
Predicting Antimicrobial Activity at the Target Site: Pharmacokinetic/Pharmacodynamic Indices versus Time–Kill Approaches


Abstract
1. Introduction
2. PK/PD Index Targets and PTA Analyses
2.1. Selecting PK/PD Targets and Performing PTA Analyses
2.2. Influence of the Shape of the PK Profile and Study Design
2.3. Plasma PK and Infection Site PD
2.4. Informative Value of PD Endpoints
2.5. Reliance on the MIC
3. Time–Kill Curve Approaches
3.1. Time-Course of Effect
3.2. In Vitro Time–Kill Experiments
3.2.1. Simulating Target Site PK
3.2.2. Translational Capacity of In Vitro Environment
3.3. In Vivo Time–Kill Experiments
3.4. PK/PD Modelling of Time–Kill Data
3.4.1. Simulating Target Site PK/PD
3.4.2. Translational PK/PD Modelling
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-resistant Bacterial Infections, Including Tuberculosis; World Health Organization: Geneva, Switzerland, 2017; Available online: https://apps.who.int/iris/handle/10665/311820 (accessed on 27 October 2021).
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Review of Antimicrobial Resistance: London, UK, 2016; Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 27 October 2021).
- European Centre for Disease Prevention and Control; European Medicines Agency. The bacterial challenge: Time to react. In A Call to Narrow the Gap between Multidrug-Resistant Bacteria in the EU and Development of New Antibacterial Agents; European Centre for Disease Prevention and Control: Stockholm, Sweden, 2009; Available online: https://www.ecdc.europa.eu/en/publications-data/ecdcemea-joint-technical-report-bacterial-challenge-time-react (accessed on 27 October 2021).
- World Health Organization. 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/publications/i/item/9789240021303 (accessed on 24 November 2021).
- Liu, P.; Müller, M.; Derendorf, H. Rational dosing of antibiotics: The use of plasma concentrations versus tissue concentrations. Int. J. Antimicrob. Agents 2002, 19, 285–290. [Google Scholar] [CrossRef]
- Lonsdale, D.O.; Udy, A.A.; Roberts, J.A.; Lipman, J. Antibacterial therapeutic drug monitoring in cerebrospinal fluid: Difficulty in achieving adequate drug concentrations. J. Neurosurg. 2013, 118, 297–301. [Google Scholar] [CrossRef]
- Zeitlinger, M.A.; Dehghanyar, P.; Mayer, B.X.; Schenk, B.S.; Neckel, U.; Heinz, G.; Georgopoulos, A.; Müller, M.; Joukhadar, C. Relevance of Soft-Tissue Penetration by Levofloxacin for Target Site Bacterial Killing in Patients with Sepsis. Antimicrob. Agents Chemother. 2003, 47, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Joukhadar, C.; Frossard, M.; Mayer, B.X.; Brunner, M.; Klein, N.; Siostrzonek, P.; Eichler, H.G.; Müller, M. Impaired target site penetration of beta-lactams may account for therapeutic failure in patients with septic shock. Crit. Care Med. 2001, 29, 385–391. [Google Scholar] [CrossRef]
- Joukhadar, C.; Klein, N.; Mayer, B.X.; Kreischitz, N.; Delle-Karth, G.; Palkovits, P.; Heinz, G.; Müller, M. Plasma and tissue pharmacokinetics of cefpirome in patients with sepsis. Crit. Care Med. 2002, 30, 1478–1482. [Google Scholar] [CrossRef]
- Sauermann, R.; Delle-Karth, G.; Marsik, C.; Steiner, I.; Zeitlinger, M.; Mayer-Helm, B.X.; Georgopoulos, A.; Müller, M.; Joukhadar, C. Pharmacokinetics and pharmacodynamics of cefpirome in subcutaneous adipose tissue of septic patients. Antimicrob. Agents Chemother. 2005, 49, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; Plock, N.; Dehghanyar, P.; Joukhadar, C.; Kloft, C. Pharmacokinetics of unbound linezolid in plasma and tissue interstitium of critically ill patients after multiple dosing using microdialysis. Antimicrob. Agents Chemother. 2006, 50, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Sinnollareddy, M.G.; Roberts, M.S.; Williams, P.; Peake, S.L.; Lipman, J.; Roberts, J.A. Plasma and interstitial fluid population pharmacokinetics of vancomycin in critically ill patients with sepsis. Int. J. Antimicrob. Agents 2019, 53, 137–142. [Google Scholar] [CrossRef]
- Minichmayr, I.K.; Schaeftlein, A.; Kuti, J.L.; Zeitlinger, M.; Kloft, C. Clinical Determinants of Target Non-Attainment of Linezolid in Plasma and Interstitial Space Fluid: A Pooled Population Pharmacokinetic Analysis with Focus on Critically Ill Patients. Clin. Pharmacokinet. 2017, 56, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Sinnollareddy, M.G.; Roberts, M.S.; Lipman, J.; Lassig-Smith, M.; Starr, T.; Robertson, T.; Peake, S.L.; Roberts, J.A. In Vivo Microdialysis To Determine Subcutaneous Interstitial Fluid Penetration and Pharmacokinetics of Fluconazole in Intensive Care Unit Patients with Sepsis. Antimicrob. Agents Chemother. 2016, 60, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Schmidtko, A.; Bräutigam, L.; Kirschbaum, A.; Geisslinger, G.; Lötsch, J. Tissue distribution of imipenem in critically ill patients. Clin. Pharmacol. Ther. 2002, 71, 325–333. [Google Scholar] [CrossRef]
- Dahyot-Fizelier, C.; Lefeuvre, S.; Laksiri, L.; Marchand, S.; Sawchuk, R.J.; Couet, W.; Mimoz, O. Kinetics of imipenem distribution into the peritoneal fluid of patients with severe peritonitis studied by microdialysis. Clin. Pharmacokinet. 2010, 49, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, U.M.; Brunner, M.; Schmid, R.; Müller, M. Soft tissue concentrations of ciprofloxacin in obese and lean subjects following weight-adjusted dosing. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, L.; Simon, P.; Busse, D.; Petroff, D.; Dorn, C.; Huisinga, W.; Dietrich, A.; Zeitlinger, M.; Wrigge, H.; Kloft, C. Risk of target non-attainment in obese compared to non-obese patients in calculated linezolid therapy. Clin. Microbiol. Infect. 2020, 26, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Wittau, M.; Paschke, S.; Kurlbaum, M.; Scheele, J.; Ly, N.S.; Hemper, E.; Kornmann, M.; Henne-Bruns, D.; Bulitta, J.B. Population Pharmacokinetics and Target Attainment of Ertapenem in Plasma and Tissue Assessed via Microdialysis in Morbidly Obese Patients after Laparoscopic Visceral Surgery. Antimicrob. Agents Chemother. 2017, 61, e00952-16. [Google Scholar] [CrossRef]
- Wittau, M.; Scheele, J.; Kurlbaum, M.; Brockschmidt, C.; Wolf, A.M.; Hemper, E.; Henne-Bruns, D.; Bulitta, J.B. Population Pharmacokinetics and Target Attainment of Meropenem in Plasma and Tissue of Morbidly Obese Patients after Laparoscopic Intraperitoneal Surgery. Antimicrob. Agents Chemother. 2015, 59, 6241–6247. [Google Scholar] [CrossRef]
- Brill, M.J.E.; Houwink, A.P.I.; Schmidt, S.; Van Dongen, E.P.A.; Hazebroek, E.J.; van Ramshorst, B.; Deneer, V.H.; Mouton, J.W.; Knibbe, C.A.J. Reduced subcutaneous tissue distribution of cefazolin in morbidly obese versus non-obese patients determined using clinical microdialysis. J. Antimicrob. Chemother. 2014, 69, 715–723. [Google Scholar] [CrossRef]
- Toma, O.; Suntrup, P.; Stefanescu, A.; London, A.; Mutch, M.; Kharasch, E. Pharmacokinetics and tissue penetration of cefoxitin in obesity: Implications for risk of surgical site infection. Anesth. Analg. 2011, 113, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.; Schmidt, S.; Rout, W.R.; Ben-David, K.; Burkhardt, O.; Derendorf, H. Soft tissue penetration of cefuroxime determined by clinical microdialysis in morbidly obese patients undergoing abdominal surgery. Int. J. Antimicrob. Agents 2009, 34, 231–235. [Google Scholar] [CrossRef]
- Skhirtladze, K.; Hutschala, D.; Fleck, T.; Thalhammer, F.; Ehrlich, M.; Vukovich, T.; Müller, M.; Tschernko, E.M. Impaired target site penetration of vancomycin in diabetic patients following cardiac surgery. Antimicrob. Agents Chemother. 2006, 50, 1372–1375. [Google Scholar] [CrossRef] [PubMed]
- Joukhadar, C.; Stass, H.; Müller-Zellenberg, U.; Lackner, E.; Kovar, F.; Minar, E.; Müller, M. Penetration of moxifloxacin into healthy and inflamed subcutaneous adipose tissues in humans. Antimicrob. Agents Chemother. 2003, 47, 3099–3103. [Google Scholar] [CrossRef]
- Kim, A.; Suecof, L.A.; Sutherland, C.A.; Gao, L.; Kuti, J.L.; Nicolau, D.P. In vivo microdialysis study of the penetration of daptomycin into soft tissues in diabetic versus healthy volunteers. Antimicrob. Agents Chemother. 2008, 52, 3941–3946. [Google Scholar] [CrossRef] [PubMed]
- Brunner, M.; Pernerstorfer, T.; Mayer, B.X.; Eichler, H.G.; Müller, M. Surgery and intensive care procedures affect the target site distribution of piperacillin. Crit. Care Med. 2000, 28, 1754–1759. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Antibacterial Therapies for Patients with an Unmet Medical Need for the Treatment of Serious Bacterial Diseases. Guidance for Industry. Available online: https://www.fda.gov/media/86250/download (accessed on 27 October 2021).
- European Medicines Agency. Guideline on the Use of Pharmacokinetics and Pharmacodynamics in the Development of Antimicrobial Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-pharmacokinetics-pharmacodynamics-development-antimicrobial-medicinal-products_en.pdf (accessed on 27 October 2021).
- Gaynes, R.; Edwards, J.R. Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis. 2005, 41, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Stamey, T.A.; Fair, W.R.; Timothy, M.M.; Millar, M.A.; Mihara, G.; Lowery, Y.C. Serum versus Urinary Antimicrobial Concentrations in Cure of Urinary-Tract Infections. N. Engl. J. Med. 1974, 291, 1159–1163. [Google Scholar] [CrossRef]
- Frimodt-Møller, N. Correlation between pharmacokinetic/pharmacodynamic parameters and efficacy for antibiotics in the treatment of urinary tract infection. Int. J. Antimicrob. Agents 2002, 19, 546–553. [Google Scholar] [CrossRef]
- Müller, M. Microdialysis in clinical drug delivery studies. Adv. Drug Deliv. Rev. 2000, 45, 255–269. [Google Scholar] [CrossRef]
- de la Peña, A.; Liu, P.; Derendorf, H. Microdialysis in peripheral tissues. Adv. Drug Deliv. Rev. 2000, 45, 189–216. [Google Scholar] [CrossRef]
- Joukhadar, C.; Müller, M. Microdialysis: Current applications in clinical pharmacokinetic studies and its potential role in the future. Clin. Pharmacokinet. 2005, 44, 895–913. [Google Scholar] [CrossRef]
- Joukhadar, C.; Derendorf, H.; Müller, M. Microdialysis. A novel tool for clinical studies of anti-infective agents. Eur. J. Clin. Pharmacol. 2001, 57, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Rodvold, K.A.; George, J.M.; Yoo, L. Penetration of anti-infective agents into pulmonary epithelial lining fluid: Focus on antibacterial agents. Clin. Pharmacokinet. 2011, 50, 637–664. [Google Scholar] [CrossRef] [PubMed]
- Mouton, J.W.; Theuretzbacher, U.; Craig, W.A.; Tulkens, P.M.; Derendorf, H.; Cars, O. Tissue concentrations: Do we ever learn? J. Antimicrob. Chemother. 2008, 61, 235–237. [Google Scholar] [CrossRef]
- Andes, D.; Craig, W.A. Animal model pharmacokinetics and pharmacodynamics: A critical review. Int. J. Antimicrob. Agents 2002, 19, 261–268. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Hope, W.W.; Eakin, A.E.; Guina, T.; Tam, V.H.; Louie, A.; Drusano, G.L.; Hoover, J.L. Generating Robust and Informative Nonclinical In Vitro and In Vivo Bacterial Infection Model Efficacy Data to Support Translation to Humans. Antimicrob. Agents Chemother. 2019, 63, e02307-18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Lepak, A.J.; Andes, D.R. Animal models in the pharmacokinetic/pharmacodynamic evaluation of antimicrobial agents. Bioorg. Med. Chem. 2016, 24, 6390–6400. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Bergen, P.J.; Lora-Tamayo, J.; Landersdorfer, C.B.; Li, J. PK/PD models in antibacterial development. Curr. Opin. Microbiol. 2013, 16, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.A. Pharmacokinetic/pharmacodynamic parameters: Rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 1998, 26, 1–10. [Google Scholar] [CrossRef]
- Craig, W.A. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn. Microbiol. Infect. Dis. 1995, 22, 89–96. [Google Scholar] [CrossRef]
- Vogelman, B.; Gudmundsson, S.; Leggett, J.; Turnidge, J.; Ebert, S.; Craig, W.A. Correlation of antimicrobial pharmacokinetic parameters with therapeutic efficacy in an animal model. J. Infect. Dis. 1988, 158, 831–847. [Google Scholar] [CrossRef]
- Ambrose, P.G.; Bhavnani, S.M.; Rubino, C.M.; Louie, A.; Gumbo, T.; Forrest, A.; Drusano, G.L. Pharmacokinetics-Pharmacodynamics of Antimicrobial Therapy: It’s Not Just for Mice Anymore. Clin. Infect. Dis. 2007, 44, 79–86. [Google Scholar] [CrossRef]
- Drusano, G.L.; Preston, S.L.; Hardalo, C.; Hare, R.; Banfield, C.; Andes, D.; Vesga, O.; Craig, W.A. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 2001, 45, 13–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bulik, C.; Bhavnani, S.; Hammel, J.; Forrest, A.; Dudley, M.; Ellis-Grosse, E.; Drusano, G.; Ambrose, P. Relationship between regulatory approval and pharmacokinetic-pharmacodynamic target attainment: Focus on community-and hospital-acquired pneumonia. In Proceedings of the 53rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Denver, CO, USA, 10–13 September 2013. [Google Scholar]
- Nielsen, E.I.; Otto, C.; Friberg, L.E. Pharmacokinetic/Pharmacodynamic (PK/PD) Indices of Antibiotics Predicted by a Semimechanistic PKPD Model: A Step toward Model-Based Dose Optimization. Antimicrob. Agents Chemother. 2011, 55, 4619–4630. [Google Scholar] [CrossRef]
- Dhaese, S.; Heffernan, A.; Liu, D.; Abdul-Aziz, M.H.; Stove, V.; Tam, V.H.; Lipman, J.; Roberts, J.A.; De Waele, J.J. Prolonged Versus Intermittent Infusion of β-Lactam Antibiotics: A Systematic Review and Meta-Regression of Bacterial Killing in Preclinical Infection Models. Clin. Pharmacokinet. 2020, 59, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.U.; Brugger, H.-P.; Feller, C.; Stritzko, T.; Stalder, B. Antibiotic Therapy of Infections Due to Pseudomonas aeruginosa in Normal and Granulocytopenic Mice: Comparison of Murine and Human Pharmacokinetics. J. Infect. Dis. 1986, 153, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J.; Holford, N.H.G. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 303–332. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.A.; Redington, J.; Ebert, S.C. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J. Antimicrob. Chemother. 1991, 27, 29–40. [Google Scholar] [CrossRef]
- Andes, D.; Craig, W.A. In vivo activities of amoxicillin and amoxicillin-clavulanate against Streptococcus pneumoniae: Application to breakpoint determinations. Antimicrob. Agents Chemother. 1998, 42, 2375–2379. [Google Scholar] [CrossRef]
- Crandon, J.L.; Nicolau, D.P. Human simulated studies of aztreonam and aztreonam-avibactam to evaluate activity against challenging gram-negative organisms, including metallo-β-lactamase producers. Antimicrob. Agents Chemother. 2013, 57, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Keel, R.A.; Crandon, J.L.; Nicolau, D.P. Efficacy of human simulated exposures of ceftaroline administered at 600 milligrams every 12 hours against phenotypically diverse Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 2011, 55, 4028–4032. [Google Scholar] [CrossRef][Green Version]
- Blatter, M.; Fluckiger, U.; Entenza, J.; Glauser, M.P.; Francioli, P. Simulated human serum profiles of one daily dose of ceftriaxone plus netilmicin in treatment of experimental streptococcal endocarditis. Antimicrob. Agents Chemother. 1993, 37, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Piroth, L.; Martin, L.; Coulon, A.; Lequeu, C.; Duong, M.; Buisson, M.; Portier, H.; Chavanet, P. Development of a new experimental model of penicillin-resistant Streptococcus pneumoniae pneumonia and amoxicillin treatment by reproducing human pharmacokinetics. Antimicrob. Agents Chemother. 1999, 43, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Robaux, M.A.; Dube, L.; Caillon, J.; Bugnon, D.; Kergueris, M.F.; Navas, D.; Le Conte, P.; Baron, D.; Potel, G. In vivo efficacy of continuous infusion versus intermittent dosing of ceftazidime alone or in combination with amikacin relative to human kinetic profiles in a Pseudomonas aeruginosa rabbit endocarditis model. J. Antimicrob. Chemother. 2001, 47, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Sou, T.; Hansen, J.; Liepinsh, E.; Backlund, M.; Ercan, O.; Grinberga, S.; Cao, S.; Giachou, P.; Petersson, A.; Tomczak, M.; et al. Model-Informed Drug Development for Antimicrobials: Translational PK and PK/PD Modeling to Predict an Efficacious Human Dose for Apramycin. Clin. Pharmacol. Ther. 2021, 109, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersson, A.N.; David-Pierson, P.; Parrott, N.J.; Kuhlmann, O.; Lave, T.; Friberg, L.E.; Nielsen, E.I. Simulation-Based Evaluation of PK/PD Indices for Meropenem Across Patient Groups and Experimental Designs. Pharm. Res. 2016, 33, 1115–1125. [Google Scholar] [CrossRef]
- Jumbe, N.; Louie, A.; Leary, R.; Liu, W.; Deziel, M.R.; Tam, V.H.; Bachhawat, R.; Freeman, C.; Kahn, J.B.; Bush, K.; et al. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 2003, 112, 275–285. [Google Scholar] [CrossRef]
- Udekwu, K.I.; Parrish, N.; Ankomah, P.; Baquero, F.; Levin, B.R. Functional relationship between bacterial cell density and the efficacy of antibiotics. J. Antimicrob. Chemother. 2009, 63, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Maglio, D.; Capitano, B.; Banevicius, M.A.; Geng, Q.; Nightingale, C.H.; Nicolau, D.P. Differential Efficacy of Clarithromycin in Lung versus Thigh Infection Models. Chemotherapy 2004, 50, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Rodvold, K.A.; Nicolau, D.P.; Lodise, T.P.; Khashab, M.; Noel, G.J.; Kahn, J.B.; Gotfried, M.; Murray, S.A.; Nicholson, S.; Laohavaleeson, S.; et al. Identifying exposure targets for treatment of staphylococcal pneumonia with ceftobiprole. Antimicrob. Agents Chemother. 2009, 53, 3294–3301. [Google Scholar] [CrossRef]
- Trang, M.; Dudley, M.N.; Bhavnani, S.M. Use of Monte Carlo simulation and considerations for PK-PD targets to support antibacterial dose selection. Curr. Opin. Pharmacol. 2017, 36, 107–113. [Google Scholar] [CrossRef]
- Ambrose, P.G. Antibacterial drug development program successes and failures: A pharmacometric explanation. Curr. Opin. Pharmacol. 2017, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, P.G.; Bhavnani, S.M.; Ellis-Grosse, E.J.; Drusano, G.L. Pharmacokinetic-Pharmacodynamic Considerations in the Design of Hospital-Acquired or Ventilator-Associated Bacterial Pneumonia Studies: Look before You Leap! Clin. Infect. Dis. 2010, 51, S103–S110. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L.; Lodise, T.P.; Melnick, D.; Liu, W.; Oliver, A.; Mena, A.; VanScoy, B.; Louie, A. Meropenem penetration into epithelial lining fluid in mice and humans and delineation of exposure targets. Antimicrob. Agents Chemother. 2011, 55, 3406–3412. [Google Scholar] [CrossRef] [PubMed]
- Louie, A.; Fregeau, C.; Liu, W.; Kulawy, R.; Drusano, G.L. Pharmacodynamics of levofloxacin in a murine pneumonia model of Pseudomonas aeruginosa infection: Determination of epithelial lining fluid targets. Antimicrob. Agents Chemother. 2009, 53, 3325–3330. [Google Scholar] [CrossRef] [PubMed]
- Landersdorfer, C.B.; Bulitta, J.B.; Kinzig, M.; Holzgrabe, U.; Sörgel, F. Penetration of antibacterials into bone: Pharmacokinetic, pharmacodynamic and bioanalytical considerations. Clin. Pharmacokinet. 2009, 48, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.H.; Louie, A.; Deziel, M.R.; Liu, W.; Drusano, G.L. The relationship between quinolone exposures and resistance amplification is characterized by an inverted U: A new paradigm for optimizing pharmacodynamics to counterselect resistance. Antimicrob. Agents Chemother. 2007, 51, 744–747. [Google Scholar] [CrossRef]
- Tam, V.H.; Louie, A.; Deziel, M.R.; Liu, W.; Leary, R.; Drusano, G.L. Bacterial-Population Responses to Drug-Selective Pressure: Examination of Garenoxacin’s Effect on Pseudomonas aeruginosa. J. Infect. Dis. 2005, 192, 420–428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tam, V.H.; Louie, A.; Fritsche, T.R.; Deziel, M.; Liu, W.; Brown, D.L.; Deshpande, L.; Leary, R.; Jones, R.N.; Drusano, G.L. Impact of drug-exposure intensity and duration of therapy on the emergence of Staphylococcus aureus resistance to a quinolone antimicrobial. J. Infect. Dis. 2007, 195, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Louie, A.; Maynard, M.; Duncanson, B.; Nole, J.; Vicchiarelli, M.; Drusano, G.L. Determination of the Dynamically Linked Indices of Fosfomycin for Pseudomonas aeruginosa in the Hollow Fiber Infection Model. Antimicrob. Agents Chemother. 2018, 62, e02627-17. [Google Scholar] [CrossRef]
- Cheah, S.E.; Li, J.; Tsuji, B.T.; Forrest, A.; Bulitta, J.B.; Nation, R.L. Colistin and Polymyxin B Dosage Regimens against Acinetobacter baumannii: Differences in Activity and the Emergence of Resistance. Antimicrob. Agents Chemother. 2016, 60, 3921–3933. [Google Scholar] [CrossRef]
- Sandoval-Motta, S.; Aldana, M. Adaptive resistance to antibiotics in bacteria: A systems biology perspective. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; He, X.; Zou, X.; Wang, G.; Li, S.; Wu, Y. Gradual increase in antibiotic concentration affects persistence of Klebsiella pneumoniae. J. Antimicrob. Chemother. 2015, 70, 3267–3272. [Google Scholar] [CrossRef][Green Version]
- Mouton, J.W.; Meletiadis, J.; Voss, A.; Turnidge, J. Variation of MIC measurements: The contribution of strain and laboratory variability to measurement precision. J. Antimicrob. Chemother. 2018, 73, 2374–2379. [Google Scholar] [CrossRef]
- Mouton, J.W.; Muller, A.E.; Canton, R.; Giske, C.G.; Kahlmeter, G.; Turnidge, J. MIC-based dose adjustment: Facts and fables. J. Antimicrob. Chemother. 2017, 73, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Friberg, L.E. Pivotal Role of Translation in Anti-Infective Development. Clin. Pharm. Therap. 2021, 109, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Ly, N.; Diep, J.K.; Rao, G.G. Moving From Point-Based Analysis to Systems-Based Modeling: Integration of Knowledge to Address Antimicrobial Resistance Against MDR Bacteria. Clin. Pharmacol. Ther. 2021, 110, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.G.; Landersdorfer, C.B. Antibiotic pharmacokinetic/pharmacodynamic modelling: MIC, pharmacodynamic indices and beyond. Int. J. Antimicrob. Agents 2021, 58, 106368. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.I.; Friberg, L.E. Pharmacokinetic-Pharmacodynamic Modeling of Antibacterial Drugs. Pharmacol. Rev. 2013, 65, 1053–1090. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; de la Peña, A.; Derendorf, H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: Kill curves versus MIC. Antimicrob. Agents Chemother. 2004, 48, 369–377. [Google Scholar] [CrossRef]
- Rayner, C.R.; Smith, P.F.; Andes, D.; Andrews, K.; Derendorf, H.; Friberg, L.E.; Hanna, D.; Lepak, A.; Mills, E.; Polasek, T.M.; et al. Model-Informed Drug Development for Anti-Infectives: State of the Art and Future. Clin. Pharmacol. Ther. 2021, 109, 867–891. [Google Scholar] [CrossRef] [PubMed]
- Gloede, J.; Scheerans, C.; Derendorf, H.; Kloft, C. In vitro pharmacodynamic models to determine the effect of antibacterial drugs. J. Antimicrob. Chemother. 2010, 65, 186–201. [Google Scholar] [CrossRef]
- Brunner, M.; Hollenstein, U.; Delacher, S.; Jäger, D.; Schmid, R.; Lackner, E.; Georgopoulos, A.; Eichler, H.G.; Müller, M. Distribution and antimicrobial activity of ciprofloxacin in human soft tissues. Antimicrob. Agents Chemother. 1999, 43, 1307–1309. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frossard, M.; Joukhadar, C.; Erovic, B.M.; Dittrich, P.; Mrass, P.E.; Van Houte, M.; Burgmann, H.; Georgopoulos, A.; Müller, M. Distribution and antimicrobial activity of fosfomycin in the interstitial fluid of human soft tissues. Antimicrob. Agents Chemother. 2000, 44, 2728–2732. [Google Scholar] [CrossRef]
- Zeitlinger, M.A.; Marsik, C.; Georgopoulos, A.; Müller, M.; Heinz, G.; Joukhadar, C. Target site bacterial killing of cefpirome and fosfomycin in critically ill patients. Int. J. Antimicrob. Agents 2003, 21, 562–567. [Google Scholar] [CrossRef]
- Joukhadar, C.; Klein, N.; Dittrich, P.; Zeitlinger, M.; Geppert, A.; Skhirtladze, K.; Frossard, M.; Heinz, G.; Müller, M. Target site penetration of fosfomycin in critically ill patients. J. Antimicrob. Chemother. 2003, 51, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Sauermann, R.; Zeitlinger, M.; Erovic, B.M.; Marsik, C.; Georgopoulos, A.; Müller, M.; Brunner, M.; Joukhadar, C. Pharmacodynamics of piperacillin in severely ill patients evaluated by using a PK/PD model. Int. J. Antimicrob. Agents 2003, 22, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Zeitlinger, M.A.; Erovic, B.M.; Sauermann, R.; Georgopoulos, A.; Müller, M.; Joukhadar, C. Plasma concentrations might lead to overestimation of target site activity of piperacillin in patients with sepsis. J. Antimicrob. Chemother. 2005, 56, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Joukhadar, C.; Dehghanyar, P.; Traunmüller, F.; Sauermann, R.; Mayer-Helm, B.; Georgopoulos, A.; Müller, M. Increase of microcirculatory blood flow enhances penetration of ciprofloxacin into soft tissue. Antimicrob. Agents Chemother. 2005, 49, 4149–4153. [Google Scholar] [CrossRef] [PubMed]
- Delacher, S.; Derendorf, H.; Hollenstein, U.; Brunner, M.; Joukhadar, C.; Hofmann, S.; Georgopoulos, A.; Eichler, H.G.; Müller, M. A combined in vivo pharmacokinetic-in vitro pharmacodynamic approach to simulate target site pharmacodynamics of antibiotics in humans. J. Antimicrob. Chemother. 2000, 46, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Harigaya, Y.; Bulitta, J.B.; Forrest, A.; Sakoulas, G.; Lesse, A.J.; Mylotte, J.M.; Tsuji, B.T. Pharmacodynamics of vancomycin at simulated epithelial lining fluid concentrations against methicillin-resistant Staphylococcus aureus (MRSA): Implications for dosing in MRSA pneumonia. Antimicrob. Agents Chemother. 2009, 53, 3894–3901. [Google Scholar] [CrossRef] [PubMed]
- Satta, G.; Cornaglia, G.; Foddis, G.; Pompei, R. Evaluation of ceftriaxone and other antibiotics against Escherichia coli, Pseudomonas aeruginosa, and Streptococcus pneumoniae under in vitro conditions simulating those of serious infections. Antimicrob. Agents Chemother. 1988, 32, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Fan, H.W.; Sutherland, C.; DeRyke, A.C.; Nicolau, D.P. Antibacterial effects of moxifloxacin and levofloxacin simulating epithelial lining fluid concentrations against community-acquired methicillin-resistant Staphylococcus aureus. Drugs R D 2007, 8, 69–77. [Google Scholar] [CrossRef]
- Deryke, C.A.; Du, X.; Nicolau, D.P. Evaluation of bacterial kill when modelling the bronchopulmonary pharmacokinetic profile of moxifloxacin and levofloxacin against parC-containing isolates of Streptococcus pneumoniae. J. Antimicrob. Chemother. 2006, 58, 601–609. [Google Scholar] [CrossRef][Green Version]
- Florea, N.R.; Tessier, P.R.; Zhang, C.; Nightingale, C.H.; Nicolau, D.P. Pharmacodynamics of moxifloxacin and levofloxacin at simulated epithelial lining fluid drug concentrations against Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2004, 48, 1215–1221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jain, J.G.; Housman, S.T.; Nicolau, D.P. Humanized tissue pharmacodynamics of cefazolin against commonly isolated pathogens in skin and skin structure infections. J. Antimicrob. Chemother. 2014, 69, 2443–2447. [Google Scholar] [CrossRef] [PubMed]
- MacVane, S.H.; So, W.; Nicolau, D.P.; Kuti, J.L. In vitro activity of human-simulated epithelial lining fluid exposures of ceftaroline, ceftriaxone, and vancomycin against methicillin-susceptible and -resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 7520–7526. [Google Scholar] [CrossRef]
- So, W.; Crandon, J.L.; Hamada, Y.; Nicolau, D.P. Antibacterial activity of achievable epithelial lining fluid exposures of Amikacin Inhale with or without meropenem. J. Antimicrob. Chemother. 2016, 71, 428–437. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hamada, Y.; Kuti, J.L.; Nicolau, D.P. In Vitro Pharmacodynamics of Vancomycin against Methicillin-Susceptible and -Resistant Staphylococcus aureus: Considering the Variability in Observed Tissue Exposure. Antimicrob. Agents Chemother. 2015, 60, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, I.M.; Grupper, M.; Nicolau, D.P. Anti-staphylococcal activity resulting from epithelial lining fluid (ELF) concentrations of amikacin inhale administered via the pulmonary drug delivery system. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, I.M.; Grupper, M.; Nicolau, D.P. Antibacterial activity of human simulated epithelial lining fluid concentrations of amikacin inhale alone and in combination with meropenem against Acinetobacter baumannii. Infect. Dis. 2017, 49, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Almarzoky Abuhussain, S.S.; Kuti, J.L.; Nicolau, D.P. Antibacterial Activity of Human Simulated Epithelial Lining Fluid Concentrations of Ceftazidime-Avibactam Alone or in Combination with Amikacin Inhale (BAY41-6551) against Carbapenem-Resistant Pseudomonas aeruginosa and Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2018, 62, e00113-18. [Google Scholar] [CrossRef] [PubMed]
- Noreddin, A.M.; Roberts, D.; Nichol, K.; Wierzbowski, A.; Hoban, D.J.; Zhanel, G.G. Pharmacodynamic modeling of clarithromycin against macrolide-resistant [PCR-positive mef(A) or erm(B)] Streptococcus pneumoniae simulating clinically achievable serum and epithelial lining fluid free-drug concentrations. Antimicrob. Agents Chemother. 2002, 46, 4029–4034. [Google Scholar] [CrossRef]
- Sevillano, D.; Alou, L.; Aguilar, L.; Echevarría, O.; Giménez, M.J.; Prieto, J. Azithromycin iv pharmacodynamic parameters predicting Streptococcus pneumoniae killing in epithelial lining fluid versus serum: An in vitro pharmacodynamic simulation. J. Antimicrob. Chemother. 2006, 57, 1128–1133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alou, L.; Giménez, M.J.; Sevillano, D.; Aguilar, L.; Cafini, F.; Echeverría, O.; Pérez-Trallero, E.; Prieto, J. A pharmacodynamic approach to antimicrobial activity in serum and epithelial lining fluid against in vivo-selected Streptococcus pneumoniae mutants and association with clinical failure in pneumonia. J. Antimicrob. Chemother. 2006, 58, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, N.; Sevillano, D.; Alou, L.; Cafini, F.; Gimenez, M.J.; Gomez-Lus, M.L.; Prieto, J.; Aguilar, L. Influence of the MBC/MIC ratio on the antibacterial activity of vancomycin versus linezolid against methicillin-resistant Staphylococcus aureus isolates in a pharmacodynamic model simulating serum and soft tissue interstitial fluid concentrations reported in diabetic patients. J. Antimicrob. Chemother. 2013, 68, 2291–2295. [Google Scholar] [CrossRef] [PubMed]
- Sime, F.B.; Johnson, A.; Whalley, S.; Santoyo-Castelazo, A.; Montgomery, A.B.; Walters, K.A.; Lipman, J.; Hope, W.W.; Roberts, J.A. Pharmacodynamics of Aerosolized Fosfomycin and Amikacin against Resistant Clinical Isolates of Pseudomonas aeruginosa and Klebsiella pneumoniae in a Hollow-Fiber Infection Model: Experimental Basis for Combination Therapy. Antimicrob. Agents Chemother. 2016, 61, e01763-16. [Google Scholar] [CrossRef]
- Zhanel, G.G.; DeCorby, M.; Noreddin, A.; Mendoza, C.; Cumming, A.; Nichol, K.; Wierzbowski, A.; Hoban, D.J. Pharmacodynamic activity of azithromycin against macrolide-susceptible and -resistant Streptococcus pneumoniae simulating clinically achievable free serum, epithelial lining fluid and middle ear fluid concentrations. J. Antimicrob. Chemother. 2003, 52, 83–88. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Johanson, C.; Hisanaga, T.; Mendoza, C.; Laing, N.; Noreddin, A.; Wierzbowski, A.; Hoban, D.J. Pharmacodynamic activity of telithromycin against macrolide-susceptible and macrolide-resistant Streptococcus pneumoniae simulating clinically achievable free serum and epithelial lining fluid concentrations. J. Antimicrob. Chemother. 2004, 54, 1072–1077. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhanel, G.G.; Johanson, C.; Laing, N.; Hisanaga, T.; Wierzbowski, A.; Hoban, D.J. Pharmacodynamic activity of telithromycin at simulated clinically achievable free-drug concentrations in serum and epithelial lining fluid against efflux (mefE)-producing macrolide-resistant Streptococcus pneumoniae for which telithromycin MICs vary. Antimicrob. Agents Chemother. 2005, 49, 1943–1948. [Google Scholar] [CrossRef]
- Zinner, S.H.; Husson, M.; Klastersky, J. An artificial capillary in vitro kinetic model of antibiotic bactericidal activity. J. Infect. Dis. 1981, 144, 583–587. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, F.; Pennington, J.H. Bacterial growth in an in vitro system simulating conditions in the urinary bladder. Br. J. Exp. Pathol. 1966, 47, 152–157. [Google Scholar]
- Greenwood, D.; O’Grady, F. An in vitro model of the urinary bladder. J. Antimicrob. Chemother. 1978, 4, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, D. An in-vitro model simulating the hydrokinetic aspects of the treatment of bacterial cystitis. J. Antimicrob. Chemother. 1985, 15, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Abbott, I.J.; Meletiadis, J.; Belghanch, I.; Wijma, R.A.; Kanioura, L.; Roberts, J.A.; Peleg, A.Y.; Mouton, J.W. Fosfomycin efficacy and emergence of resistance among Enterobacteriaceae in an in vitro dynamic bladder infection model. J. Antimicrob. Chemother. 2018, 73, 709–719. [Google Scholar] [CrossRef]
- Payne, C.J.; Thomson, A.H.; Stearns, A.T.; Watson, D.G.; Zhang, T.; Kingsmore, D.B.; Byrne, D.S.; Binning, A.S. Pharmacokinetics and tissue penetration of vancomycin continuous infusion as prophylaxis for vascular surgery. J. Antimicrob. Chemother. 2011, 66, 2624–2627. [Google Scholar] [CrossRef] [PubMed]
- Housman, S.T.; Bhalodi, A.A.; Shepard, A.; Nugent, J.; Nicolau, D.P. Vancomycin Tissue Pharmacokinetics in Patients with Lower-Limb Infections via In Vivo Microdialysis. J. Am. Podiatr. Med. Assoc. 2015, 105, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Kuti, J.L.; Nicolau, D.P. Vancomycin serum concentrations do not adequately predict tissue exposure in diabetic patients with mild to moderate limb infections. J. Antimicrob. Chemother. 2015, 70, 2064–2067. [Google Scholar] [CrossRef]
- Bue, M.; Tøttrup, M.; Hanberg, P.; Langhoff, O.; Birke-Sørensen, H.; Thillemann, T.M.; Andersson, T.L.; Søballe, K. Bone and subcutaneous adipose tissue pharmacokinetics of vancomycin in total knee replacement patients. Acta Orthop. 2018, 89, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Differences between bacteria grown in vitro and in vivo. J. Antimicrob. Chemother. 1985, 15 (Suppl. A), 175–195. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, C.R.; Friedrich, L.V.; Bosso, J.A.; White, R.L. Determination of antibiotic effect in an in vitro pharmacodynamic model: Comparison with an established animal model of infection. Antimicrob. Agents Chemother. 2002, 46, 3574–3579. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, B.V.; Diniz, A.; Palma, E.C.; Buffé, C.; Dalla Costa, T. PK-PD modeling of β-lactam antibiotics: In vitro or in vivo models? J. Antibiot. 2011, 64, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Aulin, L.B.S.; Koumans, C.I.M.; Haakman, Y.; van Os, W.; Kraakman, M.E.M.; Gooskens, J.; Smits, W.K.; Liakopoulos, A.; van Hasselt, J.G.C. Distinct evolution of colistin resistance associated with experimental resistance evolution models in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2020, 76, 533–535. [Google Scholar] [CrossRef]
- Nussbaumer-Pröll, A.; Zeitlinger, M. Use of Supplemented or Human Material to Simulate PD Behavior of Antibiotics at the Target Site In Vitro. Pharmaceutics 2020, 12, 773. [Google Scholar] [CrossRef]
- van Os, W.; Wulkersdorfer, B.; Eberl, S.; Oesterreicher, Z.; Paternostro, R.; Schwabl, P.; Willinger, B.; Weber, M.; Reiberger, T.; Zeitlinger, M. Impact of individual ascitic fluids on bacterial growth and antimicrobial effect of ceftriaxone. In Proceedings of the European Congress of Clinical Microbiology and Infectious Diseases, Online. 9–12 July 2021. Abstract 03105. [Google Scholar]
- Yadav, R.; Bulitta, J.B.; Wang, J.; Nation, R.L.; Landersdorfer, C.B. Evaluation of Pharmacokinetic/Pharmacodynamic Model-Based Optimized Combination Regimens against Multidrug-Resistant Pseudomonas aeruginosa in a Murine Thigh Infection Model by Using Humanized Dosing Schemes. Antimicrob. Agents Chemother. 2017, 61, e01268-17. [Google Scholar] [CrossRef]
- Lin, Y.W.; Zhou, Q.T.; Han, M.L.; Onufrak, N.J.; Chen, K.; Wang, J.; Forrest, A.; Chan, H.K.; Li, J. Mechanism-Based Pharmacokinetic/Pharmacodynamic Modeling of Aerosolized Colistin in a Mouse Lung Infection Model. Antimicrob. Agents Chemother. 2018, 62, e01965-17. [Google Scholar] [CrossRef] [PubMed]
- Lepak, A.J.; Marchillo, K.; Craig, W.A.; Andes, D.R. In vivo pharmacokinetics and pharmacodynamics of the lantibiotic NAI-107 in a neutropenic murine thigh infection model. Antimicrob. Agents Chemother. 2015, 59, 1258–1264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wicha, S.G.; Clewe, O.; Svensson, R.J.; Gillespie, S.H.; Hu, Y.; Coates, A.R.M.; Simonsson, U.S.H. Forecasting Clinical Dose-Response From Preclinical Studies in Tuberculosis Research: Translational Predictions With Rifampicin. Clin. Pharmacol. Ther. 2018, 104, 1208–1218. [Google Scholar] [CrossRef]
- Nielsen, E.I.; Cars, O.; Friberg, L.E. Predicting in vitro antibacterial efficacy across experimental designs with a semimechanistic pharmacokinetic-pharmacodynamic model. Antimicrob. Agents Chemother. 2011, 55, 1571–1579. [Google Scholar] [CrossRef]
- Nielsen, E.I.; Khan, D.D.; Cao, S.; Lustig, U.; Hughes, D.; Andersson, D.I.; Friberg, L.E. Can a pharmacokinetic/pharmacodynamic (PKPD) model be predictive across bacterial densities and strains? External evaluation of a PKPD model describing longitudinal in vitro data. J. Antimicrob. Chemother. 2017, 72, 3108–3116. [Google Scholar] [CrossRef]
- Martinez, M.N.; Papich, M.G.; Drusano, G.L. Dosing regimen matters: The importance of early intervention and rapid attainment of the pharmacokinetic/pharmacodynamic target. Antimicrob. Agents Chemother. 2012, 56, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Nolting, A.; Dalla Costa, T.; Rand, K.H.; Derendorf, H. Pharmacokinetic-pharmacodynamic modeling of the antibiotic effect of piperacillin in vitro. Pharm. Res. 1996, 13, 91–96. [Google Scholar] [CrossRef]
- de la Peña, A.; Gräbe, A.; Rand, K.H.; Rehak, E.; Gross, J.; Thyroff-Friesinger, U.; Müller, M.; Derendorf, H. PK-PD modelling of the effect of cefaclor on four different bacterial strains. Int. J. Antimicrob. Agents 2004, 23, 218–225. [Google Scholar] [CrossRef]
- Sadiq, M.W.; Nielsen, E.I.; Khachman, D.; Conil, J.M.; Georges, B.; Houin, G.; Laffont, C.M.; Karlsson, M.O.; Friberg, L.E. A whole-body physiologically based pharmacokinetic (WB-PBPK) model of ciprofloxacin: A step towards predicting bacterial killing at sites of infection. J. Pharmacokinet. Pharmacodyn. 2017, 44, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Broeker, A.; Nowak, H.; Rahmel, T.; Nussbaumer-Pröll, A.; Österreicher, Z.; Zeitlinger, M.; Wicha, S.G. A pharmacometric approach to define target site-specific breakpoints for bacterial killing and resistance suppression integrating microdialysis, time-kill curves and heteroresistance data: A case study with moxifloxacin. Clin. Microbiol. Infect. 2020, 26, 1255.e1–1255.e8. [Google Scholar] [CrossRef]
- Barbour, A.M.; Schmidt, S.; Zhuang, L.; Rand, K.; Derendorf, H. Application of pharmacokinetic/pharmacodynamic modelling and simulation for the prediction of target attainment of ceftobiprole against meticillin-resistant Staphylococcus aureus using minimum inhibitory concentration and time-kill curve based approaches. Int. J. Antimicrob. Agents 2014, 43, 60–67. [Google Scholar] [CrossRef]
- Liu, P.; Rand, K.H.; Obermann, B.; Derendorf, H. Pharmacokinetic-pharmacodynamic modelling of antibacterial activity of cefpodoxime and cefixime in in vitro kinetic models. Int. J. Antimicrob. Agents 2005, 25, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Nestorov, I. Whole body pharmacokinetic models. Clin. Pharmacokinet. 2003, 42, 883–908. [Google Scholar] [CrossRef] [PubMed]
- Theil, F.P.; Guentert, T.W.; Haddad, S.; Poulin, P. Utility of physiologically based pharmacokinetic models to drug development and rational drug discovery candidate selection. Toxicol. Lett. 2003, 138, 29–49. [Google Scholar] [CrossRef]
- Khan, D.D.; Lagerbäck, P.; Cao, S.; Lustig, U.; Nielsen, E.I.; Cars, O.; Hughes, D.; Andersson, D.I.; Friberg, L.E. A mechanism-based pharmacokinetic/pharmacodynamic model allows prediction of antibiotic killing from MIC values for WT and mutants. J. Antimicrob. Chemother. 2015, 70, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Brill, M.J.E.; Kristoffersson, A.N.; Zhao, C.; Nielsen, E.I.; Friberg, L.E. Semi-mechanistic pharmacokinetic–pharmacodynamic modelling of antibiotic drug combinations. Clin. Microbiol. Infect. 2018, 24, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.B.; Ledesma, K.R.; Chang, K.T.; Schwartz, M.S.; Motyl, M.R.; Tam, V.H. In vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 56, 3753–3757. [Google Scholar] [CrossRef] [PubMed]
- Ly, N.S.; Bulitta, J.B.; Rao, G.G.; Landersdorfer, C.B.; Holden, P.N.; Forrest, A.; Bergen, P.J.; Nation, R.L.; Li, J.; Tsuji, B.T. Colistin and doripenem combinations against Pseudomonas aeruginosa: Profiling the time course of synergistic killing and prevention of resistance. J. Antimicrob. Chemother. 2015, 70, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- VanScoy, B.D.; Trang, M.; McCauley, J.; Conde, H.; Bhavnani, S.M.; Friedrich, L.V.; Alexander, D.C.; Ambrose, P.G. Pharmacokinetics-Pharmacodynamics of a Novel β-Lactamase Inhibitor, CB-618, in Combination with Meropenem in an In Vitro Infection Model. Antimicrob. Agents Chemother. 2016, 60, 3891–3896. [Google Scholar] [CrossRef]
- Zhao, M.; Bulman, Z.P.; Lenhard, J.R.; Satlin, M.J.; Kreiswirth, B.N.; Walsh, T.J.; Marrocco, A.; Bergen, P.J.; Nation, R.L.; Li, J.; et al. Pharmacodynamics of colistin and fosfomycin: A ‘treasure trove’ combination combats KPC-producing Klebsiella pneumoniae. J. Antimicrob. Chemother. 2017, 72, 1985–1990. [Google Scholar] [CrossRef] [PubMed]
- Sadouki, Z.; McHugh, T.D.; Aarnoutse, R.; Ortiz Canseco, J.; Darlow, C.; Hope, W.; van Ingen, J.; Longshaw, C.; Manissero, D.; Mead, A.; et al. Application of the hollow fibre infection model (HFIM) in antimicrobial development: A systematic review and recommendations of reporting. J. Antimicrob. Chemother. 2021, 76, 2252–2259. [Google Scholar] [CrossRef]
- Sy, S.K.B.; Zhuang, L.; Xia, H.; Beaudoin, M.-E.; Schuck, V.J.; Nichols, W.W.; Derendorf, H. A mathematical model-based analysis of the time–kill kinetics of ceftazidime/avibactam against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 1295–1304. [Google Scholar] [CrossRef]
- Kristoffersson, A.N.; Bissantz, C.; Okujava, R.; Haldimann, A.; Walter, I.; Shi, T.; Zampaloni, C.; Nielsen, E.I. A novel mechanism-based pharmacokinetic–pharmacodynamic (PKPD) model describing ceftazidime/avibactam efficacy against β-lactamase-producing Gram-negative bacteria. J. Antimicrob. Chemother. 2019, 75, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.K.B.; Zhuang, L.; Xia, H.; Schuck, V.J.; Nichols, W.W.; Derendorf, H. A model-based analysis of pharmacokinetic-pharmacodynamic (PK/PD) indices of avibactam against Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2019, 25, 904.e9–904.e16. [Google Scholar] [CrossRef]
- Wicha, S.G.; Huisinga, W.; Kloft, C. Translational Pharmacometric Evaluation of Typical Antibiotic Broad-Spectrum Combination Therapies Against Staphylococcus Aureus Exploiting In Vitro Information. CPT Pharmacom. Syst. Pharmacol. 2017, 6, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.D.; Friberg, L.E.; Nielsen, E.I. A pharmacokinetic-pharmacodynamic (PKPD) model based on in vitro time-kill data predicts the in vivo PK/PD index of colistin. J. Antimicrob. Chemother. 2016, 71, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Thorsted, A.; Nielsen, E.I.; Friberg, L.E. Pharmacodynamics of immune response biomarkers of interest for evaluation of treatment effects in bacterial infections. Int. J. Antimicrob. Agents 2020, 56, 106059. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L.; Fregeau, C.; Liu, W.; Brown, D.L.; Louie, A. Impact of burden on granulocyte clearance of bacteria in a mouse thigh infection model. Antimicrob. Agents Chemother. 2010, 54, 4368–4372. [Google Scholar] [CrossRef]
- Drusano, G.L.; Vanscoy, B.; Liu, W.; Fikes, S.; Brown, D.; Louie, A. Saturability of granulocyte kill of Pseudomonas aeruginosa in a murine model of pneumonia. Antimicrob. Agents Chemother. 2011, 55, 2693–2695. [Google Scholar] [CrossRef]
- Guo, B.; Abdelraouf, K.; Ledesma, K.R.; Chang, K.T.; Nikolaou, M.; Tam, V.H. Quantitative impact of neutrophils on bacterial clearance in a murine pneumonia model. Antimicrob. Agents Chemother. 2011, 55, 4601–4605. [Google Scholar] [CrossRef] [PubMed]
- Boisson, M.; Jacobs, M.; Grégoire, N.; Gobin, P.; Marchand, S.; Couet, W.; Mimoz, O. Comparison of intrapulmonary and systemic pharmacokinetics of colistin methanesulfonate (CMS) and colistin after aerosol delivery and intravenous administration of CMS in critically ill patients. Antimicrob. Agents Chemother. 2014, 58, 7331–7339. [Google Scholar] [CrossRef] [PubMed]


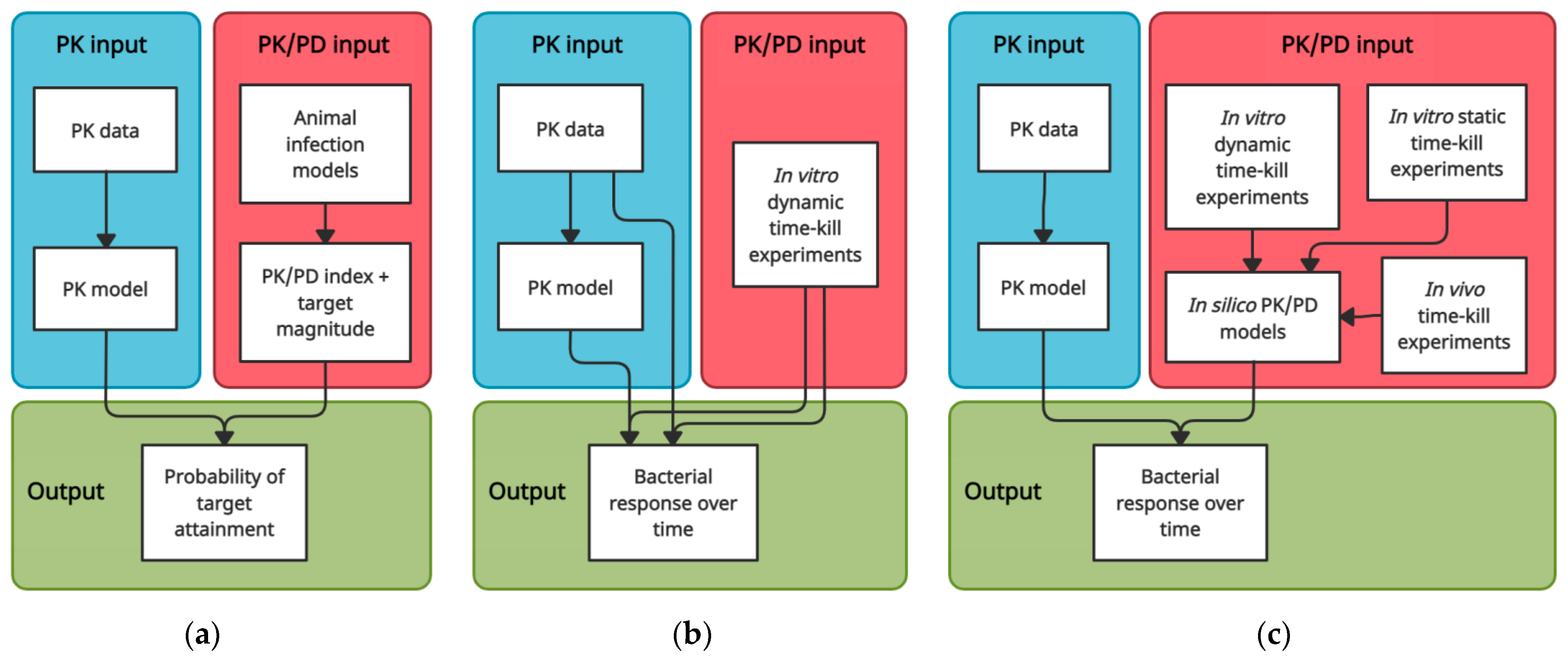{kind=link}
| PTA Analyses Using PK/PD Index Targets | In Vitro Dynamic Time–Kill Experiments | In Vivo Time–Kill Experiments | Computational PK/PD Modelling | |||||
|---|---|---|---|---|---|---|---|---|
| + | − | + | − | + | − | + | − | |
| PK aspects | Variability in PK is considered | PK/PD target set based on animal PK Based on plasma PK Assumes equal tissue distribution in animals and humans Ignores shape of PK profile | PK profiles can be exactly mimicked | Variability in PK is often not considered | - | Animal PK drives effect Often plasma PK is linked to drug effect | Bacterial response to any PK profile can be simulated Variability in PK can be taken into account | - |
| PD aspects | PK/PD target set based on in vivo data Can be calculated across MICs | PK/PD target set based on PD readout at 24 h Provides no detailed PD information (e.g., rate or extent of kill) Resistance development usually not considered Relies heavily on MIC Within-MIC variability in PD is not considered | Depicts PD time-course Resistance development can be followed | Performed in in vitro environment | Performed in in vivo environment Depicts PD time-course Resistance development can be followed | PD time-course cannot be followed within the same animal | Depicts PD time-course Resistance development can be simulated Variability in PD can be considered Data from multiple sources (e.g., in vivo) can be incorporated | Mostly based on in vitro data, often from static time–kill experiments Dependent on MIC to predict effect on untested strains |
| Practical aspects | PK/PD targets are available for most antibiotics Economical approach | If no PK/PD target is available, animal studies must usually be performed | Can be performed without a PK model | Setups can be complex and resource-intensive Limited number of strains, PK profiles and conditions can be investigated | Can be performed without a PK model | Requires large number of animals, ethical considerations Resource-intensive Limited number of strains, doses and conditions can be investigated | Can be based on data from simple and cheap experiments Untested scenarios can be simulated; these can also inform optimal design of further studies | May be complex and time-consuming Dependent on availability and quality of experimental data |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Os, W.; Zeitlinger, M. Predicting Antimicrobial Activity at the Target Site: Pharmacokinetic/Pharmacodynamic Indices versus Time–Kill Approaches. Antibiotics 2021, 10, 1485. https://doi.org/10.3390/antibiotics10121485
van Os W, Zeitlinger M. Predicting Antimicrobial Activity at the Target Site: Pharmacokinetic/Pharmacodynamic Indices versus Time–Kill Approaches. Antibiotics. 2021; 10(12):1485. https://doi.org/10.3390/antibiotics10121485
Chicago/Turabian Stylevan Os, Wisse, and Markus Zeitlinger. 2021. "Predicting Antimicrobial Activity at the Target Site: Pharmacokinetic/Pharmacodynamic Indices versus Time–Kill Approaches" Antibiotics 10, no. 12: 1485. https://doi.org/10.3390/antibiotics10121485
APA Stylevan Os, W., & Zeitlinger, M. (2021). Predicting Antimicrobial Activity at the Target Site: Pharmacokinetic/Pharmacodynamic Indices versus Time–Kill Approaches. Antibiotics, 10(12), 1485. https://doi.org/10.3390/antibiotics10121485