagnostic biomarker for Alzheimer’s disease in a mixed memory clinic population. Alzheimer’s Dement. 2021, 17, e05214. [Google Scholar] [CrossRef]
- Sorensen, M.; Sorensen, S.P.L. The proteins in whey. Compte Rendu Des Trav. Lab. Carlsberg Ser. Chim. 1940, 23, 55–99. [Google Scholar]
- Groves, M.L. The Isolation of a Red Protein from Milk2. J. Am. Chem. Soc. 1960, 82, 3345–3350. [Google Scholar] [CrossRef]
- Johanson, B. Isolation of an Iron containing red protein from Human milk. Acta Chem. Scand. 1960, 14, 510–512. [Google Scholar] [CrossRef]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Scotti, M.J.; Conte, M.P.; Paesano, R.; Valenti, P. Physico-chemical properties influence the functions and efficacy of commercial bovine lactoferrins. Biometals 2018, 31, 301–312. [Google Scholar] [CrossRef]
- Wang, B.; Timilsena, Y.P.; Blanch, E.; Adhikari, B. Lactoferrin: Structure, function, denaturation and digestion. Crit. Rev. Food Sci. Nutr. 2019, 59, 580–596. [Google Scholar] [CrossRef]
- Kawamata, T.; Tooyama, I.; Yamada, T.; Walker, D.G.; McGeer, P.L. Lactotransferrin immunocytochemistry in Alzheimer and normal human brain. Am. J. Pathol. 1993, 142, 1574–1585. [Google Scholar]
- Orsi, N. The antimicrobial activity of lactoferrin: Current status and perspectives. Biometals 2004, 17, 189–196. [Google Scholar] [CrossRef]
- van der Strate, B.W.; Beljaars, L.; Molema, G.; Harmsen, M.C.; Meijer, D.K. Antiviral activities of lactoferrin. Antivir. Res. 2001, 52, 225–239. [Google Scholar] [CrossRef]
- Beljaars, L.; van der Strate, B.W.; Bakker, H.I.; Rekersmit, C.; Vanloenenweemaes, A.; Wiegmans, F.; Harmsen, M.; Molema, G.; Meijer, D. Inhibition of cytomegalovirus infection by lactoferrin in vitro and in vivo. Antivir. Res. 2004, 63, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.L.; Hunter, H.N.; Vogel, H.J. Lactoferricin: A lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell. Mol. Life Sci. 2005, 62, 2588–2598. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.M.; Fagan, A.M.; Williams, M.M.; Ghoshal, N.; Aeschleman, M.; Grant, E.A.; Marcus, D.S.; Mintun, M.A.; Holtzman, D.M.; Morris, J.C. Improving CSF biomarker accuracy in predicting prevalent and incident Alzheimer disease. Neurology 2011, 76, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ohtsuki, S.; Kamiie, J.; Nezu, Y.; Terasaki, T. Cerebral clearance of human amyloid-beta peptide (1–40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J. Neurochem. 2007, 103, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, S.; Pietrzik, C.U. Functional role of lipoprotein receptors in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 15–25. [Google Scholar]
- Osmand, A.P.; Switzer, R.C. III. Differential distribution of lactoferrin and Alz-50 immunoreactivities in neuritic plaques and neurofibrillary tangles in Alzheimer’s disease. Alzheimer’s Disease: Basic Mechanisms. In Diagnosis and Therapeutic Strategies; Lqbal, K., McLachlan, D.R.C., Winblad, B., Wisniewski, H.M., Eds.; Wiley: Chichester, UK, 1991; pp. 219–228. [Google Scholar]
- An, L.; Sato, H.; Konishi, Y.; Walker, D.G.; Beach, T.G.; Rogers, J.; Tooyama, I. Expression and localization of lactotransferrin messenger RNA in the cortex of Alzheimer’s disease. Neurosci. Lett. 2009, 452, 277–280. [Google Scholar] [CrossRef]
- Guo, C.; Yang, Z.H.; Zhang, S.; Chai, R.; Xue, H.; Zhang, Y.H.; Li, J.Y.; Wang, Z.Y. Intranasal lactoferrin enhances α-secretase-dependent amyloid precursor protein processing via the ERK1/2-CREB and HIF-1α pathways in an Alzheimer’s disease mouse model. Neuropsychopharmacology 2017, 42, 2504–2515. [Google Scholar] [CrossRef]
- Mohamed, W.A.; Salama, R.M.; Schaalan, M.F. A pilot study on the effect of lactoferrin on Alzheimer’s disease pathological sequelae: Impact of the p-Akt/PTEN pathway. Biomed. Pharmacother. 2019, 111, 714–723. [Google Scholar] [CrossRef]
- González-Sánchez, M.; Bartolome, F.; Antequera, D.; Puertas-Martín, V.; González, P.; Gómez-Grande, A.; Llamas-Velasco, S.; Herrero-San Martín, A.; Pérez-Martínez, D.; Villarejo-Galende, A.; et al. Decreased salivary lactoferrin levels are specific to Alzheimer’s disease. EBioMedicine 2020, 57, 102834. [Google Scholar] [CrossRef]
- Kazakos, E.I.; Kountouras, J.; Polyzos, S.A.; Deretzi, G. Novel aspects of defensins’ involvement in virus-induced autoimmunity in the central nervous system. Med. Hypotheses 2017, 102, 33–36. [Google Scholar] [CrossRef]
- Amerikova, M.; Pencheva El-Tibi, I.; Maslarska, V.; Bozhanov, S.; Tachkov, K. Antimicrobial activity, mechanism of action, and methods for stabilisation of defensins as new therapeutic agents. Biotechnol. Biotechnol. Equip. 2019, 33, 671–682. [Google Scholar] [CrossRef]
- Watt, A.D.; Perez, K.A.; Ang, C.S.; O’Donnell, P.; Rembach, A.; Pertile, K.K.; Rumble, R.L.; Trounson, B.O.; Fowler, C.J.; Faux, N.G.; et al. Peripheral α-defensins 1 and 2 are elevated in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, M.; Ivitz, E.; Datki, Z.; Kálmán, J.; Pákáski, M.; Várhelyi, Z.P.; Klivényi, P.; Zadori, D.; Somogyvári, F.; Szolnoki, Z.; et al. Relevance of defensin β-2 and α defensins (HNP1–3) in Alzheimer’s disease. Psychiatry Res. 2016, 239, 342–345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shah, A.; Bano, B. Cystatins in health and diseases. Int. J. Pept. Res. Ther. 2009, 15, 43–48. [Google Scholar] [CrossRef]
- Ochieng, J.; Chaudhuri, G. Cystatin superfamily. J. Health Care Poor Underserved 2010, 21 (Suppl. S1), 51. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Rinne, R.; Kirschke, H.; Jarvinen, M.; Knofel, B.; Rinne, A. Cystatin A-like immunoreactivity is widely distributed in human brain and accumulates in neuritic plaques of Alzheimer disease subjects. Brain Res. Bull. 1994, 33, 477–481. [Google Scholar] [CrossRef]
- Ii, K.; Ito, H.; Kominami, E.; Hirano, A. Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer’s disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 185–194. [Google Scholar] [CrossRef]
- Levy, E.; Sastre, M.; Kumar, A.; Gallo, G.; Piccardo, P.; Ghetti, B.; Tagliavini, F. Codeposition of cystatin C with amyloid-beta protein in the brain of Alzheimer disease patients. J. Neuropathol. Exp. Neurol. 2001, 60, 94–104. [Google Scholar] [CrossRef]
- Skerget, K.; Taler-Vercic, A.; Bavdek, A.; Hodnik, V.; Ceru, S.; Tusek-Znidaric, M.; Kumm, T.; Pitsi, D.; Pompe-Novak, M.; Palumaa, P.; et al. Interaction between oligomers of stefin B and amyloid-β in vitro and in cells. J. Biol. Chem. 2010, 285, 3201–3210. [Google Scholar] [CrossRef]
- Sastre, M.; Calero, M.; Pawlik, M.; Mathews, P.M.; Kumar, A.; Danilov, V.; Schmidt, S.D.; Nixon, R.A.; Frangione, B.; Levy, E. Binding of cystatin C to Alzheimer’s amyloid beta inhibits in vitro amyloid fibril formation. Neurobiol. Aging 2004, 25, 1033–1043. [Google Scholar] [CrossRef]
- Magister, Š.; Kos, J. Cystatins in immune system. J. Cancer 2013, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Soond, S.M.; Kozhevnikova, M.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Cysteine cathepsin protease inhibition: An update on its diagnostic, prognostic and therapeutic potential in cancer. Pharmaceuticals 2019, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef]
- Wu, Z.; Ni, J.; Liu, Y.; Teeling, J.L.; Takayama, F.; Collcutt, A.; Ibbett, P.; Nakanishi, H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 2017, 65, 350–361. [Google Scholar] [CrossRef]
- Kaur, G.; Levy, E. Cystatin C in Alzheimer’s disease. Front. Mol. Neurosci. 2012, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Olafsson, I.; Grubb, A. Hereditary cystatin C amyloid angiopathy. Amyloid 2000, 7, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Nagai, A.; Ryu, J.K.; Kobayash, S.; Kim, S.U. Cystatin C induces neuronal cell death in vivo. Ann. N. Y. Acad. Sci. 2002, 977, 315–321. [Google Scholar] [CrossRef]
- Nagai, A.; Ryu, J.K.; Terashima, M.; Tanigawa, Y.; Wakabayashi, K.; McLarnon, J.G.; Kobayashi, S.; Masuda, J.; Kim, S.U. Neuronal cell death induced by cystatin C in vivo and in cultured human CNS neurons is inhibited with cathepsin B. Brain Res. 2005, 1066, 120–128. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, Y.X.; Song, J.W.; Ma, Q.M. Thymosin β4 inhibits microglia activation through microRNA 146a in neonatal rats following hypoxia injury. Neuroreport 2015, 26, 1032–1038. [Google Scholar] [CrossRef]
- Le Pera, M.; Urso, E.; Sprovieri, T.; Bossio, S.; Aguglia, U.; Manna, I.; Cupidi, C.; Ferraro, T.; Gambardella, A.; Qualtieri, A.; et al. Contribution of cerebrospinal fluid thymosin β4 levels to the clinical differentiation of Creutzfeldt-Jakob disease. Arch. Neurol. 2012, 69, 868–872. [Google Scholar] [CrossRef][Green Version]
- Khurshid, Z.; Naseem, M.; Asiri, Y.I.; Mali, M.; Sannam Khan, R.; Sahibzada, H.A.; Zafar, M.S.; Moin, S.F.; Khan, E. Significance and diagnostic role of antimicrobial cathelicidins (LL-37) peptides in oral health. Biomolecules 2017, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.O.; Varoga, D.; Nicolaeva, N.; Leib, S.L.; Wilms, H.; Podschun, R.; Lucius, R. Role of glial cells in the functional expression of LL-37/rat cathelin-related antimicrobial peptide in meningitis. J. Neuropathol. Exp. Neurol. 2008, 67, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Shi, X.; Barron, A.E.; McGeer, E.; McGeer, P.L. Human antimicrobial peptide LL-37 induces glial-mediated neuroinflammation. Biochem. Pharmacol. 2015, 94, 130–141. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzi, E.; Chiari, M.; Colombo, R.; Cretich, M.; Sola, L.; Vanna, R.; Gagni, P.; Bisceglia, F.; Morasso, C.; Lin, J.S.; et al. Evidence that the human innate immune peptide LL-37 may be a binding partner of amyloid-β and inhibitor of fibril assembly. J. Alzheimer’s Dis. 2017, 59, 1213–1226. [Google Scholar] [CrossRef]
- Keikha, M.; Rahdar, H.A.; Karami-Zarandi, M.; Azadi, D. The New Insight for Novel Antimicrobial Peptides Designing by Computational Design and Improvement of an Antimicrobial Peptide Derivate of LL-37. Avicenna J. Clin. Microbiol. Infect. 2019, 6, 15–20. [Google Scholar] [CrossRef]
- Kavanagh, K.; Dowd, S. Histatins: Antimicrobial peptides with therapeutic potential. J. Pharm. Pharmacol. 2004, 56, 285–289. [Google Scholar] [CrossRef]
- Van’t Hof, W.; Veerman, E.C.; Amerongen, A.V.N.; Ligtenberg, A.J. Antimicrobial defense systems in saliva. Saliva Secret. Funct. 2014, 24, 40–51. [Google Scholar]


{kind=link}
| Antimicrobial Peptide | Source | Description | Reference |
|---|---|---|---|
| Aβ1-42 | Saliva | Increased in AD | [73] |
| CFS | Reduced in AD | [71] | |
| Lactoferrin | Saliva | Increased in AD | [41] |
| α-defensin 1 | Saliva | Increased in AD | [43] |
| Blood | Increased in AD | [94] | |
| Serum | Increased in AD | [95] | |
| CFS | Increased in AD | [95] | |
| α-defensin 2 | Saliva | Increased in AD | [43] |
| Blood | Increased in AD | [94] | |
| Serum | Increased in AD | [95] | |
| CFS | Increased in AD | [95] | |
| α-defensin 3 | Saliva | Increased in AD | [43] |
| Serum | Increased in AD | [95] | |
| CFS | Increased in AD | [95] | |
| α-defensin 4 | Saliva | Increased in AD | [43] |
| β-defensin 2 | Serum | Increased in AD | [95] |
| CFS | Increased in AD | [95] | |
| Cystatin A | Saliva | Increased in AD | [43] |
| Cystatin B | Saliva | Increased in AD | [43] |
| Cystatin C | CFS | Decreased in AD | [107] |
| Thymosin β4 | Saliva | Increased in AD | [43] |
| Histatin 1 | Saliva | Increased in AD | [43] |
| Statherin | Saliva | Increased in AD | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, F.; Malvaso, A.; Canterini, S.; Bruni, A.C. Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics 2022, 11, 726. https://doi.org/10.3390/antibiotics11060726
Bruno F, Malvaso A, Canterini S, Bruni AC. Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics. 2022; 11(6):726. https://doi.org/10.3390/antibiotics11060726
Chicago/Turabian StyleBruno, Francesco, Antonio Malvaso, Sonia Canterini, and Amalia Cecilia Bruni. 2022. "Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment" Antibiotics 11, no. 6: 726. https://doi.org/10.3390/antibiotics11060726
APA StyleBruno, F., Malvaso, A., Canterini, S., & Bruni, A. C. (2022). Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics, 11(6), 726. https://doi.org/10.3390/antibiotics11060726