
Distinct Resistomes and Microbial Communities of Soils, Wastewater Treatment Plants and Households Suggest Development of Antibiotic Resistances Due to Distinct Environmental Conditions in Each Environment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Bacterial Diversity and Richness of Species, ARG and MGE
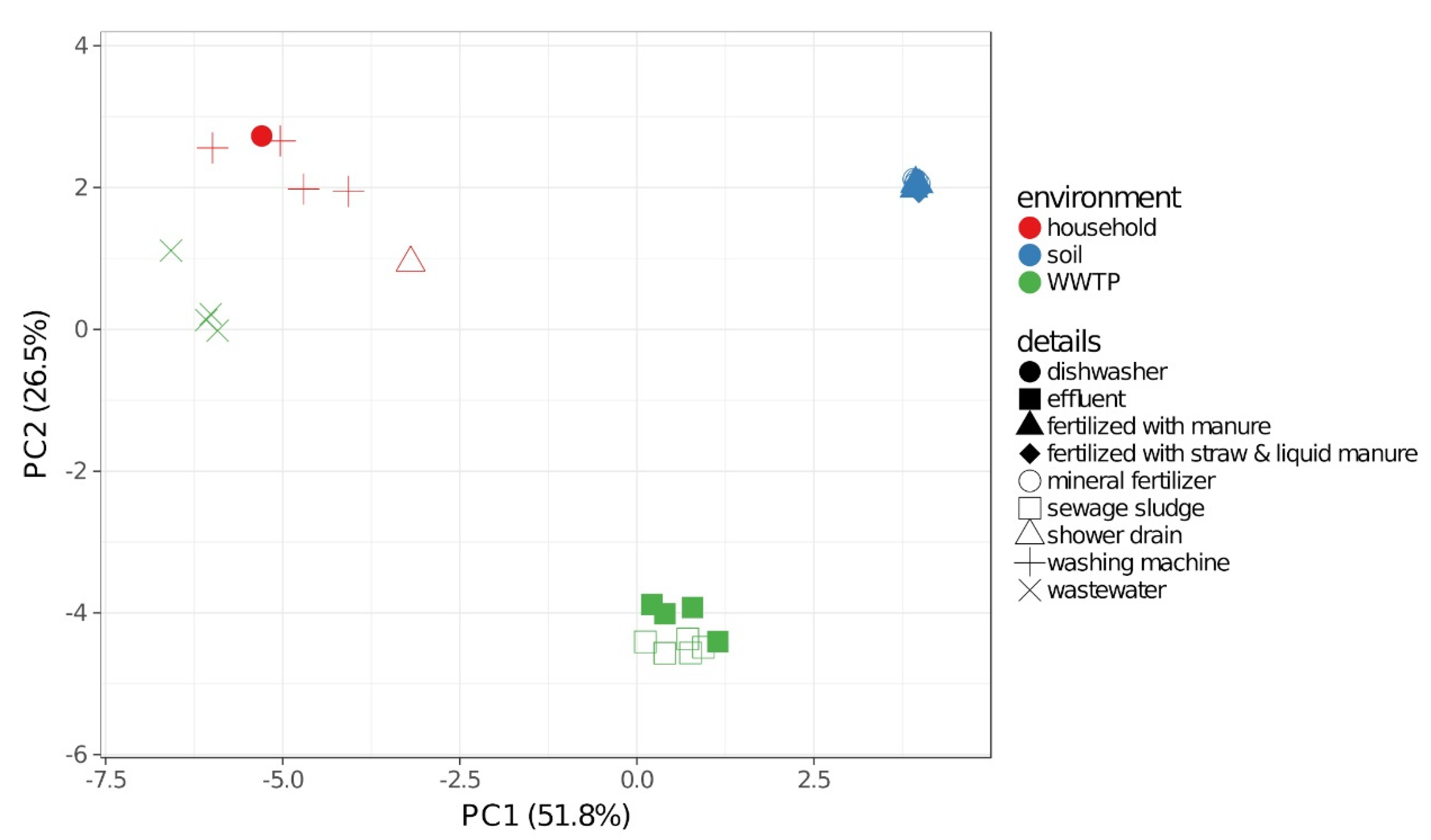
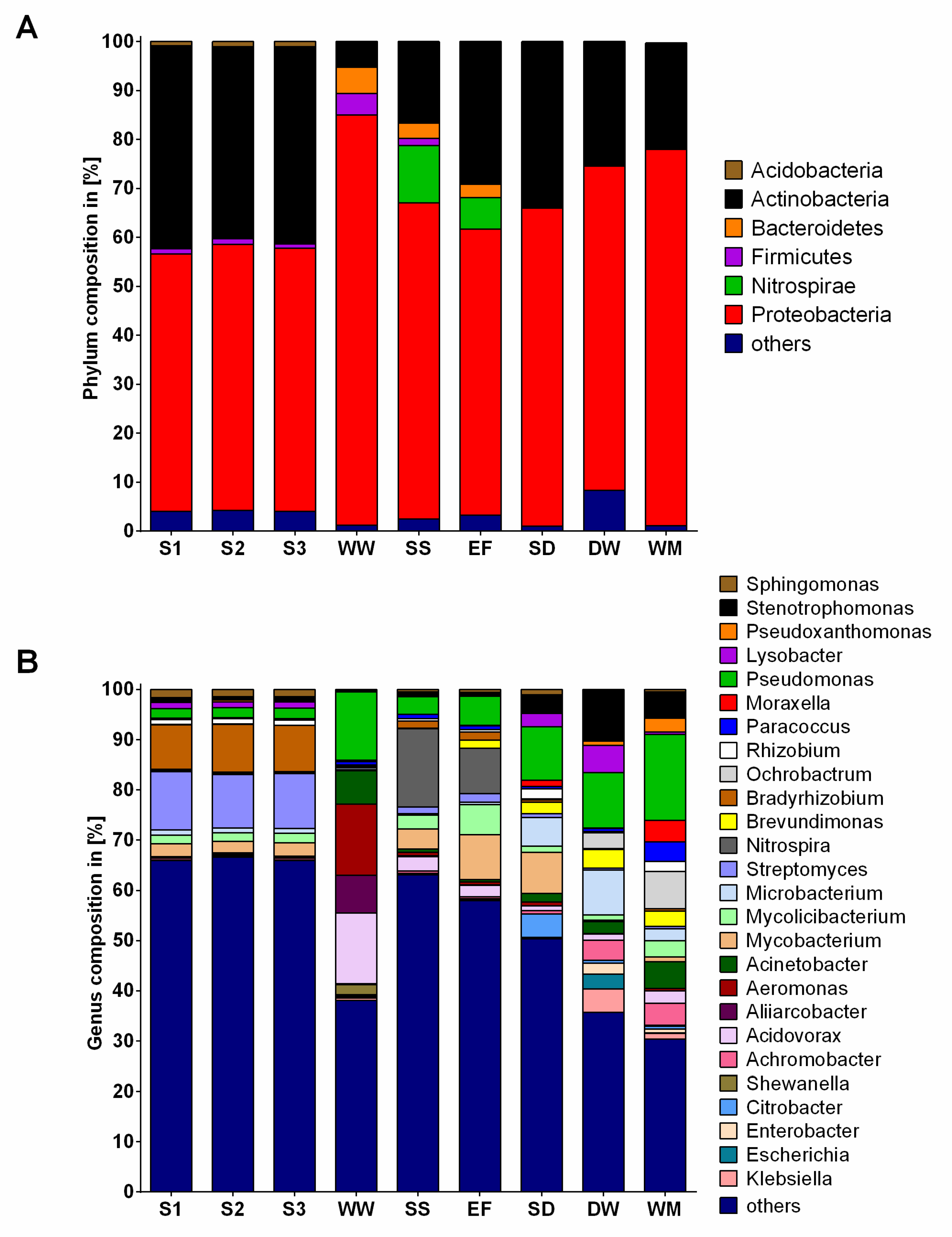
2.2. Community Composition
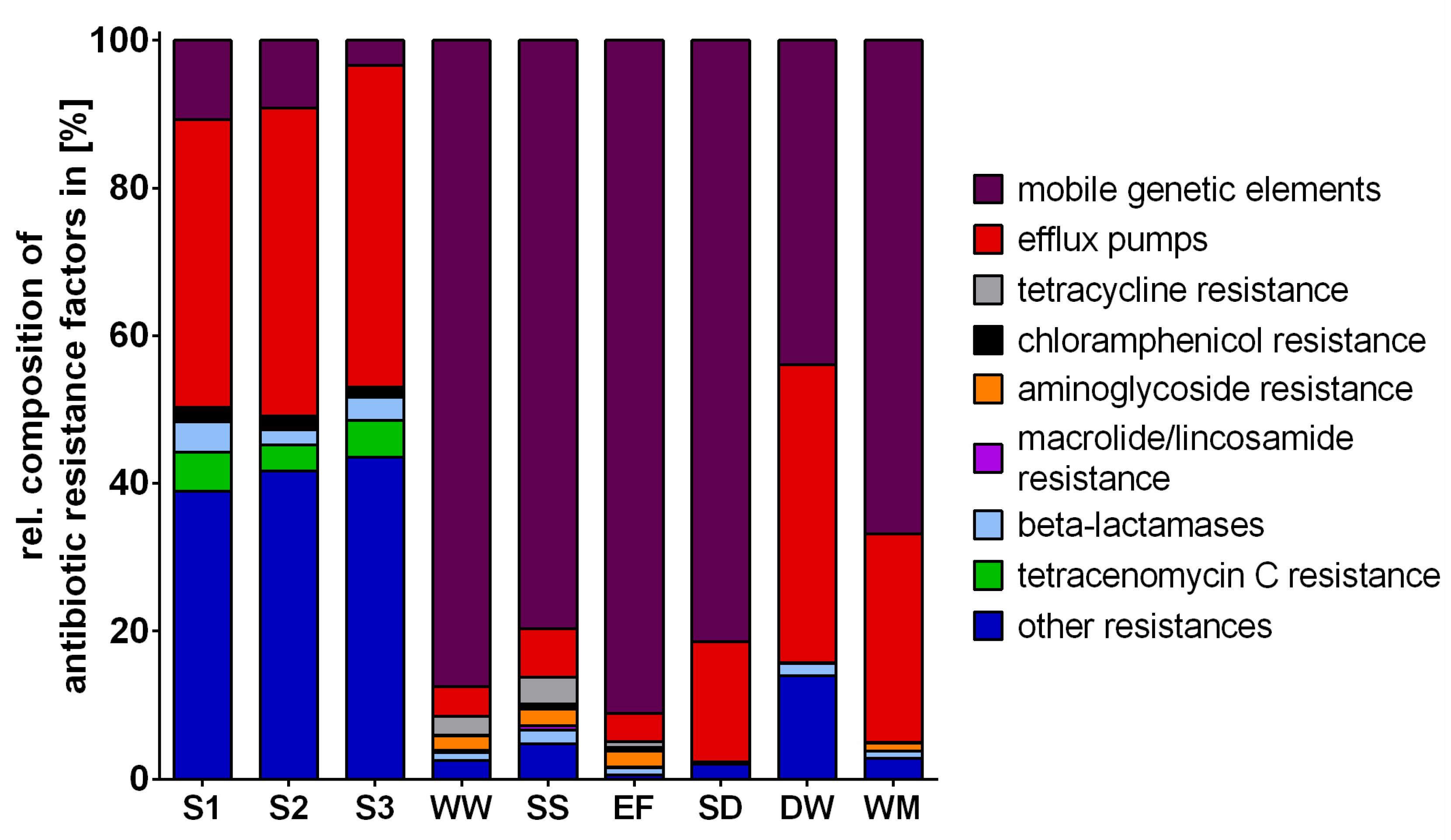
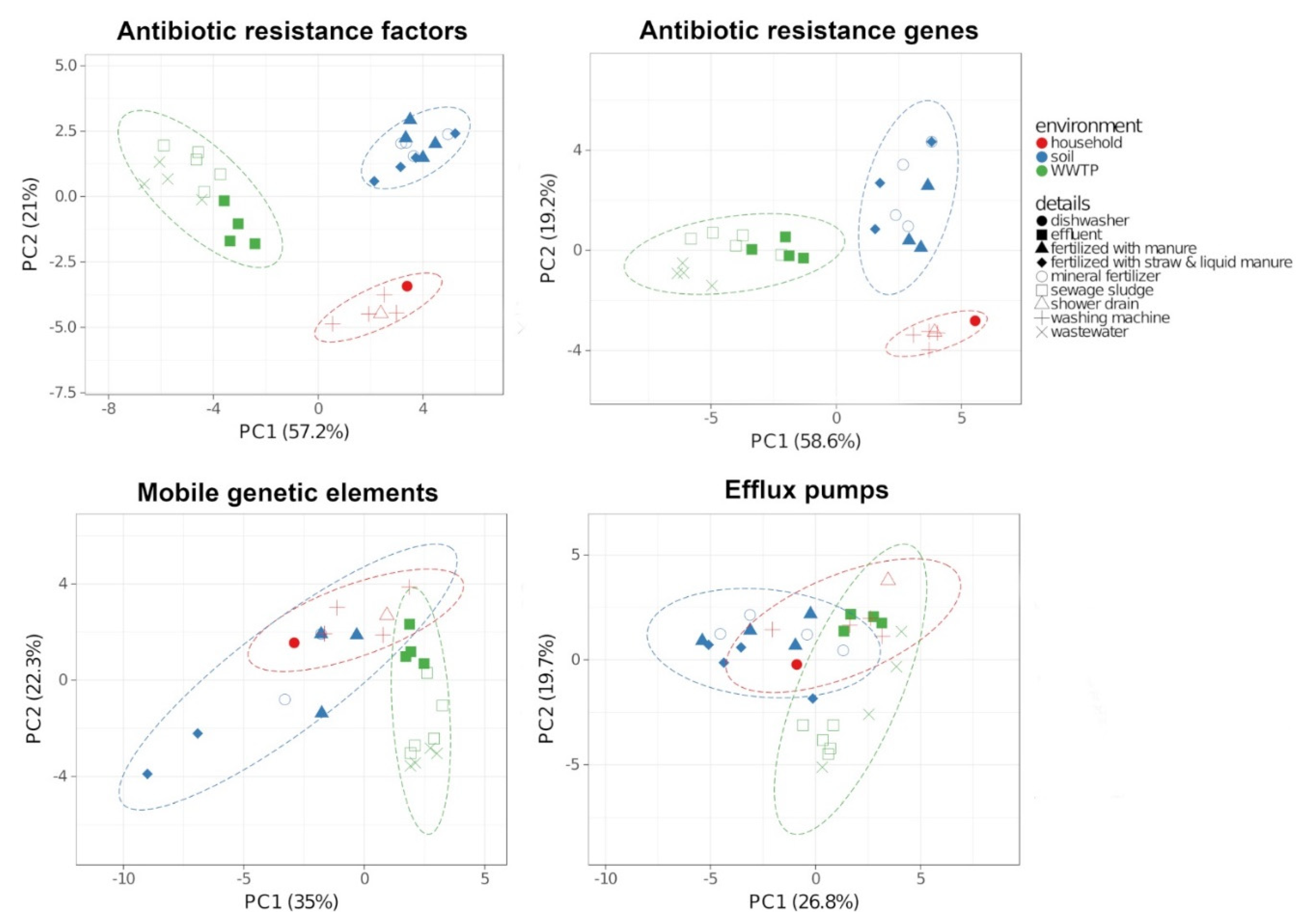
2.3. Antibiotic Resistance Factors
3. Discussion
3.1. Bacterial Communities and Resistomes in the Different Environments
3.2. Transfer Between the Environments
4. Materials and Methods
4.1. Sample Collection and Preparation
4.2. DNA Extraction
4.3. Metagenomic Analysis
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE 2012, 7, e0034953. [Google Scholar] [CrossRef]
- Woolhouse, M.; Ward, M.; van Bunnik, B.; Farrar, J. Antimicrobial resistance in humans, livestock and the wider environment. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140083. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. P T A peer-reviewed. J. Formul. Manag. 2015, 40, 277–283. [Google Scholar]
- McKinney, C.W.; Dungan, R.S.; Moore, A.; Leytem, A.B. Occurrence and abundance of antibiotic resistance genes in agricultural soil receiving dairy manure. FEMS Microbiol. Ecol. 2018, 94, fiy010. [Google Scholar] [CrossRef]
- Pärnänen, K.M.M.; Narciso-Da-Rocha, C.; Kneis, D.; Berendonk, T.U.; Cacace, D.; Do, T.T.; Elpers, C.; Fatta-Kassinos, D.; Henriques, I.; Jaeger, T.; et al. Antibiotic resistance in European wastewater treatment plants mirrors the pattern of clinical antibiotic resistance prevalence. Sci. Adv. 2019, 5, eaau9124. [Google Scholar] [CrossRef]
- Irrgang, A.; Roschanski, N.; Tenhagen, B.A.; Grobbel, M.; Skladnikiewicz-Ziemer, T.; Thomas, K.; Roesler, U.; Käsbohrer, A. Prevalence of mcr-1 in E. coli from livestock and food in Germany, 2010–2015. PLoS ONE 2016, 11, e0159863. [Google Scholar] [CrossRef]
- Morrison, B.J.; Rubin, J.E. Carbapenemase producing bacteria in the food supply escaping detection. PLoS ONE 2015, 10, e0126717. [Google Scholar] [CrossRef]
- Rolain, J.M. Food and human gut as reservoirs of transferable antibiotic resistance encoding genes. Front. Microbiol. 2013, 4, 173. [Google Scholar] [CrossRef]
- Schages, L.; Wichern, F.; Kalscheuer, R.; Bockmühl, D. Winter is coming—Impact of temperature on the variation of beta-lactamase and mcr genes in a wastewater treatment plant. Sci. Total Environ. 2020, 712, 136499. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Morten, O.A. The shared antibiotic resistome of soil bacteria and human pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Ju, F.; Beck, K.; Yin, X.; Maccagnan, A.; McArdell, C.S.; Singer, H.P.; Johnson, D.R.; Zhang, T.; Bürgmann, H. Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 2019, 13, 346–360. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef]
- Pehrsson, E.C.; Tsukayama, P.; Patel, S.; Mejía-Bautista, M.; Sosa-Soto, G.; Navarrete, K.M.; Calderon, M.; Cabrera, L.; Hoyos-Arango, W.; Bertoli, M.T.; et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature 2016, 533, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Smith, M.B.; Friedman, J.; Cordero, O.X.; David, L.A.; Alm, E.J. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011, 480, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 2011, 35, 790–819. [Google Scholar] [CrossRef]
- Giedraitienė, A.; Vitkauskienė, A.; Naginienė, R.; Pavilonis, A. Antibiotic resistance mechanisms of clinically important bacteria. Medicina 2011, 47, 19. [Google Scholar] [CrossRef]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef]
- Wellington, E.M.H.; Boxall, A.B.A.; Cross, P.; Feil, E.J.; Gaze, W.H.; Hawkey, P.M.; Johnson-Rollings, A.S.; Jones, D.L.; Lee, N.M.; Otten, W.; et al. The role of the natural environment in the emergence of antibiotic resistance in Gram-negative bacteria. Lancet Infect. Dis. 2013, 13, 155–165. [Google Scholar] [CrossRef]
- Wiedenbeck, J.; Cohan, F.M. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 2011, 35, 957–976. [Google Scholar] [CrossRef]
- Sabri, N.A.; Schmitt, H.; Van der Zaan, B.; Gerritsen, H.W.; Zuidema, T.; Rijnaarts, H.H.M.; Langenhoff, A.A.M. Prevalence of antibiotics and antibiotic resistance genes in a wastewater effluent-receiving river in the Netherlands. J. Environ. Chem. Eng. 2020, 8, 102245. [Google Scholar] [CrossRef]
- Sanganyado, E.; Gwenzi, W. Antibiotic resistance in drinking water systems: Occurrence, removal, and human health risks. Sci. Total Environ. 2019, 669, 785–797. [Google Scholar] [CrossRef]
- Schwartz, T.; Kohnen, W.; Jansen, B.; Obst, U. Detection of antibiotic resistant bacteria and their resistance genes in wastewater, surface water, and drinking water biofilms. Microbiol. Ecol. 2003, 43, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Knapp, C.W.; Dolfing, J.; Ehlert, P.A.I.; Graham, D.W. Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940. Environ. Sci. Technol. 2010, 44, 580–587. [Google Scholar] [CrossRef]
- Nesme, J.; Simonet, P. The soil resistome: A critical review on antibiotic resistance origins, ecology and dissemination potential in telluric bacteria. Environ. Microbiol. 2015, 17, 913–930. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.K.; Moe, L.A.; Rodbumrer, J.; Gaarder, A.; Handelsman, J. Functional metagenomics reveals diverse Β-lactamases in a remote Alaskan soil. ISME J. 2009, 3, 243–251. [Google Scholar] [CrossRef]
- Martin, J.F.; Liras, P. Organization and expression of genes involved in the biosynthesis of antibiotics and other secondary metabolites. Annu. Rev. Microbiol. 1989, 43, 173–206. [Google Scholar] [CrossRef]
- Martínez, J.L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef]
- Pazda, M.; Kumirska, J.; Stepnowski, P.; Mulkiewicz, E. Antibiotic resistance genes identified in wastewater treatment plant systems—A review. Sci. Total Environ. 2019, 697, 134023. [Google Scholar] [CrossRef]
- Aristizábal-Hoyos, A.M.; Rodríguez, E.A.; Arias, L.; Jiménez, J.N. High clonal diversity of multidrug-resistant and extended spectrum beta-lactamase-producing Escherichia coli in a wastewater treatment plant. J. Environ. Manag. 2019, 245, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Bouki, C.; Venieri, D.; Diamadopoulos, E. Detection and fate of antibiotic resistant bacteria in wastewater treatment plants: A review. Ecotoxicol. Environ. Saf. 2013, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Caltagirone, M.; Nucleo, E.; Spalla, M.; Zara, F.; Novazzi, F.; Marchetti, V.M.; Piazza, A.; Bitar, I.; De Cicco, M.; Paolucci, S.; et al. Occurrence of extended spectrum β-lactamases, KPC-Type, and MCR-1.2-producing enterobacteriaceae from wells, river water, and wastewater treatment plants in Oltrepò Pavese area, Northern Italy. Front. Microbiol. 2017, 8, 2232. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, L.; Manaia, C.; Merlin, C.; Schwartz, T.; Dagot, C.; Ploy, M.C.; Michael, I.; Fatta-Kassinos, D. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: A review. Sci. Total Environ. 2013, 447, 345–360. [Google Scholar] [CrossRef]
- Lucassen, R.; Rehberg, L.; Heyden, M.; Bockmühl, D.P. Strong correlation of total phenotypic resistance of samples from household environments and the prevalence of class 1 integrons suggests for the use of the relative prevalence of intI1 as a screening tool for multi-resistance. PLoS ONE 2019, 14, e0218277. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.M.; Robleto, E.; Dumont, T.; Levy, S.B. The frequency of antibiotic-resistant bacteria in homes differing in their use of surface antibacterial agents. Curr. Microbiol. 2012, 65, 407–415. [Google Scholar] [CrossRef]
- Rehberg, L.; Frontzek, A.; Melhus, Å.; Bockmühl, D.P. Prevalence of β-lactamase genes in domestic washing machines and dishwashers and the impact of laundering processes on antibiotic-resistant bacteria. J. Appl. Microbiol. 2017, 123, 3218–3221. [Google Scholar] [CrossRef]
- Schages, L.; Lucassen, R.; Wichern, F.; Kalscheuer, R.; Bockmühl, D. The household resistome—frequency of β-lactamases, class 1 integron and antibiotic resistant bacteria in the domestic environment and their reduction during automated dishwashing/laundering. Appl. Environ. Microbiol. 2020, 86, e02062-20. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, P.M.; Loureiro, L.; Matos, A.J.F. Transfer of multidrug-resistant bacteria between intermingled ecological niches: The interface between humans, animals and the environment. Int. J. Environ. Res. Public Health 2013, 10, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. The structure and diversity of human, animal and environmental resistomes. Microbiome 2016, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Huse, S.M.; Mueller-Spitz, S.R.; Andreishcheva, E.N.; Sogin, M.L. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 2010, 12, 378–392. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Munck, C.; Albertsen, M.; Telke, A.; Ellabaan, M.; Nielsen, P.H.; Sommer, M.O.A. Limited dissemination of the wastewater treatment plant core resistome. Nat. Commun. 2015, 6, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Savage, A.M.; Hills, J.; Driscoll, K.; Fergus, D.J.; Grunden, A.M.; Dunn, R.R. Microbial diversity of extreme habitats in human homes. PeerJ 2016, 4, e2376. [Google Scholar] [CrossRef] [PubMed]
- Novo, A.; André, S.; Viana, P.; Nunes, O.C.; Manaia, C.M. Antibiotic resistance, Antimicrobial residues and bacterial community composition in urban wastewater. Water Res. 2013, 47, 1875–1887. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Yu, S.; Rysz, M.; Luo, Y.; Yang, F.; Li, F.; Hou, J.; Mu, Q.; Alvarez, P.J.J. Prevalence and proliferation of antibiotic resistance genes in two municipal wastewater treatment plants. Water Res. 2015, 85, 458–466. [Google Scholar] [CrossRef]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.N.; et al. Tackling antibiotic resistance: The environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef]
- Manaia, C.M.; Rocha, J.; Scaccia, N.; Marano, R.; Radu, E.; Biancullo, F.; Cerqueira, F.; Fortunato, G.; Iakovides, I.C.; Zammit, I.; et al. Antibiotic resistance in wastewater treatment plants: Tackling the black box. Environ. Int. 2018, 115, 312–324. [Google Scholar] [CrossRef]
- Levy, S.B. Antibacterial household products: Cause for concern. Emerg. Infect. Dis. 2001, 7, 512–515. [Google Scholar] [CrossRef]
- Joergensen, R.G.; Wichern, F. Alive and kicking: Why dormant soil microorganisms matter. Soil Biol. Biochem. 2018, 116, 419–430. [Google Scholar] [CrossRef]
- Joergensen, R.G.; Wichern, F. Quantitative assessment of the fungal contribution to microbial tissue in soil. Soil Biol. Biochem. 2008, 40, 2977–2991. [Google Scholar] [CrossRef]
- Geisen, S.; Mitchell, E.A.D.; Adl, S.; Bonkowski, M.; Dunthorn, M.; Ekelund, F.; Fernández, L.D.; Jousset, A.; Krashevska, V.; Singer, D.; et al. Soil protists: A fertile frontier in soil biology research. FEMS Microbiol. Rev. 2018, 42, 293–323. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.G. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Tennstedt, T.; Szczepanowski, R.; Braun, S.; Pühler, A.; Schlüter, A. Occurrence of integron-associated resistance gene cassettes located on antibiotic resistance plasmids isolated from a wastewater treatment plant. FEMS Microbiol. Ecol. 2003, 45, 239–252. [Google Scholar] [CrossRef]
- Karkman, A.; Do, T.T.; Walsh, F.; Virta, M.P.J. Antibiotic-Resistance Genes in Waste Water. Trends Microbiol. 2018, 26, 220–228. [Google Scholar] [CrossRef]
- Jutkina, J.; Marathe, N.P.; Flach, C.F.; Larsson, D.G.J. Antibiotics and common antibacterial biocides stimulate horizontal transfer of resistance at low concentrations. Sci. Total Environ. 2018, 616–617, 172–178. [Google Scholar] [CrossRef]
- Chen, G.H.; Leung, D.H.W.; Hung, J.C. Biofilm in the sediment phase of a sanitary gravity sewer. Water Res. 2003, 37, 2784–2788. [Google Scholar] [CrossRef]
- Gattlen, J.; Amberg, C.; Zinn, M.; Mauclaire, L. Biofilms isolated from washing machines from three continents and their tolerance to a standard detergent. Biofouling 2010, 26, 873–882. [Google Scholar] [CrossRef]
- Raghupathi, P.K.; Zupančič, J.; Brejnrod, A.D.; Jacquiod, S.; Houf, K.; Burmølle, M.; Gunde-Cimerman, N.; Sørensen, S.J. Microbial Diversity and Putative Opportunistic Pathogens in Dishwasher Biofilm Communities. Appl. Environ. Microbiol. 2018, 84, e02755-17. [Google Scholar] [CrossRef]
- McBain, A.J.; Bartolo, R.G.; Catrenich, C.E.; Charbonneau, D.; Ledder, R.G.; Rickard, A.H.; Symmons, S.A.; Gilbert, P. Microbial characterization of biofilms in domestic drains and the establishment of stable biofilm microcosms. Appl. Environ. Microbiol. 2003, 69, 177–185. [Google Scholar] [CrossRef]
- Amos, G.C.A.; Hawkey, P.M.; Gaze, W.H.; Wellington, E.M. Waste water effluent contributes to the dissemination of CTX-M-15 in the natural environment. J. Antimicrob. Chemother. 2014, 69, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Jäger, T.; Hembach, N.; Elpers, C.; Wieland, A.; Alexander, J.; Hiller, C.; Krauter, G.; Schwartz, T. Reduction of Antibiotic Resistant Bacteria During Conventional and Advanced Wastewater Treatment, and the Disseminated Loads Released to the Environment. Front. Microbiol. 2018, 9, 2599. [Google Scholar] [CrossRef]
- Quach-Cu, J.; Herrera-Lynch, B.; Marciniak, C.; Adams, S.; Simmerman, A.; Reinke, R.A. The effect of primary, secondary, and tertiary wastewater treatment processes on antibiotic resistance gene (ARG) concentrations in solid and dissolved wastewater fractions. Water 2018, 10, 37. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, 68–80. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics 2015, 16, 964. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Potron, A.; Nordmann, P. OXA-48-like carbapenemases: The phantom menace. J. Antimicrob. Chemother. 2012, 67, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, P.M.; Jones, A.M. The changing epidemiology of resistance. J. Antimicrob. Chemother. 2009, 64, i3–i10. [Google Scholar] [CrossRef] [PubMed]
- Cantón, R.; Coque, T.M. The CTX-M β-lactamase pandemic. Curr. Opin. Microbiol. 2006, 9, 466–475. [Google Scholar] [CrossRef]
- Udikovic-Kolic, N.; Wichmann, F.; Broderick, N.A.; Handelsman, J. Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization. Proc. Natl. Acad. Sci. USA 2014, 111, 15202–15207. [Google Scholar] [CrossRef]
- Hemkemeyer, M.; Christensen, B.T.; Martens, R.; Tebbe, C.C. Soil particle size fractions harbour distinct microbial communities and differ in potential for microbial mineralisation of organic pollutants. Soil Biol. Biochem. 2015, 90, 255–265. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schages, L.; Wichern, F.; Geisen, S.; Kalscheuer, R.; Bockmühl, D. Distinct Resistomes and Microbial Communities of Soils, Wastewater Treatment Plants and Households Suggest Development of Antibiotic Resistances Due to Distinct Environmental Conditions in Each Environment. Antibiotics 2021, 10, 514. https://doi.org/10.3390/antibiotics10050514
Schages L, Wichern F, Geisen S, Kalscheuer R, Bockmühl D. Distinct Resistomes and Microbial Communities of Soils, Wastewater Treatment Plants and Households Suggest Development of Antibiotic Resistances Due to Distinct Environmental Conditions in Each Environment. Antibiotics. 2021; 10(5):514. https://doi.org/10.3390/antibiotics10050514
Chicago/Turabian StyleSchages, Laura, Florian Wichern, Stefan Geisen, Rainer Kalscheuer, and Dirk Bockmühl. 2021. "Distinct Resistomes and Microbial Communities of Soils, Wastewater Treatment Plants and Households Suggest Development of Antibiotic Resistances Due to Distinct Environmental Conditions in Each Environment" Antibiotics 10, no. 5: 514. https://doi.org/10.3390/antibiotics10050514
APA StyleSchages, L., Wichern, F., Geisen, S., Kalscheuer, R., & Bockmühl, D. (2021). Distinct Resistomes and Microbial Communities of Soils, Wastewater Treatment Plants and Households Suggest Development of Antibiotic Resistances Due to Distinct Environmental Conditions in Each Environment. Antibiotics, 10(5), 514. https://doi.org/10.3390/antibiotics10050514