
The Perfect Condition for the Rising of Superbugs: Person-to-Person Contact and Antibiotic Use Are the Key Factors Responsible for the Positive Correlation between Antibiotic Resistance Gene Diversity and Virulence Gene Diversity in Human Metagenomes

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Methods
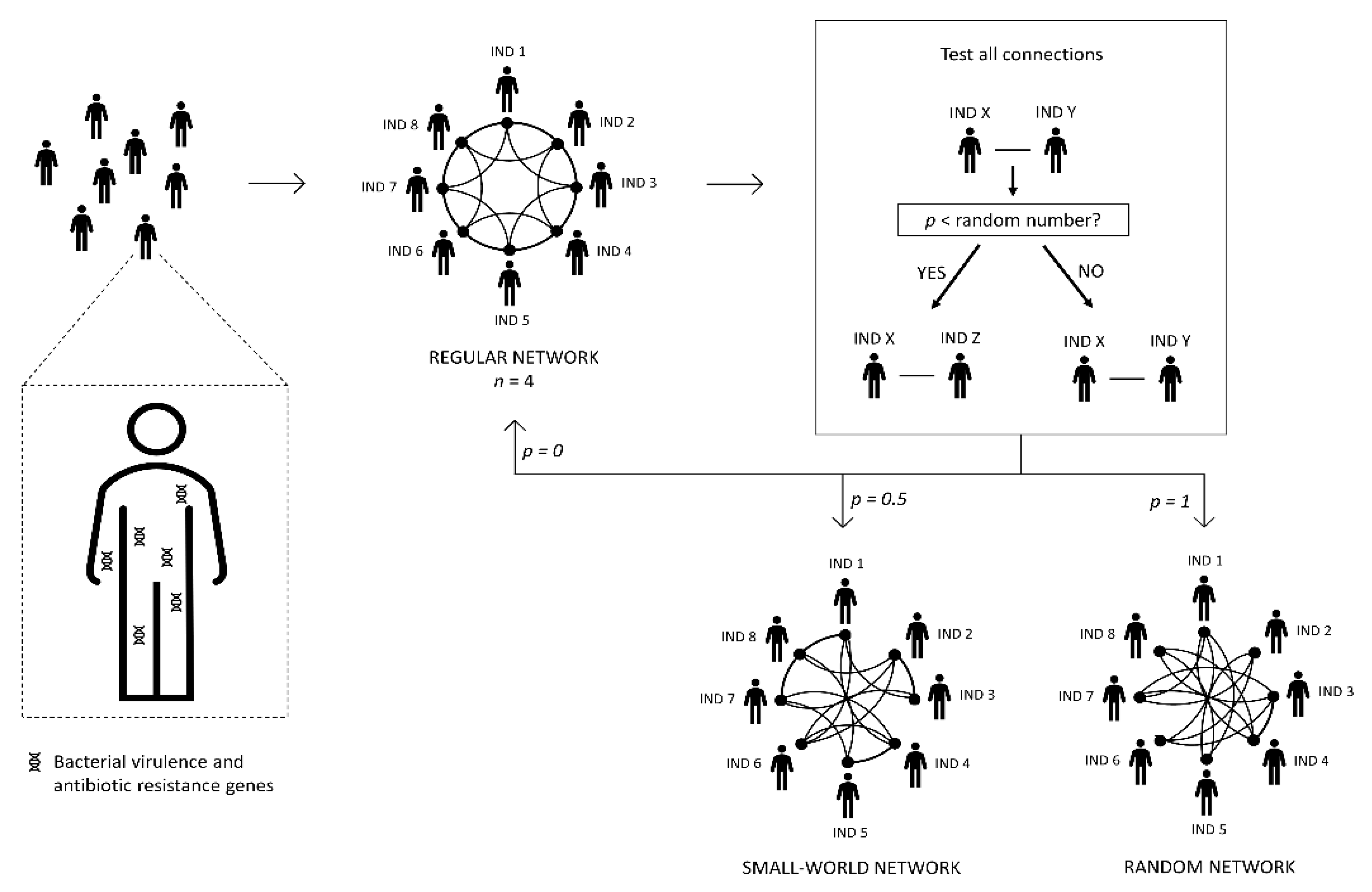
2.1. Building the Human Network
2.2. The Metagenome, Pathogenic Bacteria, and Antibiotic Administration
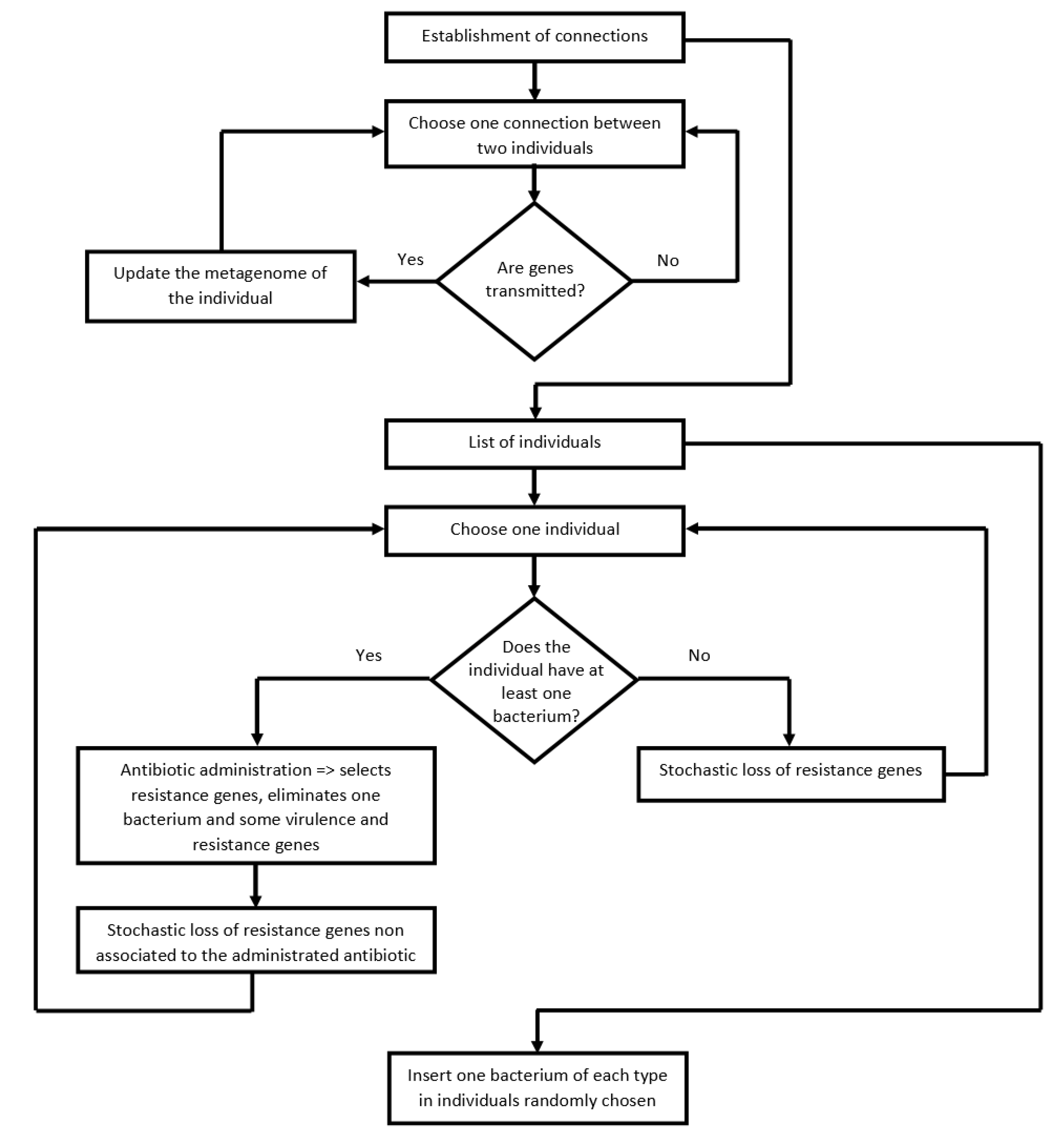
2.3. Algorithm of the Program
- (i)
- Transfer of pathogenic bacteria, virulence, and resistance genes between people (i.e., between linked nodes), according to specific transmission probabilities (Table 2). With this process, the diversity of genes present in the recipient metagenome increases.
- (ii)
- Select people infected by at least one pathogenic bacterium. These people take antibiotics (chosen according to the pathogen). The antibiotic clears the pathogen and selects for resistance genes for the antibiotic used. According to a certain probability (Table 2), the antibiotic also eliminates virulence genes and resistance genes unrelated to the administered antibiotic. Finally, the metagenome loses a few more resistance genes not associated with the antibiotic, according to the loss rate probability (Table 2). The cause of this loss is the fitness cost of resistance genes.
- (iii)
- The metagenomes of people that did not take an antibiotic in this cycle also lose resistance genes according to the loss rate probability (Table 2). As above, this loss is a consequence of the fitness cost imposed by resistance genes on their hosts, which is not happening with virulence genes.
- (iv)
- Add the five bacterial pathogens in five randomly-chosen individuals of the community.
2.4. Statistical analysis
3. Results
3.1. The Number of Diseases and the Probability of Transmission
3.2. Calibration of the Transmission Probability
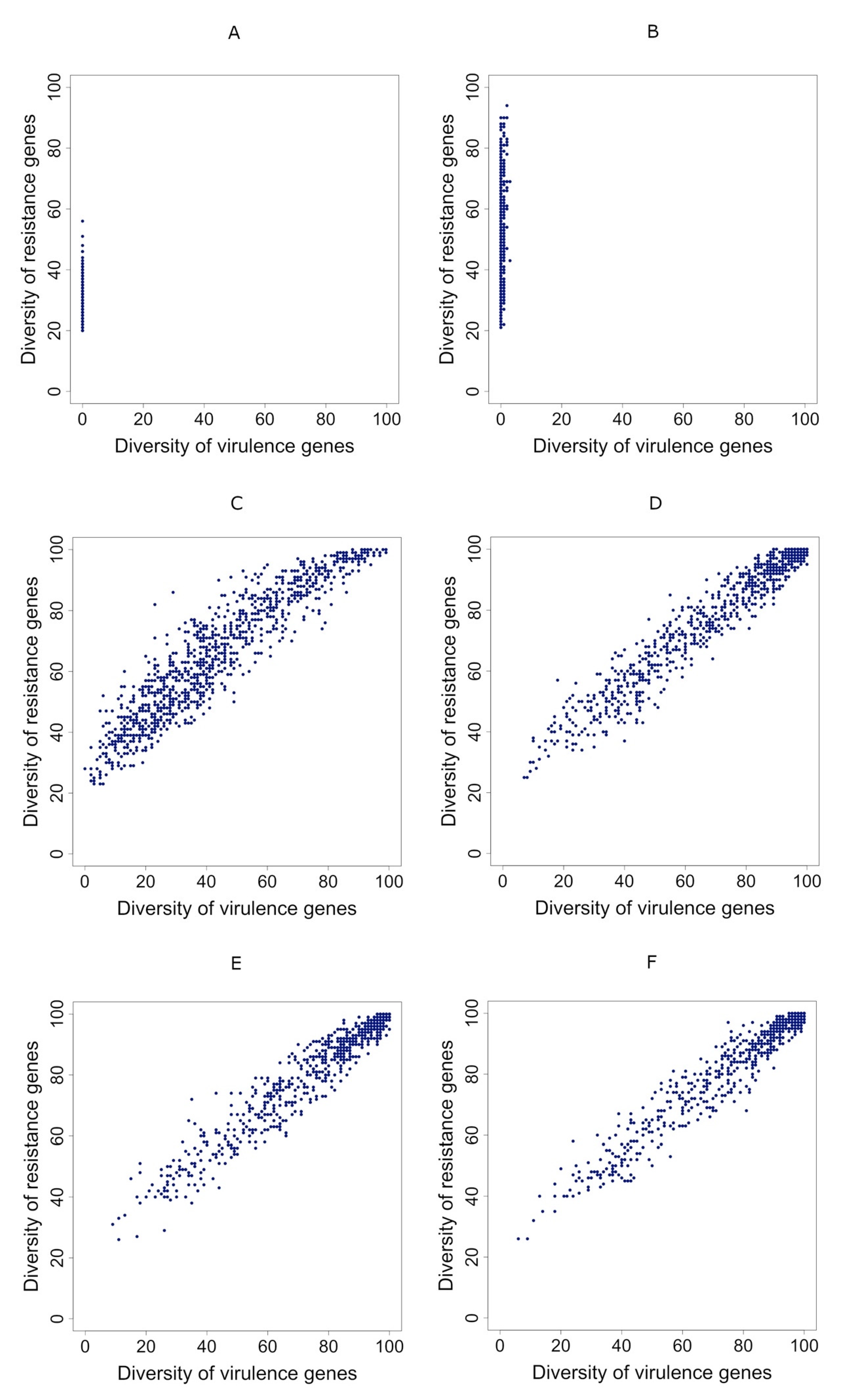
3.3. Correlation between Diversities Is Positive if Gene Transmission Probability Is Higher Than the Resistance Gene Loss Rate
3.4. Correlations Maintain Sign Even when People Take Antibiotics Randomly
3.5. Taking Antibiotics Is Crucial for a Positive Correlation between Virulence and Resistance Genes’ Diversity in Metagenomes
3.6. Positive Correlations Are Robust under Changes in the Main Simulated System’s Properties
3.6.1. Positive Correlations Are Robust under Changes in the Population Size
3.6.2. Positive Correlations Are Robust under Changes in the Ratios between Virulence and Antibiotic Resistance Genes Diversities
3.6.3. Positive Correlations Are Robust under Changes in the Gene Elimination Probability when People Take Antibiotics
3.6.4. Positive Correlations Are Robust under Changes in the Initial Proportion of Metagenomes Containing Antibiotic-Resistance Genes
3.6.5. Positive Correlations Are Robust under Changes in the Network Type
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castanon, J.I.R. History of the Use of Antibiotic as Growth Promoters in European Poultry Feeds. Poult. Sci. 2007, 86, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, S.H. The evolving response to antibiotic resistance (1945–2018). Palgrave Commun. 2018, 4, 124. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Genet. 2014, 12, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- WHO. New Report Calls for Urgent-Action to Avert Antimicrobial Resistance Crisis 2019.
- Alanis, A.J. Resistance to Antibiotics: Are We in the Post-Antibiotic Era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2011, 40, D641–D645. [Google Scholar] [CrossRef]
- Johnson, T.J.; Nolan, L.K. Pathogenomics of the Virulence Plasmids of Escherichia coli. Microbiol. Mol. Biol. Rev. 2009, 73, 750–774. [Google Scholar] [CrossRef]
- Kim, K.S. Strategy ofEscherichia colifor Crossing the Blood-Brain Barrier. J. Infect. Dis. 2002, 186, S220–S224. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Gotoh, Y.; Itoh, T.; Sato, M.P.; Seto, K.; Yoshino, S.; Isobe, J.; Etoh, Y.; Kurogi, M.; Kimata, K.; et al. Population structure of Escherichia coli O26: H11 with recent and repeated stx2 acquisition in multiple lineages. Microb. Genom. 2017, 3. [Google Scholar] [CrossRef]
- Rankin, D.J.; Mc Ginty, S.E.; Nogueira, T.; Touchon, M.; Taddei, F.; Rocha, E.P.C.; Brown, S.P. Bacterial cooperation controlled by mobile elements: Kin selection and infectivity are part of the same process. Heredity 2011, 107, 279–281. [Google Scholar] [CrossRef][Green Version]
- Escudeiro, P.; Pothier, J.; Dionisio, F.; Nogueira, T. Antibiotic Resistance Gene Diversity and Virulence Gene Diversity Are Correlated in Human Gut and Environmental Microbiomes. mSphere 2019, 4, e00135-19. [Google Scholar] [CrossRef] [PubMed]
- Beceiro, A.; Tomás, M.; Bou, G. Antimicrobial Resistance and Virulence: Aa Successful or Deleterious Association in the Bacterial World? Clin. Microbiol. Rev. 2013, 26, 185–230. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, L.; Liu, S.; Wang, Q.; Guo, Z.; Chen, C.; Feng, J. What drives changes in the virulence and antibiotic resistance of Vibrio Harveyi in the South China Sea? J. Fish Dis. 2020, 43, 853–862. [Google Scholar] [CrossRef]
- Pan, Y.; Zeng, J.; Li, L.; Yang, J.; Tang, Z.; Xiong, W.; Li, Y.; Chen, S.; Zeng, Z. Coexistence of Antibiotic Resistance Genes and Virulence Factors Deciphered by Large-Scale Complete Genome Analysis. mSystems 2020, 5, e00821-19. [Google Scholar] [CrossRef]
- Canteón, R. Antibiotic resistance genes from the environment: A perspective through newly identified antibiotic resistance mechanisms in the clinical setting. Clin. Microbiol. Infect. 2009, 15, 20–25. [Google Scholar] [CrossRef]
- Martinez, J.L. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc. R. Soc. B Biol. Sci. 2009, 276, 2521–2530. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The Pervasive Effects of an Antibiotic on the Human Gut Microbiota, as Revealed by Deep 16S rRNA Sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Trindade, S.; Sousa, A.; Xavier, K.B.; Dionisio, F.; Ferreira, M.G.; Gordo, I. Positive Epistasis Drives the Acquisition of Multidrug Resistance. PLoS Genet. 2009, 5, e1000578. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.F.; Mendonça, S.C.M.; Carvalho, L.M.; Reis, A.M.; Gordo, I.; Trindade, S.; Dionisio, F. Pervasive Sign Epistasis between Conjugative Plasmids and Drug-Resistance Chromosomal Mutations. PLoS Genet. 2011, 7, e1002181. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nat. Cell Biol. 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Brito, I.L.; Gurry, T.; Zhao, S.; Huang, K.; Young, S.K.; Shea, T.P.; Naisilisili, W.; Jenkins, A.; Jupiter, S.D.; Gevers, D.; et al. Transmission of human-associated microbiota along family and social networks. Nat. Microbiol. 2019, 4, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Falkow, S. What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 2009, 7, 887–894. [Google Scholar] [CrossRef]
- Nayfach, S.; Rodriguez-Mueller, B.; Garud, N.; Pollard, K.S. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 2016, 26, 1612–1625. [Google Scholar] [CrossRef]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S.; Zolfo, M.; et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 2018, 24, 133–145.e5. [Google Scholar] [CrossRef]
- Yassour, M.; Jason, E.; Hogstrom, L.J.; Arthur, T.D.; Tripathi, S.; Siljander, H.; Selvenius, J.; Oikarinen, S.; Hyöty, H.; Virtanen, S.M.; et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 2018, 24, 146–154.e4. [Google Scholar] [CrossRef]
- Nogueira, T.; David, P.H.C.; Pothier, J. Antibiotics as both friends and foes of the human gut microbiome: The microbial community approach. Drug Dev. Res. 2019, 80, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.P.F.; Rebelo, J.S.; Dionisio, F.; Botelho, A.; Nogueira, T. The Social Distancing Imposed To Contain COVID-19 Can Affect Our Microbiome: A Double-Edged Sword in Human Health. mSphere 2020, 5, e00716-20. [Google Scholar] [CrossRef] [PubMed]
- Durrett, R.; Levin, S.A. Stochastic Spatial Models—A User’s Guide to Ecological Applications. Philos. Trans. R. Soc. B Biol. Sci. 1994, 343, 329–350. [Google Scholar] [CrossRef]
- Keeling, M.J.; Eames, K.T.D. Networks and epidemic models. J. R. Soc. Interface 2005, 2, 295–307. [Google Scholar] [CrossRef]
- Watts, D.J.; Strogatz, S. Collective dynamics of ‘small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.; Barabási, A.-L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47–97. [Google Scholar] [CrossRef]
- Kuperman, M.; Abramson, G. Small World Effect in an Epidemiological Model. Phys. Rev. Lett. 2001, 86, 2909–2912. [Google Scholar] [CrossRef]
- Klemm, K.; Eguíluz, V.M.; Toral, R.; Miguel, M.S. Nonequilibrium transitions in complex networks: A model of social interaction. Phys. Rev. E 2003, 67, 026120. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Martinez, J.L. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ. Pollut. 2009, 157, 2893–2902. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Selection and Transmission of Antibiotic-Resistant Bacteria. Microb. Transm. 2019, 5, 117–137. [Google Scholar] [CrossRef]
- Sarowska, J.; Futoma-Koloch, B.; Jama-Kmiecik, A.; Frej-Madrzak, M.; Ksiazczyk, M.; Bugla-Ploskonska, G.; Choroszy-Krol, I. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: Recent reports. Gut Pathog. 2019, 11, 10. [Google Scholar] [CrossRef]
- Horcajada, J.P.; Vila, J.; Moreno-Martínez, A.; Ruiz, J.; Martínez, J.A.; Sánchez, M.; Soriano, E.; Mensa, J. Molecular epidemiology and evolution of resistance to quinolones in Escherichia coli after prolonged administration of ciprofloxacin in patients with prostatitis. J. Antimicrob. Chemother. 2002, 49, 55–59. [Google Scholar] [CrossRef]
- Lautenbach, E.; Tolomeo, P.; Mao, X.; Fishman, N.O.; Metlay, J.P.; Bilker, W.B.; Nachamkin, I. Duration of Outpatient Fecal Colonization Due to Escherichia coli Isolates with Decreased Susceptibility to Fluoroquinolones: Longitudinal Study of Patients Recently Discharged from the Hospital. Antimicrob. Agents Chemother. 2006, 50, 3939–3943. [Google Scholar] [CrossRef]
- O’Fallon, E.; Gautam, S.; D’Agata, E.M.C. Colonization with Multidrug-Resistant Gram-Negative Bacteria: Prolonged Duration and Frequent Cocolonization. Clin. Infect. Dis. 2009, 48, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.A.; Kennedy, K.J.; Sidjabat, H.E.; Jones, M.; Collignon, P.; Paterson, D.L. Prolonged carriage of resistant E. coli by returned travellers: Clonality, risk factors and bacterial characteristics. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2413–2420. [Google Scholar] [CrossRef]
- Andersson, D.I.; Levin, B.R. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 1999, 2, 489–493. [Google Scholar] [CrossRef]
- Levin, B.R.; Lipsitch, M.; Perrot, V.; Schrag, S.; Antia, R.; Simonsen, L.; Walker, N.M.; Stewart, F.M. The Population Genetics of Antibiotic Resistance. Clin. Infect. Dis. 1997, 24, S9–S16. [Google Scholar] [CrossRef]
- Schrag, S.J.; Perrot, V.; Levin, B.R. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc. R. Soc. B Biol. Sci. 1997, 264, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Björkman, J.; Nagaev, I.; Berg, O.G.; Hughes, D.; Andersson, D.I. Effects of Environment on Compensatory Mutations to Ameliorate Costs of Antibiotic Resistance. Science 2000, 287, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Maisnier-Patin, S.; I Andersson, D. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 2004, 155, 360–369. [Google Scholar] [CrossRef]
- Nilsson, A.I.; Zorzet, A.; Kanth, A.; Dahlström, S.; Berg, O.G.; Andersson, D.I. Reducing the fitness cost of antibiotic resistance by amplification of initiator tRNA genes. Proc. Natl. Acad. Sci. USA 2006, 103, 6976–6981. [Google Scholar] [CrossRef] [PubMed]
- Bouma, J.E.; Lenski, R.E. Evolution of a bacteria/plasmid association. Nature 1988, 335, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Modi, R.I.; Adams, J. Coevolution in Bacterial-Plasmid Populations. Evol. Int. J. Org. Evol. 1991, 45, 656–667. [Google Scholar] [CrossRef]
- Dahlberg, C.; Chao, L. Amelioration of the Cost of Conjugative Plasmid Carriage in Eschericha coli K. Genetics 2003, 165, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, F.; Conceição, I.; Marques, A.C.; Fernandes, L.; Gordo, I. The evolution of a conjugative plasmid and its ability to increase bacterial fitness. Biol. Lett. 2005, 1, 250–252. [Google Scholar] [CrossRef]
- Harrison, E.; Guymer, D.; Spiers, A.J.; Paterson, S.; Brockhurst, M.A. Parallel Compensatory Evolution Stabilizes Plasmids across the Parasitism-Mutualism Continuum. Curr. Biol. 2015, 25, 2034–2039. [Google Scholar] [CrossRef]
- Hall, J.P.J.; Wright, R.C.T.; Guymer, D.; Harrison, E.; Brockhurst, M.A.Y. 2020 Extremely Fast Amelioration of Plasmid Fitness Costs by Multiple Functionally Diverse Pathways. Microbiology 2020, 166, 56–62. [Google Scholar] [CrossRef]
- Millan, A.S.; Heilbron, K.; MacLean, R.C. Positive epistasis between co-infecting plasmids promotes plasmid survival in bacterial populations. ISME J. 2014, 8, 601–612. [Google Scholar] [CrossRef]
- Gama, J.A.; Zilhão, R.; Dionisio, F. Co-resident plasmids travel together. Plasmid 2017, 93, 24–29. [Google Scholar] [CrossRef]
- Gama, J.A.; Zilhão, R.; Dionisio, F. Multiple plasmid interference—Pledging allegiance to my enemy’s enemy. Plasmid 2017, 93, 17–23. [Google Scholar] [CrossRef]
- Gama, J.A.; Zilhão, R.; Dionisio, F. Conjugation efficiency depends on intra and intercellular interactions between distinct plasmids: Plasmids promote the immigration of other plasmids but repress co-colonizing plasmids. Plasmid 2017, 93, 6–16. [Google Scholar] [CrossRef]
- Gama, J.A.; Zilhão, R.; Dionisio, F. Impact of plasmid interactions with the chromosome and other plasmids on the spread of antibiotic resistance. Plasmid 2018, 99, 82–88. [Google Scholar] [CrossRef]
- Lau, B.T.; Malkus, P.; Paulsson, J. New quantitative methods for measuring plasmid loss rates reveal unexpected stability. Plasmid 2013, 70, 353–361. [Google Scholar] [CrossRef]
- Ghigo, J.-M. Natural conjugative plasmids induce bacterial biofilm development. Nature 2001, 412, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Reisner, A.; Höller, B.M.; Molin, S.; Zechner, E.L. Synergistic Effects in Mixed Escherichia coli Biofilms: Conjugative Plasmid Transfer Drives Biofilm Expansion. J. Bacteriol. 2006, 188, 3582–3588. [Google Scholar] [CrossRef] [PubMed]
- Gama, J.A.; Fredheim, E.G.A.; Cléon, F.; Reis, A.M.; Zilhão, R.; Dionisio, F. Dominance Between Plasmids Determines the Extent of Biofilm Formation. Front. Microbiol. 2020, 11, 2070. [Google Scholar] [CrossRef] [PubMed]
- Alonso-del Valle, A.; León-Sampedro, R.; Rodríguez-Beltrán, J.; DelaFuente, J.; Hernández-García, M.; Ruiz-Garbajosa, P.; Cantón, R.; Peña-Miller, R.; San Millán, A. Variability of Plasmid Fitness Effects Contributes to Plasmid Persistence in Bacterial Communities. Nat. Commun. 2021, 12, 2653. [Google Scholar] [CrossRef] [PubMed]
- Combadão, J.; Campos, P.R.A.; Dionisio, F.; Gordo, I. Small-world networks decrease the speed of Muller’s ratchet. Genet. Res. 2007, 89, 7–18. [Google Scholar] [CrossRef]
- Fararo, T.J.; Sunshine, M.H. A Study of a Biased Friendship Net; Youth Development Center Syracuse University: Syracuse, NY, USA, 1964. [Google Scholar]
- Amaral, L.A.N.; Scala, A.; Barthélémy, M.; Stanley, H.E. Classes of small-world networks. Proc. Natl. Acad. Sci. USA 2000, 97, 11149–11152. [Google Scholar] [CrossRef]
- Liljeros, F.; Edling, C.; Amaral, L.A.N.; Stanley, H.E.; Åberg, Y. The web of human sexual contacts. Nature 2001, 411, 907–908. [Google Scholar] [CrossRef]
- Milgram, S. The small-world problem. Psychol. Today 1967, 60–67. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Pseudo Code |
|---|---|
| Gene transfer | For each connection between two individuals do (for each individual of the connection do (get the genes present in each individual metagenome; transmit genes to the other individual of the connection according to the gene transmission probability)). |
| Transfer of bacterial pathogens | For each connection between two individuals do (for each individual of the connection do (get the pathogenic species present in each individual; transmit pathogen to the other individual of the connection according to the bacterial pathogen transmission probability)). |
| Screening of individuals | For each individual do (check if the individual has a pathogenic bacteria). |
| Antibiotic effect | Choose an antibiotic randomly. Select all resistance genes associated with the chosen antibiotic. Eliminate resistance genes not associated with the chosen antibiotic according to the probability of eliminating genes under antibiotic intake. Eliminate virulence genes according to the probability of eliminating genes under antibiotic intake. |
| Loss rate of resistance genes under antibiotic consumption | Eliminate resistance genes not associated with the chosen antibiotic due to fitness cost according to the loss rate probability. |
| Loss rate of resistance genes without antibiotic consumption | Eliminate resistance genes according to the loss rate probability. |
| Immigration of bacterial pathogen into the network | For each bacterial species do (select a random individual; insert the bacterial pathogen in the individual). |
| Parameters | Default Values | Other Values |
|---|---|---|
| Rewiring connectivity probability p | 0.5 | 0 or 1 |
| Number of individuals | 1000 | 3000 |
| Number of virulence genes | 100 | 200, 400 |
| Number of resistance genes | 100 | 200, 400 |
| Number of pathogenic bacterial species | 5 | NA |
| Number of antibiotics | 5 | NA |
| Gene transmission probability | 0.005, 0.01 | 0.0005, 0.0025, 0.015, 0.02 |
| Bacterial pathogen transmission probability | 0.15 | 0.05, 0.1, 0.2, 0.25 |
| Probability of eliminating genes under antibiotic intake | 0.7 | 0.3, 0.5 |
| The loss rate of resistance genes | 0, 0.005, 0.01, 0.015, 0.02, 0.025, 0.03 | NA |
| Number of Pathogenic Species (in 2,000,000 Possibilities) | ||||||
|---|---|---|---|---|---|---|
| Bacterial Pathogen Transmission Probability | 0 | 1 | 2 | 3 | 4 | 5 |
| 0.05 | 1,987,473 | 12,496 | 31 | 0 | 0 | 0 |
| 0.1 | 1,982,852 | 17,094 | 54 | 0 | 0 | 0 |
| 0.15 | 1,973,053 | 26,763 | 184 | 0 | 0 | 0 |
| 0.2 | 1,940,458 | 58,759 | 779 | 4 | 0 | 0 |
| 0.25 | 104,967 | 262,575 | 527,204 | 705,479 | 399,253 | 522 |
| Bacterial Pathogen Transmission Probability | Number of Times that All Pathogenic Bacterial Species Disappeared in a Cycle (in 2000 Possibilities) |
|---|---|
| 0.05 | 570 |
| 0.1 | 70 |
| 0.15 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingues, C.P.F.; Rebelo, J.S.; Pothier, J.; Monteiro, F.; Nogueira, T.; Dionisio, F. The Perfect Condition for the Rising of Superbugs: Person-to-Person Contact and Antibiotic Use Are the Key Factors Responsible for the Positive Correlation between Antibiotic Resistance Gene Diversity and Virulence Gene Diversity in Human Metagenomes. Antibiotics 2021, 10, 605. https://doi.org/10.3390/antibiotics10050605
Domingues CPF, Rebelo JS, Pothier J, Monteiro F, Nogueira T, Dionisio F. The Perfect Condition for the Rising of Superbugs: Person-to-Person Contact and Antibiotic Use Are the Key Factors Responsible for the Positive Correlation between Antibiotic Resistance Gene Diversity and Virulence Gene Diversity in Human Metagenomes. Antibiotics. 2021; 10(5):605. https://doi.org/10.3390/antibiotics10050605
Chicago/Turabian StyleDomingues, Célia P. F., João S. Rebelo, Joël Pothier, Francisca Monteiro, Teresa Nogueira, and Francisco Dionisio. 2021. "The Perfect Condition for the Rising of Superbugs: Person-to-Person Contact and Antibiotic Use Are the Key Factors Responsible for the Positive Correlation between Antibiotic Resistance Gene Diversity and Virulence Gene Diversity in Human Metagenomes" Antibiotics 10, no. 5: 605. https://doi.org/10.3390/antibiotics10050605
APA StyleDomingues, C. P. F., Rebelo, J. S., Pothier, J., Monteiro, F., Nogueira, T., & Dionisio, F. (2021). The Perfect Condition for the Rising of Superbugs: Person-to-Person Contact and Antibiotic Use Are the Key Factors Responsible for the Positive Correlation between Antibiotic Resistance Gene Diversity and Virulence Gene Diversity in Human Metagenomes. Antibiotics, 10(5), 605. https://doi.org/10.3390/antibiotics10050605