
Potential Target Site for Inhibitors in MLSB Antibiotic Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
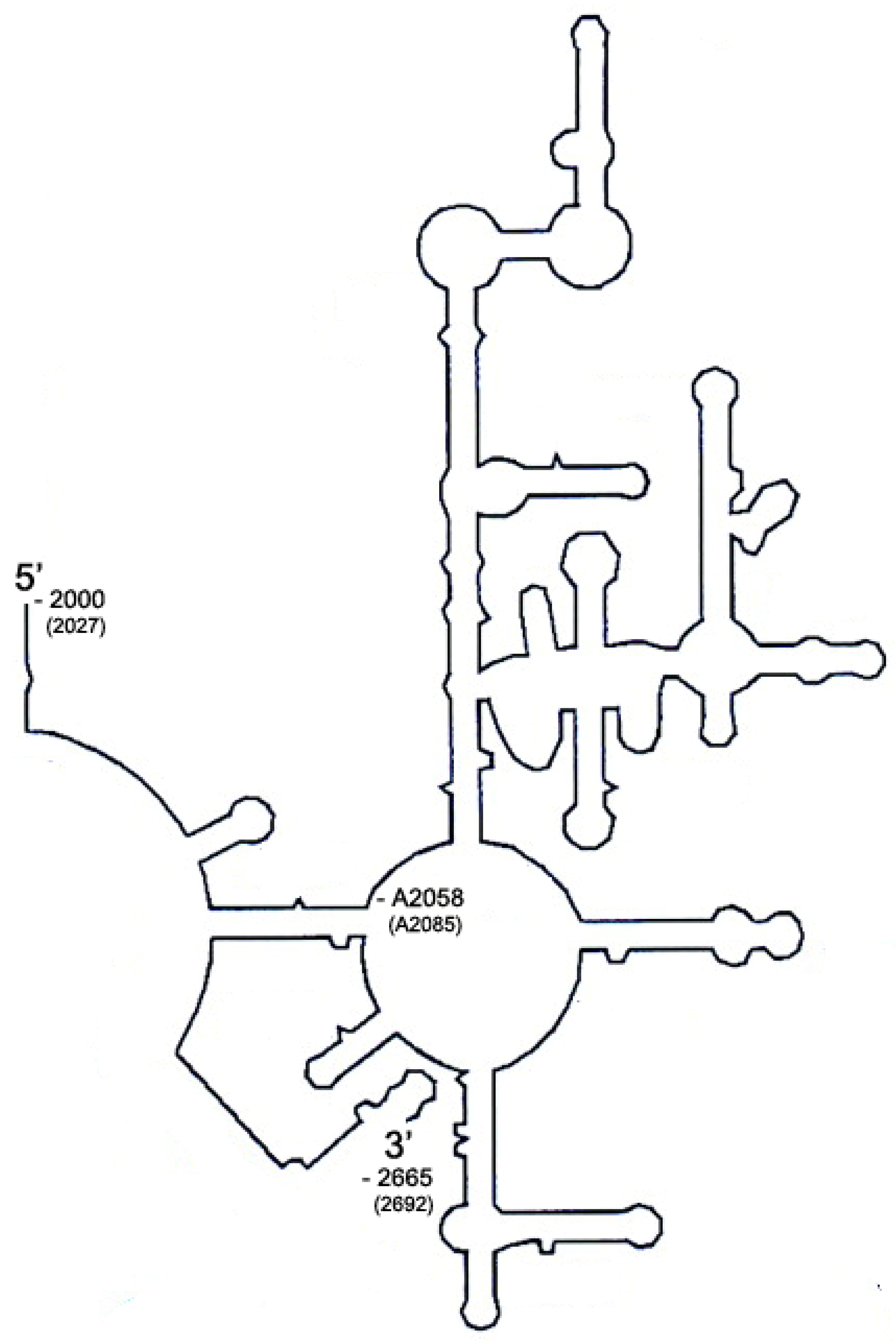
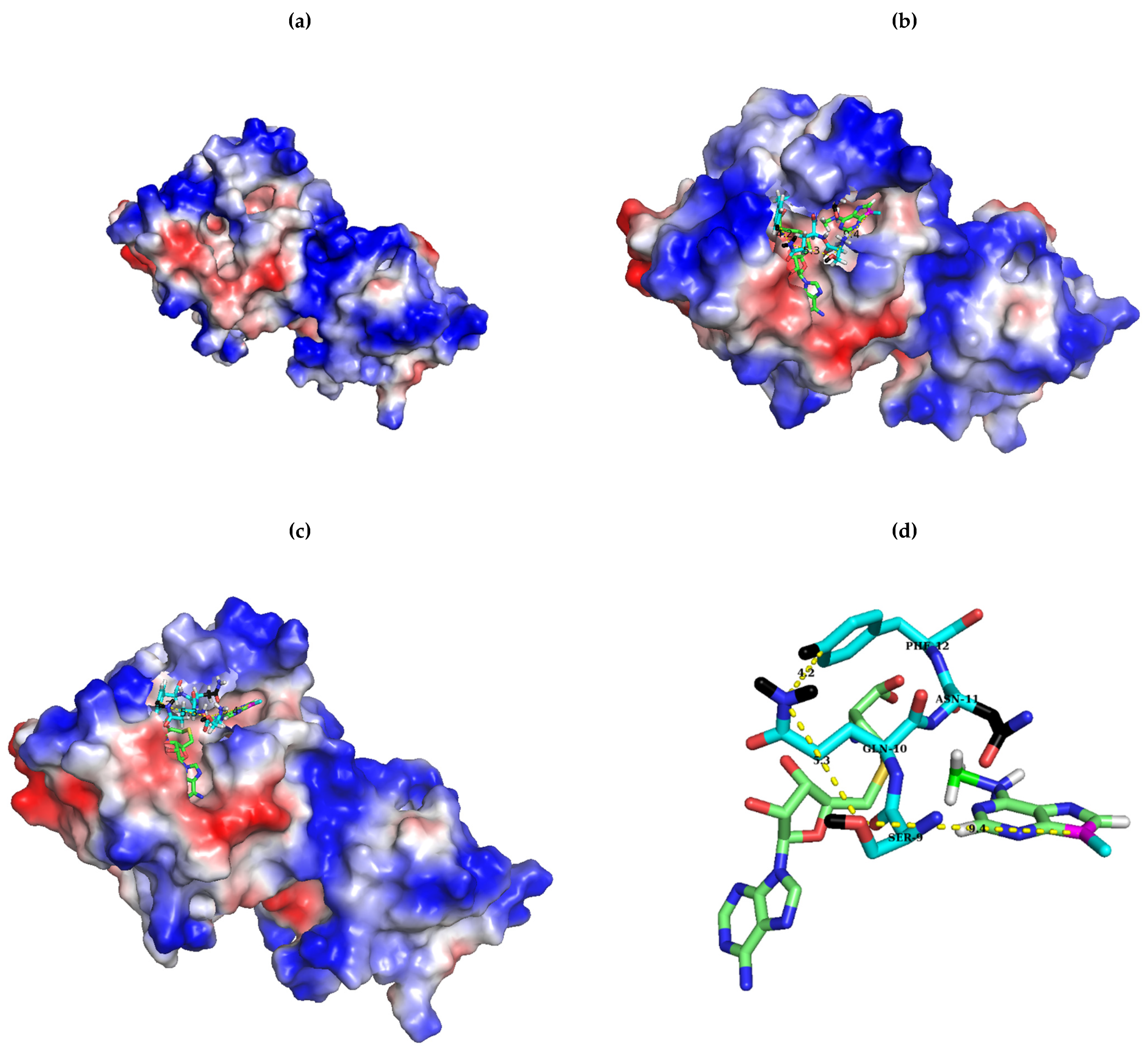
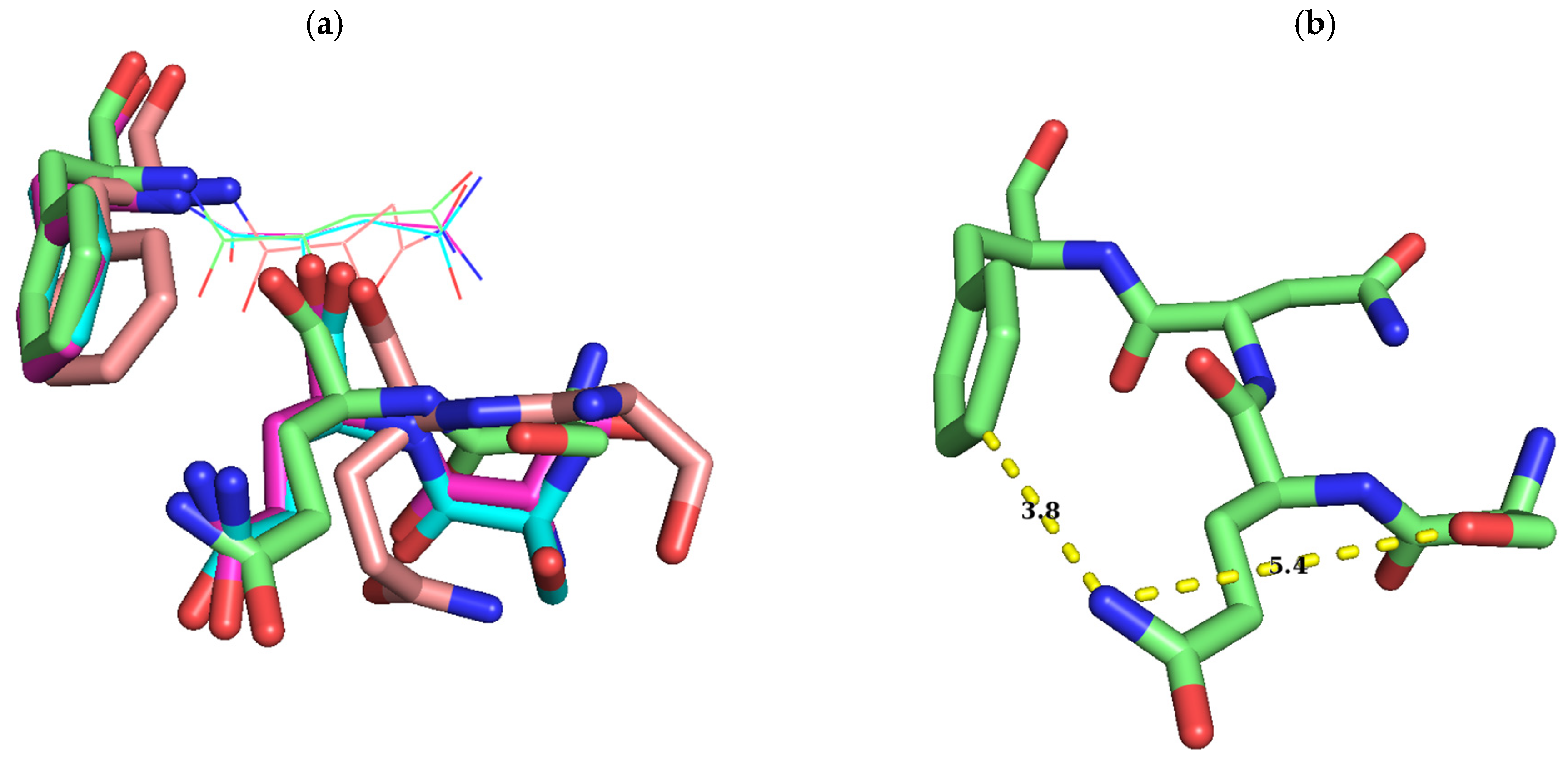
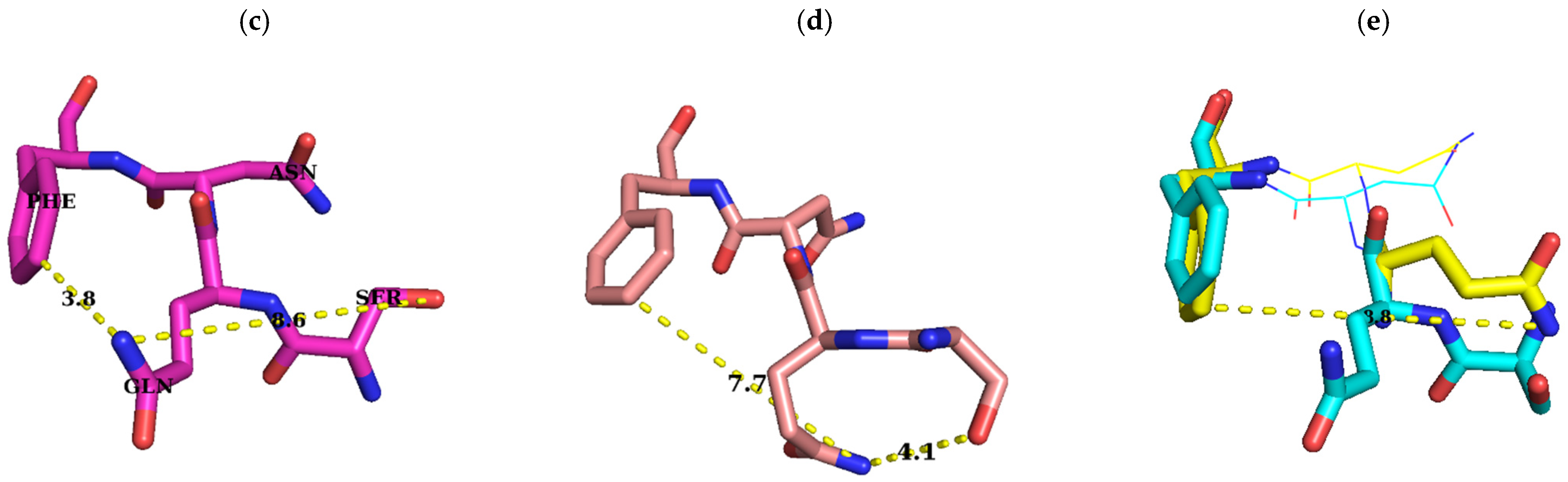
2.1. Sequence Alignment of Erm and KsgA/Dim1
2.2. Expression of Mutant Proteins in E. coli
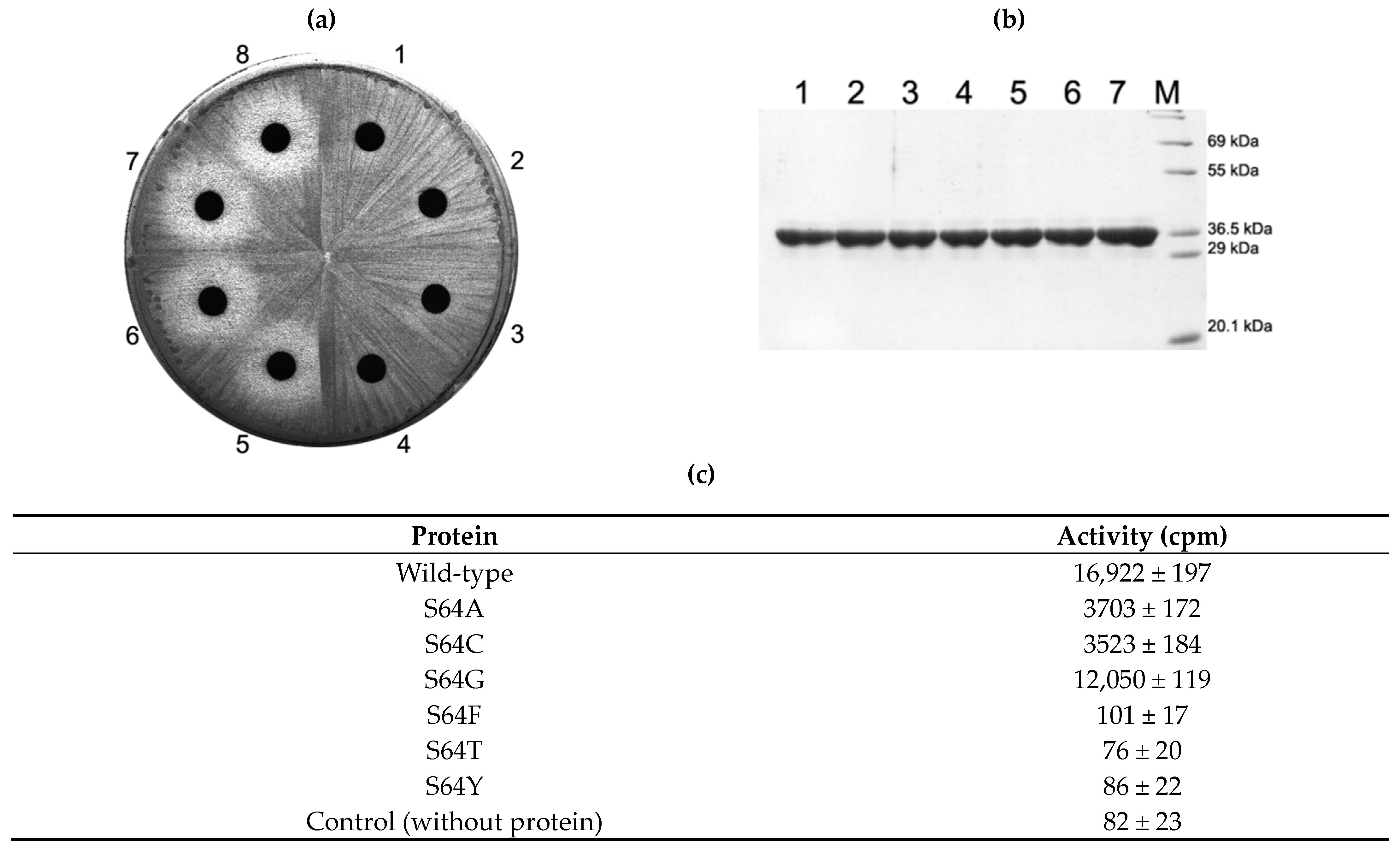
2.3. Activity of S64 Mutants
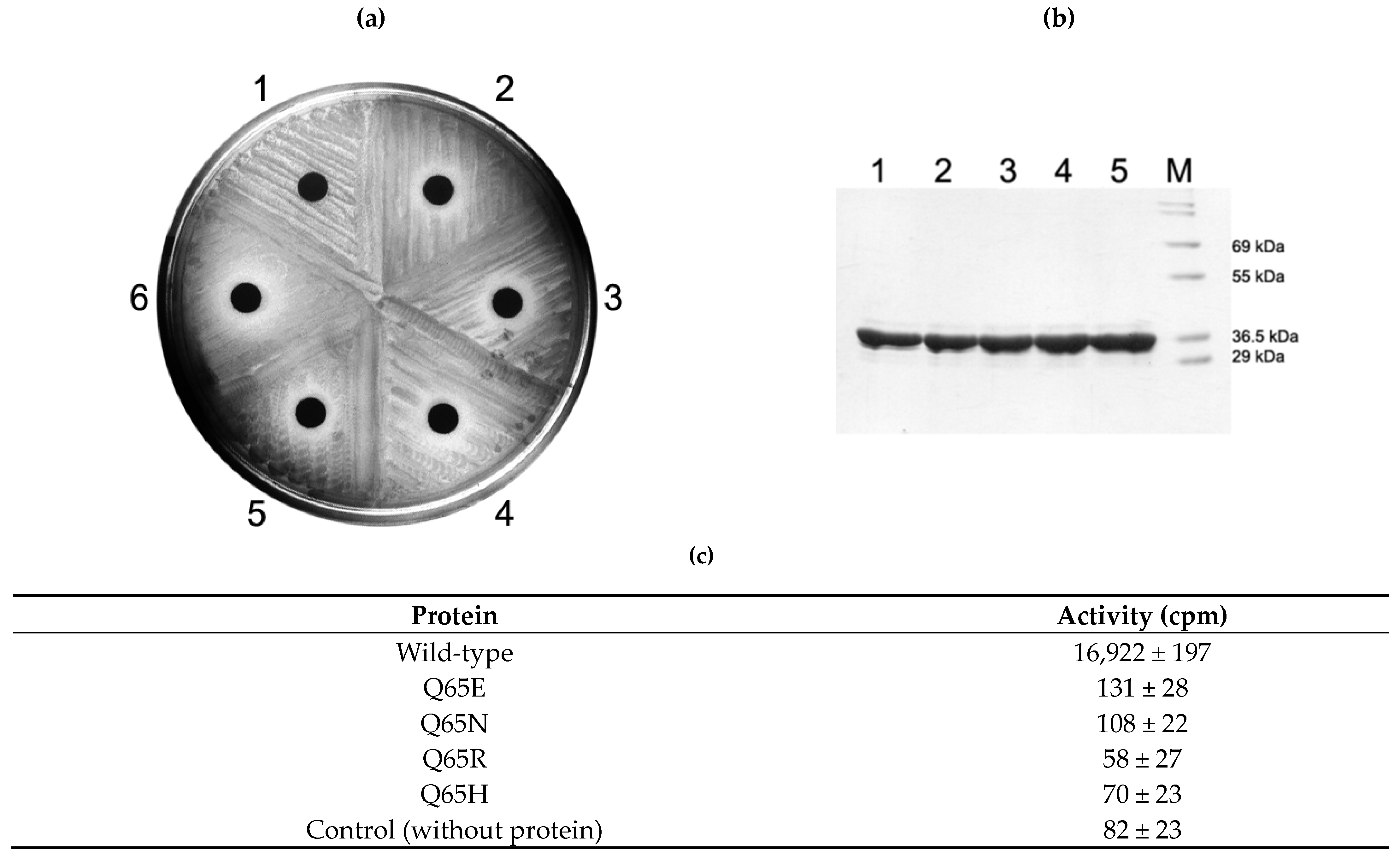
2.4. Activity of Q65 Mutants
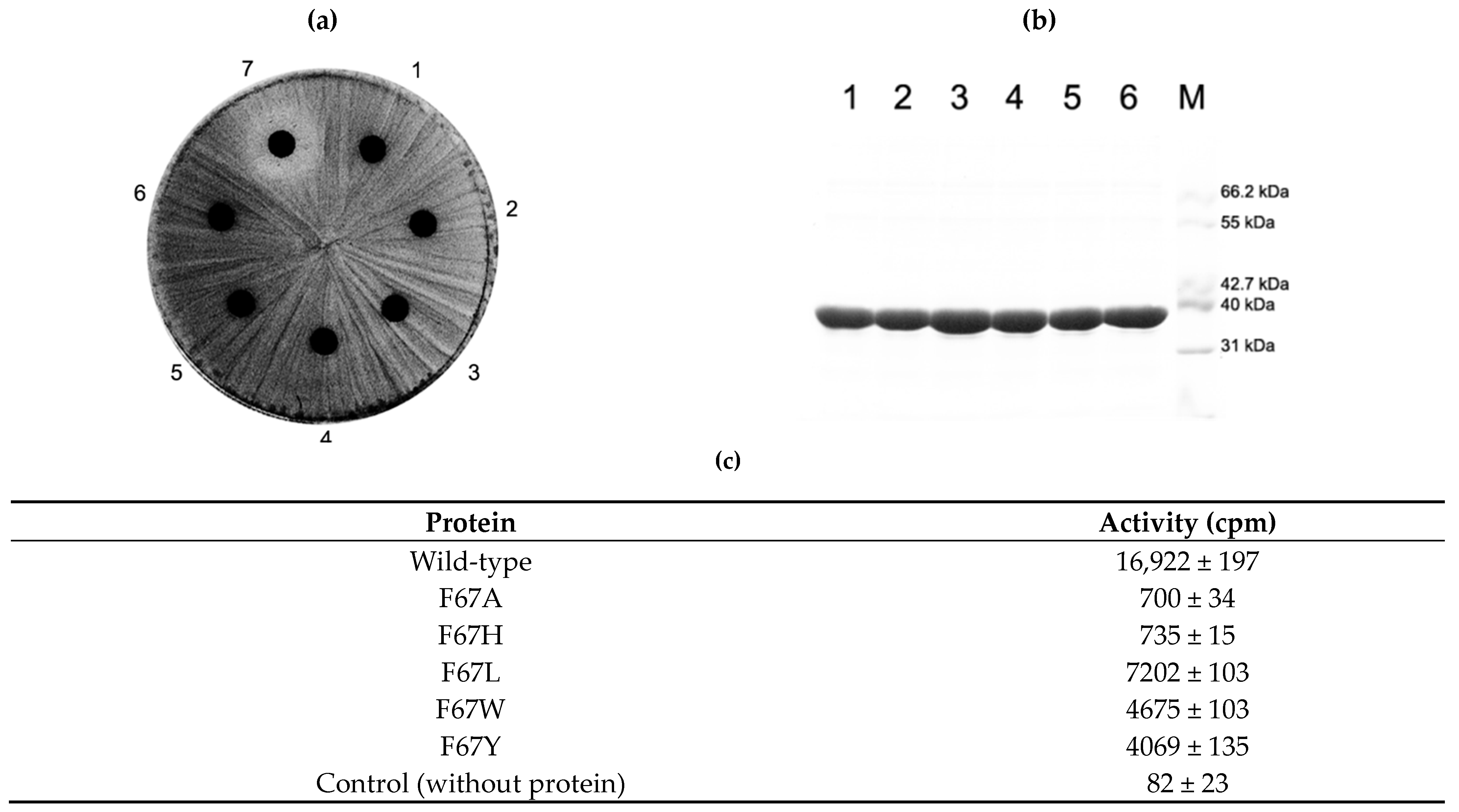
2.5. Activity of F67 Mutants
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Site-Directed Mutagenesis and Construction of Expression Vector
4.1.2. In Vivo Activity Assay for ErmS and Its Mutant Proteins (Antibiotic Susceptibility Assay)
4.1.3. Protein Expression and Purification
4.1.4. Cloning of B. subtilis Domain V DNA and Its In Vitro Transcription
4.1.5. In Vitro Methylation Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 481–511. [Google Scholar] [CrossRef]
- Spratt, B.G. Resistance to antibiotics mediated by target alterations. Science 1994, 264, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Prevention of drug access to bacterial targets: Permeability barriers and active efflux. Science 1994, 264, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Davies, J. Inactivation of antibiotics and the dissemination of resistance genes. Science 1994, 264, 375–382. [Google Scholar] [CrossRef]
- Schmitz, F.-J.; Verhoef, J.; Fluit, A.C.; The Sentry Participants Group. Prevalence of resistance to MLS antibiotics in 20 European university hospitals participating in the European SENTRY surveillance programme. J. Antimicrob. Chemother. 1999, 43, 783–792. [Google Scholar] [CrossRef]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.; Schmieder, B.J.; Petitpas, J.W.; Manousos, M.; Williams, J.A.; Faiella, J.A.; Girard, A.E.; McGuirk, P.R. Assays to Detect and Characterize Synthetic Agents that Inhibit the ErmC Methyltransferase. J. Antibiot. 1995, 48, 1273–1279. [Google Scholar] [CrossRef][Green Version]
- Bussiere, D.E.; Muchmore, S.W.; Dealwis, C.G.; Schluckebier, G.; Nienaber, V.L.; Edalji, R.P.; Walter, K.A.; Ladror, U.S.; Holzman, T.F.; Abad-Zapatero, C. Crystal Structure of ErmC‘, an rRNA Methyltransferase Which Mediates Antibiotic Resistance in Bacteria. Biochemistry 1998, 37, 7103–7112. [Google Scholar] [CrossRef]
- Yu, L.; Petros, A.M.; Schnuchel, A.; Zhong, P.; Severin, J.M.; Walter, K.; Holzman, T.F.; Fesik, S.W. Solution structure of an rRNA methyltransferase (ErmAM) that confers macrolide-lincosamide-streptogramin antibiotic resistance. Nat. Genet. 1997, 4, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Dinges, J.; Schkeryantz, J.M.; Janowick, D.; Kaminski, M.; Tufano, M.; Augeri, D.J.; Petros, A.; Nienaber, V.; Zhong, P.; et al. Novel Inhibitors of Erm Methyltransferases from NMR and Parallel Synthesis. J. Med. Chem. 1999, 42, 3852–3859. [Google Scholar] [CrossRef]
- Feder, M.; Purta, E.; Koscinski, L.; Čubrilo, S.; Vlahovicek, G.M.; Bujnicki, J. Virtual screening and experimental verification to identify potential inhibitors of the ErmC methyl-transferase responsible for bacterial resistance against macrolide antibiotics. ChemMedChem 2008, 3, 316–322. [Google Scholar] [CrossRef]
- Hansen, L.H.; Lobedanz, S.; Douthwaite, S.; Arar, K.; Wengel, J.; Kirpekar, F.; Vester, B. Minimal substrate features for Erm methyltransferases defined by using a combinatorial oligonu-cleotide library. Chembiochem 2011, 12, 610–614. [Google Scholar] [CrossRef]
- Schluckebier, G.; Zhong, P.; Stewart, K.D.; Kavanaugh, T.J.; Abad-Zapatero, C. The 2.2 Å structure of the rRNA methyltransferase ErmC′ and its complexes with cofactor and cofactor analogs: Implications for the reaction mechanism. J. Mol. Biol. 1999, 289, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Dirk, L.M.A.; Yesselman, J.D.; Nimtz, J.S.; Adhikari, U.; Mehl, R.A.; Scheiner, S.; Houtz, R.L.; Al-Hashimi, H.M.; Trievel, R.C. Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J. Am. Chem. Soc. 2013, 135, 15536–15548. [Google Scholar] [CrossRef]
- Maravic, G.; Bujnicki, J.M.; Feder, M.; Pongor, S.; Flögel, M. Alanine-scanning mutagenesis of the predicted rRNA-binding domain of ErmC’ redefines the sub-strate-binding site and suggests a model for protein-RNA interactions. Nucleic Acids Res. 2003, 31, 4941–4949. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, H.; Jin, H.J. Phylogenetic analysis of rRNA methyltransferases, Erm and KsgA, as related to anti-biotic resistance. FEMS Microbiol. Lett. 2010, 309, 151–162. [Google Scholar] [PubMed]
- Bhujbalrao, R.; Anand, R. Deciphering Determinants in Ribosomal Methyltransferases That Confer Antimicrobial Resistance. J. Am. Chem. Soc. 2018, 141, 1425–1429. [Google Scholar] [CrossRef]
- Malone, T.; Blumenthal, R.M.; Cheng, X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.A.; Lyras, D.; Polekhina, G.; Koutsis, K.; Parker, M.W.; Rood, J.I. Identification of Essential Residues in the Erm(B) rRNA Methyltransferase of Clostridium perfringens. Antimicrob. Agents Chemother. 2002, 46, 1253–1261. [Google Scholar] [CrossRef]
- Maravic, G.; Feder, M.; Pongor, S.; Flögel, M.; Bujnicki, J.M. Mutational analysis defines the roles of conserved amino acid residues in the predicted catalytic pocket of the rRNA:m6A methyltransferase ErmC’. J. Mol. Biol. 2003, 332, 99–109. [Google Scholar] [CrossRef]
- Zalacain, M.; Cundliffe, E. Methylation of 23S rRNA caused by tlrA (ermSF), a tylosin resistance determinant from Streptomyces fradiae. J. Bacteriol. 1989, 171, 4254–4260. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Hara, O.; Hutchinson, C.R. Cloning of midecamycin (MLS)-resistance genes from Streptomyces mycarofaciens, Streptomyces lividans and Streptomyces coelicolor A3(2). J. Antibiot. (Tokyo) 1990, 43, 977–991. [Google Scholar] [CrossRef]
- Le, T.; Lee, H.J.; Jin, H.J. Recognition Site Generated by Natural Changes in Erm Proteins Leads to Unexpectedly High Susceptibility to Chymotrypsin. ACS Omega 2017, 2, 8129–8140. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Nakase, K.; Nakaminami, H.; Wajima, T.; Hayashi, N.; Noguchi, N. Transferable Multidrug-Resistance Plasmid Carrying a Novel Macrolide-Clindamycin Resistance Gene, erm(50), in Cutibacterium acnes. Antimicrob. Agents Chemother. 2019, 64. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.; Giguère, S.; Slovis, N.M.; Álvarez-Narváez, S.; Hart, K.A.; Greiter, M.; Morris, E.R.A.; Cohen, N.D. The novel and transferable erm(51) gene confers macrolides, lincosamides and streptogramins B (MLSB) resistance to clonal Rhodococcus equi in the environment. Environ. Microbiol. 2020, 22, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.T.; Jakobsen, L.; Buriánková, K.; Doucet-Populaire, F.; Pernode, J.-L.; Douthwaite, S. Methyltransferase Erm(37) slips on rRNA to confer atypical resistance in Mycobacterium tubercu-losis. J. Biol. Chem. 2005, 280, 38942–38947. [Google Scholar] [CrossRef]
- Madsen, C.T.; Jakobsen, L.; Douthwaite, S. Mycobacterium smegmatis Erm(38) Is a Reluctant Dimethyltransferase. Antimicrob. Agents Chemother. 2005, 49, 3803–3809. [Google Scholar] [CrossRef]
- Gandecha, A.R.; Cundliffe, E. Molecular analysis of tlrD, an MLS resistance determinant from the tylosin producer, Streptomyces fradiae. Gene 1996, 180, 173–176. [Google Scholar] [CrossRef]
- Jenkins, G.; Cundliffe, E. Cloning and characterization of two genes from Streptomyces lividans that confer induc-ible resistance to lincomycin and macrolide antibiotics. Gene 1991, 108, 55–62. [Google Scholar] [CrossRef]
- Zhang, H.-Z.; Schmidt, H.; Piepersberg, W. Molecular cloning and characterization of two lincomycin-resistance genes, ImrA and ImrB, from Streptomyces lincolnensls 78–11. Mol. Microbiol. 1992, 6, 2147–2157. [Google Scholar] [CrossRef]
- Almutairi, M.M.; Park, S.R.; Rose, S.; Hansen, D.A.; Vazquez-Laslop, N.; Douthwaite, S.; Sherman, D.H.; Mankin, A.S. Resistance to ketolide antibiotics by coordinated expression of rRNA methyltransferases in a bacterial producer of natural ketolides. Proc. Natl. Acad. Sci. USA 2015, 112, 12956–12961. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Jin, H.J.; Yang, Y.D. Purification and Biochemical Characterization of the ErmSF Macrolide-Lincosamide-Streptogramin B Resistance Factor Protein Expressed as a Hexahistidine-Tagged Protein in Escherichia coli. Protein Expr. Purif. 2002, 25, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kovalic, D.; Giannattasio, R.B.; Jin, H.J.; Weisblum, B. 23S rRNA domain V, a fragment that can be specifically methylated in vitro by the ErmSF (TlrA) me-thyltransferase. J. Bacteriol. 1994, 176, 6992–6998. [Google Scholar] [CrossRef] [PubMed]
- Vester, B.; Douthwaite, S. Domain V of 23S rRNA contains all the structural elements necessary for recognition by the ErmE methyltransferase. J. Bacteriol. 1994, 176, 6999–7004. [Google Scholar] [CrossRef]
- Tu, C.; Tropea, J.E.; Austin, B.P.; Court, D.L.; Waugh, D.S.; Ji, X. Structural Basis for Binding of RNA and Cofactor by a KsgA Methyltransferase. Structure 2009, 17, 374–385. [Google Scholar] [CrossRef]
- Denoya, C.; Dubnau, D. Mono- and dimethylating activities and kinetic studies of the ermC 23 S rRNA methyl-transferase. J. Biol. Chem. 1989, 264, 2615–2624. [Google Scholar] [CrossRef]
- Grove, T.L.; Benner, J.S.; Radle, M.I.; Ahlum, J.H.; Landgraf, B.J.; Krebs, C.; Booker, S.J. A Radically Different Mechanism for S-Adenosylmethionine-Dependent Methyltransferases. Science 2011, 332, 604–607. [Google Scholar] [CrossRef]
- Goedecke, K.; Pignot, M.; Goody, R.; Scheidig, A.J.; Weinhold, E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat. Genet. 2001, 8, 121–125. [Google Scholar] [CrossRef]
- Christian, T.; Evilia, C.; Hou, Y.M. Catalysis by the second class of tRNA(m1G37) methyl transferase requires a conserved proline. Biochemistry 2006, 45, 7463–7473. [Google Scholar] [CrossRef]
- Järvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The new (dis)order in RNA regulation. Cell Commun. Signal. 2016, 14, 1–22. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Van Buul, C.; Van Knippenberg, P. Nucleotide sequence of the ksgA gene of Escherichia coli: Comparison of methyltransferases effecting dimethylation of adenosine in ribosomal RNA. Gene 1985, 38, 65–72. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, Y.I.; Jin, H.J. Plausible Minimal Substrate for Erm Protein. Antimicrob. Agents Chemother. 2020, 64, 45. [Google Scholar] [CrossRef]
- Champney, W.S.; Chittum, H.S.; Tober, C.L. A 50S Ribosomal Subunit Precursor Particle Is a Substrate for the ErmC Methyltransferase in Staphylococcus aureus Cells. Curr. Microbiol. 2003, 46, 453–460. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Zhong, P.; Pratt, S.D.; Edalji, R.P.; A Walter, K.; Holzman, T.F.; Shivakumar, A.G.; Katz, L. Substrate requirements for ErmC’ methyltransferase activity. J. Bacteriol. 1995, 177, 4327–4332. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Jhang, S.T.; Jin, H.J. Potential Target Site for Inhibitors in MLSB Antibiotic Resistance. Antibiotics 2021, 10, 264. https://doi.org/10.3390/antibiotics10030264
Lee HJ, Jhang ST, Jin HJ. Potential Target Site for Inhibitors in MLSB Antibiotic Resistance. Antibiotics. 2021; 10(3):264. https://doi.org/10.3390/antibiotics10030264
Chicago/Turabian StyleLee, Hak Jin, Seong Tae Jhang, and Hyung Jong Jin. 2021. "Potential Target Site for Inhibitors in MLSB Antibiotic Resistance" Antibiotics 10, no. 3: 264. https://doi.org/10.3390/antibiotics10030264
APA StyleLee, H. J., Jhang, S. T., & Jin, H. J. (2021). Potential Target Site for Inhibitors in MLSB Antibiotic Resistance. Antibiotics, 10(3), 264. https://doi.org/10.3390/antibiotics10030264