1. Introduction
Lateral flow immunoassay (LFIA) is a rapid technique which relies on the fast interaction between an antibody and a target antigen [
1]. These devices have experienced a surge in popularity in the medical and food safety fields, since their birth as home-pregnancy tests [
2]. It is preferred for LFIAs to use purified, fast, specific and properly characterized antibodies [
3]. Although LFIAs are classified as a rapid method, they still require 10–20 min to complete [
4]. In order to create high-speed LFIAs, it is necessary to test the antibody rate of association towards the target analyte, as well as use a nitrocellulose membrane with a high flow rate. Traditional antibody selection techniques, such as enzyme linked immunosorbent assay (ELISA) and western blot, do not necessarily convert well into LFIAs due to the much faster rate of kinetics in LFIA [
5]. As trends move toward rapid on-site testing, with consumer-friendly tests such as LFIA and smartphone-based readout systems, the need for antibodies with rapid association towards their target becomes more apparent [
4]. In addition to requiring fast antibodies, it is necessary to have a rational way of quickly comparing and selecting such antibodies. One way of speeding up the antibody screening and LFIA prototyping process is to use a label-free biosensor to compare relative antibody-antigen association binding speeds to facilitate the selection process [
6,
7,
8].
Surface plasmon resonance (SPR) is one such technique. SPR allows label-free, optical monitoring of important kinetic information, such as the association and dissociation rates of antibodies, in real time [
9]. Using SPR it is possible to screen crude antibodies. Herein, the term crude refers to: a mixture of un-purified, cell culture media with variable specific antibody concentrations. Screening crude monoclonal antibodies (mAbs) saves time and money in comparison with first purifying a panel of mAbs and then testing them all for application in LFIA [
10]. Previously, true kinetic studies have been carried out to select antibodies based on their affinities, association and dissociation rates, for application in a direct SPR biosensor [
11]. In the medical sector there is interest in screening and ranking hybridomas, hybrid cells formed from the antibody producing spleen cell of an immunized animal fused with a myeloma cell, for their affinities. However, studies in the literature have not yet focused on the ranking and selection of antibodies based on their association rates towards food allergens, for application in rapid ligand-binding assays such as LFIA [
6,
12,
13]. Current antibody selection processes using SPR are affinity based and the antibodies are screened against purified analytes. By contrast, in this study, an unpurified hazelnut extract, which is a complex mixture of heterogeneous proteins of various molecular weights, is the target analyte [
14].
When developing sandwich format assays for large molecular weight proteins (e.g., food allergens) it is essential to select appropriate antibody pairs for the capture and detection of the target analyte [
15]. Hazelnut has been selected as the target for this study, as hazelnut is considered the most prevalent tree nut allergy in Europe [
16]. Sandwich pairs are antibodies that are capable of simultaneously binding an antigen. Pre-matched antibody pairs can be purchased from commercial vendors which can save time and resources, or they can be selected through sandwich pairing experiments [
17]. These pairs are often found using ELISA. However, the results obtained in ELISA do not always predict how the antibodies will perform in LFIA [
3]. Alternatively, antibody pairs can be determined by using a half-stick format LFIA [
18]. Pairwise selection can also be achieved using biosensors by epitope binning [
19]. This process assesses whether antibodies bind to overlapping epitopes on the target antigen, or whether they are capable of binding to different epitopes [
20]. Using SPR to select antibody pairs for use in LFIA saves time and can be largely automated for screening large antibody panels for their pairs [
20].
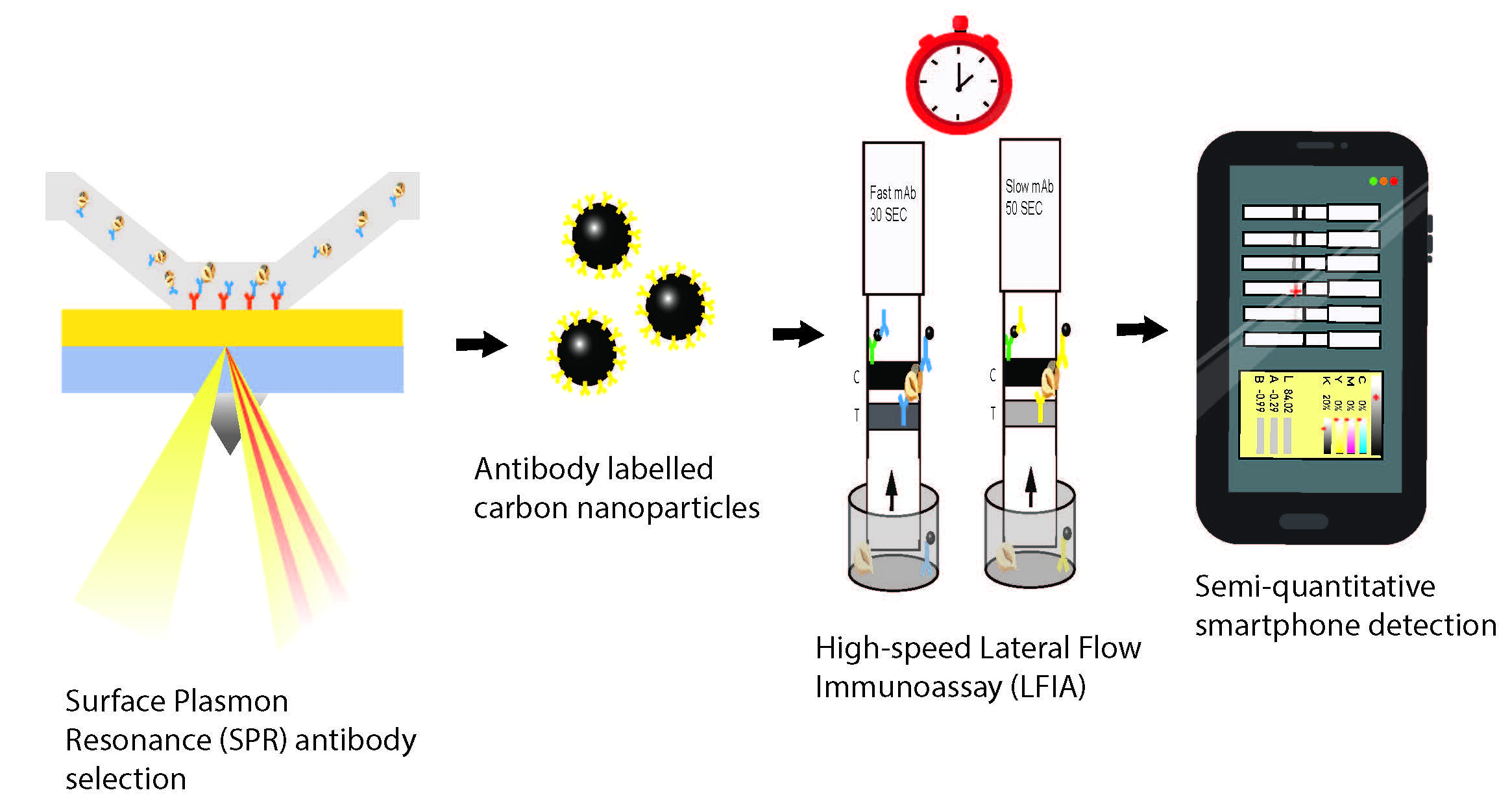
To the best of the authors’ knowledge, this is the first example of using SPR as a screening method for selection of high-quality antibodies from crude samples for application in LFIA. SPR has been utilized for selection of purified mAbs for these characteristics, illuminating its importance as a selection tool in this sector [
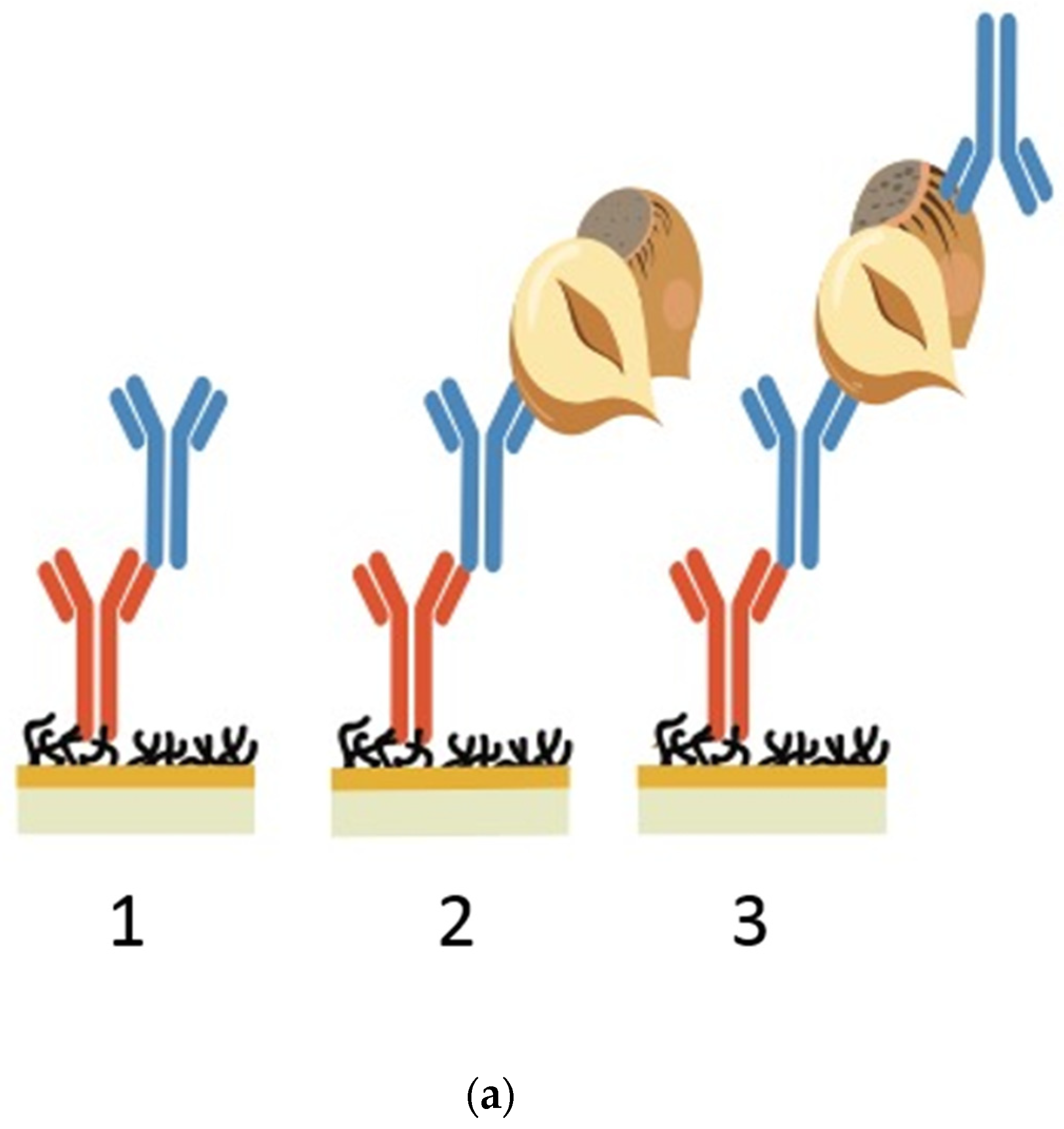
18]. A batch screening method was designed using an FC-specific anti-mouse IgG (FC-IgG) immobilised onto an SPR chip. The FC-IgG captures the anti-hazelnut antibodies of interest on the surface. Subsequently, hazelnut protein extract is injected and the binding between the antibody and hazelnut is monitored. Using an FC-IgG surface offers on chip affinity purification of the crude sample, as it captures the crude anti-hazelnut antibodies in their FC region. This allows the captured crude antibodies to be uniformly distributed, in the assumed correct orientation, without any compromise of their biological activity [
21]. Furthermore, using an FC-IgG expedites the regeneration of the chip surface between each cycle for screening subsequent antibodies. Following normalization, to compensate for differences in specific antibody concentrations in crude samples, a visual assessment can be made to compare the relative association rates of each mAb towards hazelnut. The un-purified antibodies can then be ranked based on their fast association, sensitivity, specificity and sandwich pairing. As a result, fast (F) and slow (S) antibody pairs selected by SPR were used to develop a carbon nanoparticle-based LFIA system, to identify whether similar kinetics could be observed in LFIA as was seen in SPR. Amorphous carbon nanoparticles are excellent labels in LFIA as they are easy and low-cost to prepare; have high signal to background contrast, making them easier to read with the naked eye; and can allow for increased sensitivity compared with other labels [
22]. Even lower LOD’s might be achieved for carbon nanoparticle-labelled LFIAs by using a flatbed scanner to determine grey pixel values. An alternative, more consumer-orientated method is to use smartphone apps to determine RGB/CMYK values of the test line region of the LFIAs and to convert these to LAB (where L is Luminance and A and B are color channels) values. Whilst RGB (red, green, blue) and CMYK (cyan, magenta, yellow, key) values are device dependent, LAB values provide device independent information about the darkness/lightness of a selected region of an image [
23]. In this way a calibration curve of LAB color values against allergen concentration (ppm) can be plotted for semi-quantification of LFIA results. Furthermore, there are currently no food allergen LFIAs that apply carbon nanoparticles, exemplifying the label novelty in this field [
24]. The LFIA prototypes developed were compared based on their speed and sensitivity and applied to a real food matrix of cookies as a proof-of-concept. Cookies have been selected for a matrix as a 2018 report determined that products such as cookies, chocolate and bread are responsible for the majority of accidental allergic reactions [
25]. Finally, the LFIAs were semi-quantified by a smartphone using freely downloadable color analysis apps.
2. Materials and Methods
2.1. Equipment
All SPR experiments were carried out using a BIACORE 3000 (GE Healthcare, Uppsala, Sweden). An EL x 808 BioSPX Microplate Reader was used for the determination of the Bicinchoninic acid (BCA) results (Beun De Ronde, Abcoude, The Netherlands). A NanoDrop ND-3300 (Isogen Life Sciences, De Meern, The Netherlands) or the DeNovix DS-11 spectrophotometer (DeNovix, Wilmington, DE, USA) was used for all other protein quantifications. A Braun Turbo 600 W Food Processor (Kronberg im Taunus, Germany) was used for homogenizing the food samples. All food extracts were filtered through low-binding syringe filters (5 to 0.45 µm; Pall Life Sciences, Portsmouth, UK). The LFIA strips were sprayed using a Linomat IV TLC-spotter (CAMAG, Berlin, Germany). The CM4000 BioDot Guillotine (Biodot Inc., Irvine, CA, USA) was used to cut the strips. A Bioruptor Plus Diagenode (Diagenode SA, Seraing, Belgium) was used to sonicate the carbon nanoparticle suspensions. All smartphone video recordings and photos were taken using a Google Pixel 2 XL (Google, Mountain View, CA, USA). All smartphone-based color detection was accomplished using ‘RGB Color Detector’ (version 1.0.35, The Programmer; Google Play Store) and color conversions using ‘Nix Pro Color Sensor’ (version 1.28; Nix Sensor Ltd., Hamilton, ON, Canada; Google Play Store).
2.2. Chemicals & Reagents
The SPR experiments were carried out using carboxymethylated dextran sensor chips (CM5), HBS-EP buffer (pH 7.4, consisting of 10 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, 150 mM sodium chloride, 3 mM ethyldiaminetetraacetic acid, 0.005% v/v surfactant polysorbate 20), an amine coupling kit (containing: 0.1 M N-hydroxysuccinimide (NHS), 0.4 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and 1 M ethanolamine hydrochloride (pH 8.4)), all purchased from GE Healthcare (Uppsala, Sweden). Bovine serum albumin (BSA) was purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). Analysis of all SPR results was performed using the BiaEvaluation software (Biacore, Uppsala, Sweden).
The washing buffer (WB) was composed of 5 mM borate buffer (BB) (pH 8.8) diluted from a mixture of 100 mM sodium tetraborate (VWR, Leuven, Belgium) and 100 mM boric acid (Merck, Darmstadt, Germany), and bovine serum albumin (BSA) was added to a final concentration of 1% (
w/
v). The storage buffer (SB) consisted of 100 mM BB containing BSA to a final concentration of 1% (
w/
v). The running buffer (RB) was prepared by adding 1% BSA (
w/
v) and 0.05% Tween-20 (
v/
v) (Merck, Darmstadt, Germany) to 100 mM BB. TRIS-buffered saline (TBS; pH 8.2) was prepared from 20 mM TRIS (Duchefa Biochemie, Haarlem, The Netherlands) and 300 mM NaCl (Merck, Darmstadt, Germany). Phosphate-buffered saline (PBS) pH 7.4 was purchased from Sigma-Aldrich (Sigma-Aldrich, St Louis, MO, USA). The BCA reagents were purchased from Pierce (Rockford, IL, USA). All solutions were prepared with MQ water from a MilliQ-system (>18.2 MΩ/cm) purchased from Millipore (Burlington, MA, USA). ‘Spezial Schwartz 4’ carbon nanoparticles were purchased from Degussa AG (Frankfurt, Germany). Goat anti-mouse IgG Fc specific antibody in PBS (2.4 mg/mL) used in the SPR study was purchased from ThermoFisher Scientific (Landsmeer, The Netherlands). Goat anti-mouse IgG in PBS (pH 7.6) (1.2 mg/mL; AffiniPure F(ab’)
2 Fragment GAM IgG Fcγ) used for spraying LFIA control lines was purchased from Jackson Immunoresearch Laboratories Inc (Sanbio, Uden, The Netherlands). All other antibodies were developed by RIKILT, Wageningen University & Research (Wageningen, The Netherlands), according to the procedure described in [
26,
27]. In short, the antibody panel listed in
Table 1 was produced by immunizing mice with 50 µg extracted hazelnut (mixed) protein, with booster immunizations containing 25 µg extracted hazelnut protein. Antibodies selected for LFIA were purified using a HiTrap Protein G column (GE Healthcare, Uppsala, Sweden). Briefly, antibodies were collected from 1 L of raw cell culture media by ammonium sulphate precipitation and subsequent affinity chromatography purification. Following this method, around 15–20 mg of purified antibodies was obtained from 1 L of raw cell culture medium.
2.3. Allergen Extractions
Certified standardized reference materials for food allergens are not commercially available and so antigen standards require in-house preparation. Allergen extracts were made from a ‘blank’ matrix of organic whole meal digestive biscuits (containing: flour, palm oil, sugar, barley malt extract, sodium bicarbonate, ammonium bicarbonate, salt; Dove’s Farm Organic Whole meal Digestive Biscuits; Dove’s Farm, Berkshire, UK), from hazelnut cookies (TimeOut Hazelnoot Granenbiscuits containing: 10% hazelnut, egg, milk & sesame; Albert Heijn, The Netherlands) and from hazelnuts, pecan nuts, pistachio nuts, brazil nuts, peanuts, cashew nuts, almonds, walnuts and macadamia nuts, which were all purchased from a local supermarket. All extracts were filtered through a series (5 µm, 1.2 µm, 0.45 µm) of low protein-binding syringe filters. For the SPR study, whole raw hazelnuts were frozen at −80
°C for 4 h. The frozen hazelnuts were homogenized to a fine powder using a commercial hand blender. The protein was extracted by adding 10 mL of heated TBS buffer per gram of ground hazelnut. The mixture was vortexed for 30 s before rotating end-over-end for 30 min at 37
°C. The solution was centrifuged at room temperature for 15 min at 4000×
g. The resulting liquid phase was filtered through a series of low protein-binding syringe filters. Total protein concentrations were determined according to the BCA protein assay using BSA as the standard. All hazelnut protein extracts were aliquoted and stored at −20
°C until use. For the cross-reactivity study, a universal allergen extraction procedure was applied that can be used to simultaneously extract multiple different food allergens. Extracts were made from hazelnut, peanut, pecan, pistachio, walnut, brazil nut, macadamia nut, almond and cashew following the method described by Raz [
28]. Briefly, nuts were homogenized using a Braun Turbo 600 W Food Processor, and 0.25 g sample portions were weighed out. Twenty-five millilitres of PBS (pH 7.4) was added to the ground samples and incubated at room temperature for 1 h. Following incubation, extracts were centrifuged at 3220×
g for 20 min. The extracts were then filtered through a series of low protein-binding syringe filters, aliquoted and stored at −20
°C until use. The same procedure was applied for the matrix extraction of the ‘Blank’ matrix and hazelnut cookies but using a 2.5 g ground food in 25 mL PBS. Total protein contents of all allergen/matrix extracts were determined using the NanoDrop.
2.4. Biosensor Chip Preparation
A standard amine coupling procedure was applied at 25 °C to immobilize the Fc-Specific IgG (FC-IgG) onto the CM5 surface. Immobilization pH scouting for coupling of FC-IgG to CM5 chip was performed. The FC-IgG was diluted to 20 µg/mL in 10 mM sodium acetate of varying pH’s and tested using the pH scouting wizard in the Biacore 3000 control software (Uppsala, Sweden). A high immobilization level was reached at pH 5.5, so sodium acetate pH 5.5 was selected as the immobilization buffer in the following procedure.
The four flow channels, clamped against the carboxylmethylated (CM) dextran chip surface, were simultaneously activated by injecting 35 µL of a mixture of EDC and NHS (1:1 v/v) at a flow rate of 5 µL/min. Then, FC-IgG diluted (20 µg/mL) in coupling buffer (10 mM sodium acetate, pH 5.5) was injected in flow cells 2–4, and FC-IgG was attached to the activated CM-dextran surface via its exposed primary amine groups. Flow cell 1 was used as a reference channel and was left blank and was only activated by EDC/NHS. The coupling was followed by blocking the remaining active ionic groups in all flow cells with ethanolamine (1 M) preventing electrostatic interactions with the CM-dextran surface. Around 10,000 RU of FC-IgG was immobilized in each channel (2–4) using this method; this high level was aimed for in order to properly cover the chip surface with FC-IgG for the subsequent capture of the specific anti-hazelnut mAbs of interest.
2.5. Crude Antibody Screening Assay
The screening analysis was performed at 25
°C using HBS-EP (pH 7.4) as the screening buffer. The crude antibodies were diluted 1/20 in the screening buffer. The hazelnut protein extract was diluted to 20 ppm in the screening buffer. Twenty microliters of each crude antibody dilution was injected at a flow rate of 20 µL per minute for capture. These flow conditions were selected to more accurately reflect the fast flow kinetics observed in LFIAs. Subsequently, 20 µL of 20 ppm hazelnut extract was injected at a flow rate of 20 µL per minute. The surfaces were immediately regenerated with 2 pulses of 5 µL, 5 mM NaOH to return the biosensor signal to baseline [
29]. A range of different regeneration conditions were tested, including glycine, HCl and different strengths, volumes and flow rates of NaOH. Of all the tested regeneration conditions, 2 short NaOH pulses were found to be the most appropriate for removing both strong and weak binders whilst minimising FC-IgG surface deterioration, and these were applied as the standard regeneration conditions.
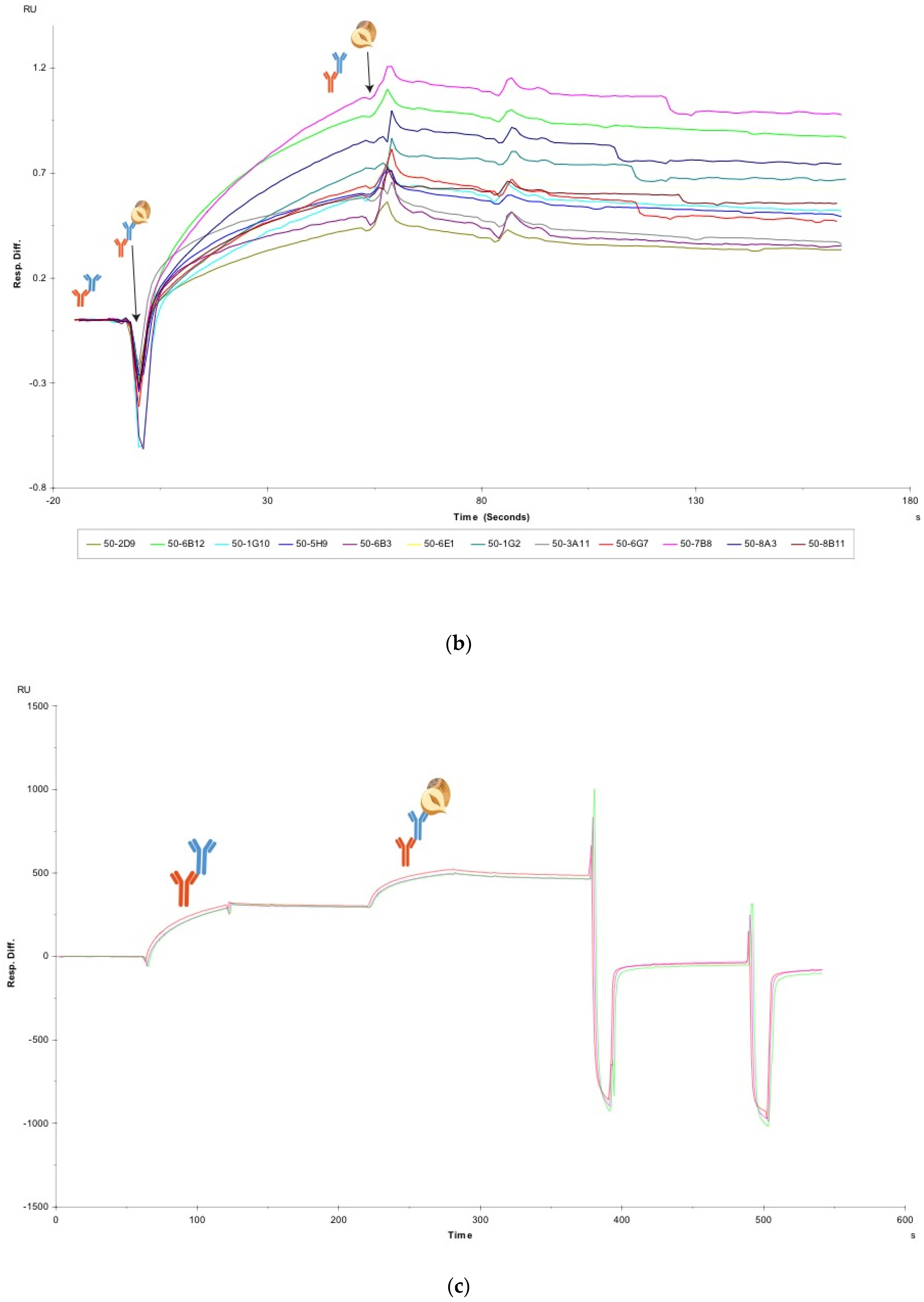
Using the Biaevaluation software (Biacore, Uppsala, Sweden), the whole sensorgrams for each crude antibody capture and antigen binding cycle were superimposed. As the antigen in this study is comprised of heterogeneous proteins, the curves do not conform to Langmuir binding models. Therefore, as this study is focused on a rapid screening process, a full kinetic curve fitting was not performed. The sensorgrams were aligned on the x-axis at the hazelnut antigen injection point. A snapshot of the relevant part of the sensorgram, containing the hazelnut association and dissociation data, was made in the software. The sensorgrams were double referenced, first by using flow cell 1 as a blank reference channel for buffer signal subtraction and subsequently by normalizing the hazelnut response by dividing the antigen response by the corresponding crude antibody capture level, as described in [
11]. All sensorgrams were y-axis zeroed to baseline. After data processing and removal of the FC-IgG capture curve and the regeneration peaks, a visual assessment of the association rates of each antibody towards hazelnut could be achieved. The visual assessment of the steepness of the association curves for the crude antibodies toward hazelnut was confirmed using the slope analysis function in Microsoft Excel.
2.6. Cross-Reactivity Testing
Total protein extracts from tree nuts and peanut (in PBS) were protein content determined using the NanoDrop and then were diluted to 100 ppm in HBS-EP. Three different antibodies were captured by the FC-IgG surface, in individual flow cells, at a flow rate of 20 µL per minute. During the first cycle, 20 ppm hazelnut extract was injected as a control to monitor the binding response of these crude antibodies towards hazelnut. Following this, the surface was regenerated with the standard regeneration conditions. Subsequently, the same antibodies were re-captured and 20 µL of one of the other tree nut/peanut protein extracts was injected over the antibodies using the same flow conditions. Following this, the surface was regenerated using 1 or 2 pulses of 5 mM NaOH, depending on the extent of tree nut/peanut binding. The procedure was repeated for all of the tree nut/peanut extracts.
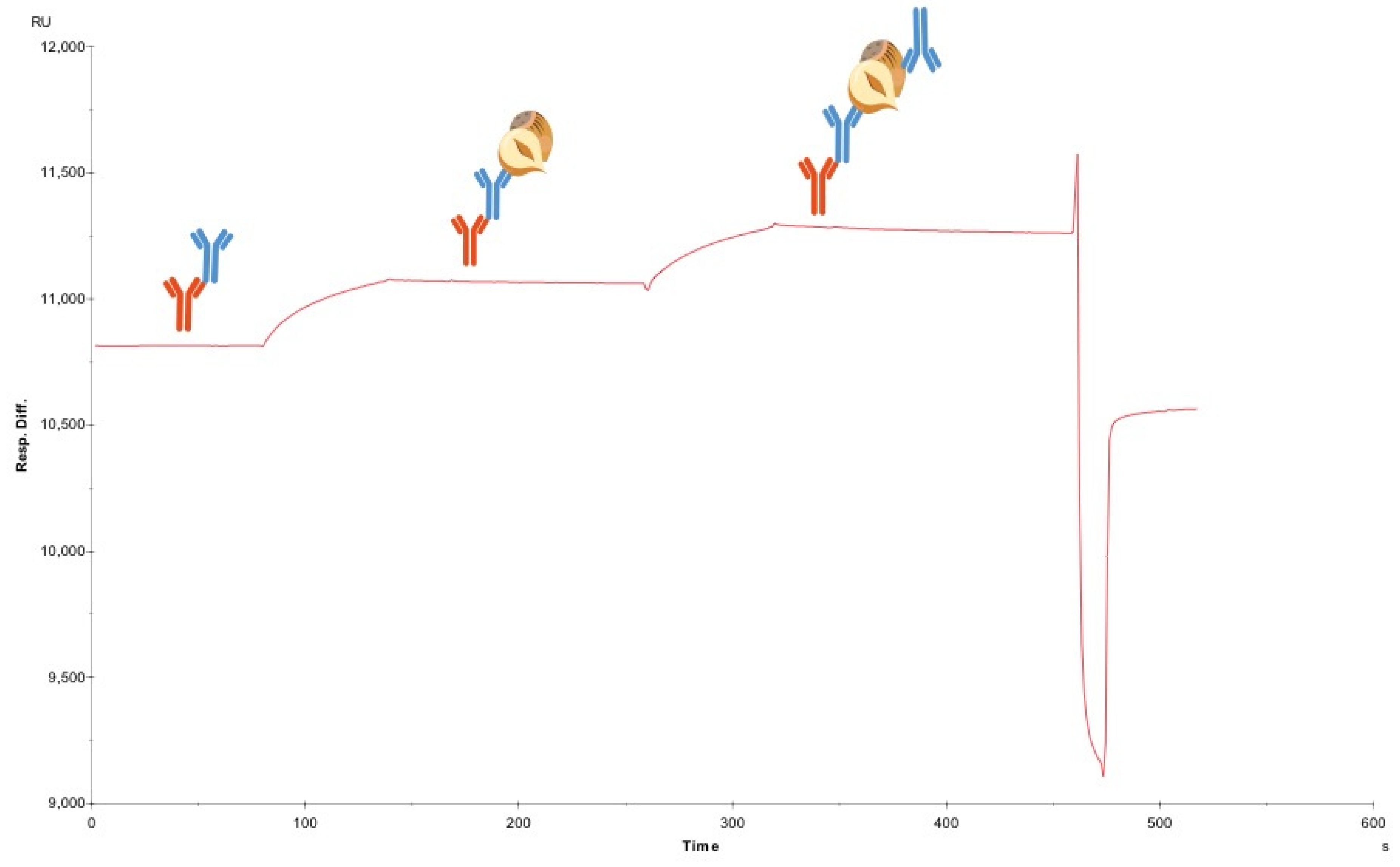
2.7. Sandwich Pairing Assay
Twenty microliters of each of three antibodies was captured in individual flow cells at a flow rate of 20 µL per minute. Next, 20 µL of 20 ppm hazelnut extract was injected over all flow cells simultaneously at flow of 20 µL per minute. Subsequently, 20 µL of one crude antibody was injected over all three flow cells, generating data for one antibody against itself and against two other antibodies. Following this, the surface was regenerated with standard conditions to return the signal to baseline.
2.8. Labelling with Carbon Black Nanoparticles
A 1% suspension of carbon nanoparticles was prepared by adding 1 mL of MilliQ Water (MQ) to 10 mg carbon and sonicating for 10 min. The resulting 1% carbon suspension was diluted five times in 5 mM BB (pH 8.8) to obtain a 0.2% suspension, which was then sonicated for a further 5 min. Next, 350 µg of purified anti-hazelnut antibody was added per 1 mL of 0.2% carbon suspension and stirred overnight at 4
°C. The suspension was divided into two aliquots and 500 µL of WB was added to each and centrifuged for 15 min at 13,636×
g at 4
°C. Following this, the supernatants were removed and the pellets re-suspended in WB, this process was repeated 3 times. After the final wash, the supernatants were discarded, and the pellets were pooled together with 1 mL storage buffer and stored at 4
°C until use. Scanning electron microscopy (SEM) images of F-50-6B12-carbon nanoparticle suspension can be seen in the
Supplementary Material (Figure S1).
2.9. Lateral Flow Immunoassay
2.9.1. Preparation of Lateral Flow Immunoassay Prototype
Lateral flow strips were manufactured using nitrocellulose (NC) membranes (HiFlow Plus HF13502; Millipore, Carrigtwohill, Co. Cork, Ireland) cut to approximately 2.5 cm in length; an SEM image of the NC membrane can be seen in the
Supplementary Material (Figure S2). The NC membrane was secured on a plastic backing (G & L, San Jose, CA, USA), with 4.5 cm of absorbent pad (Schleicher & Schuell, Dassel, Germany) overlapping one end of the NC. Four LFIAs were prepared for each antibody, with different antibody concentrations dispensed onto the test line to determine the optimum conditions. A TLC spotter was used to dispense the test line (the anti-hazelnut antibody at 0.2 mg/mL, 0.15 mg/mL, 0.1 mg/mL or 0.05 mg/mL) at 1.2 cm and the control line (Goat anti-mouse Fab Fragment at 0.1 mg/mL) at 1.5 cm from the sample application end of LFIA. The TLC spotter used 1 µL of antibody per 5 mm wide strip, at a speed of 15 µL per second. The membranes were allowed to dry at room temperature for 30 min. Finally, 5-mm-wide strips were cut using the BioDot Guillotine CM4000 (Biodot Inc., Irvine, CA, USA) and were packaged in aluminium pouches with silica desiccation packs, heat-sealed and stored at room temperature until future use.
2.9.2. Lateral Flow Immunoassay: Limit of Detection
First, the visual limit of detection (LOD) of the strip tests was determined using a decreasing concentration of hazelnut protein extract diluted in PBS. Herein, the visual LOD is defined as the lowest concentration of total hazelnut protein capable of resulting in the appearance of a test line. Both strip batches had the same amount of purified anti-hazelnut antibody immobilized on the test line, and both sets of carbon nanoparticle labelled mAbs had 350 µg of antibody immobilized per mL of carbon so that a fair comparison could be made between the two sets of antibodies. For dipstick analysis, a strip test was placed in a well of low binding microtiter plate containing 100 µL running buffer (RB), 1 µL carbon-antibody conjugate and 1 µL hazelnut extract (dilution range: 100, 50, 25, 10, 5, 2.5, 1, 0.5, 0.25, 0.1, 0 ppm) and was allowed to run for 5 min. Subsequently, the visual LOD of the dipsticks in a spiked commodity was determined. To test for matrix LOD’s, total hazelnut protein extract was spiked into a blank cookie extract in the range of 100 ppm to 0.5 ppm (100 ppm, 50 ppm, 25 ppm, 10 ppm, 5ppm, 2.5 ppm, 1 ppm, 0.5 ppm, 0 ppm). The testing procedure was the same as that described above. Additional matrix LOD determinations were made using 50 µL RB, 50 µL spiked commodity and 1 µL carbon conjugated-mAb in order to reduce further dilution that was caused by adding 100 µL of running buffer to 1 µL of sample. In order to establish the real life applicability of the optimal LFIA, the real life matrix of a hazelnut cookie extract was also tested (1 µL sample in 100 µL RB) spiked into a decreasing dilution in a blank cookie extract in the range of 1:1 to 1:1,000,000.
2.9.3. Lateral Flow Immunoassay: Test Line Kinetics
To compare the antibody to hazelnut association rates in LFIA, it is necessary to time the appearance of the test line. The strips were tested by inserting a test strip into a microwell containing 100 µL RB, 1 µL carbon-mAb and 1 µL of 50 ppm hazelnut protein extract (in PBS). A higher concentration of hazelnut extract was used for the kinetic study, as a higher analyte level results in the appearance of a darker line with a high contrast, making it easy to visualize the line as soon as it forms. Instead of allowing the strips to run for 5 min, as soon as a test line appeared on the strip, this time was recorded. The kinetic experiments were repeated multiple times (n = 8) and were assessed visually and by smartphone video recording for the test line formations.
2.9.4. Semi-quantitative Smartphone Lateral Flow Readout
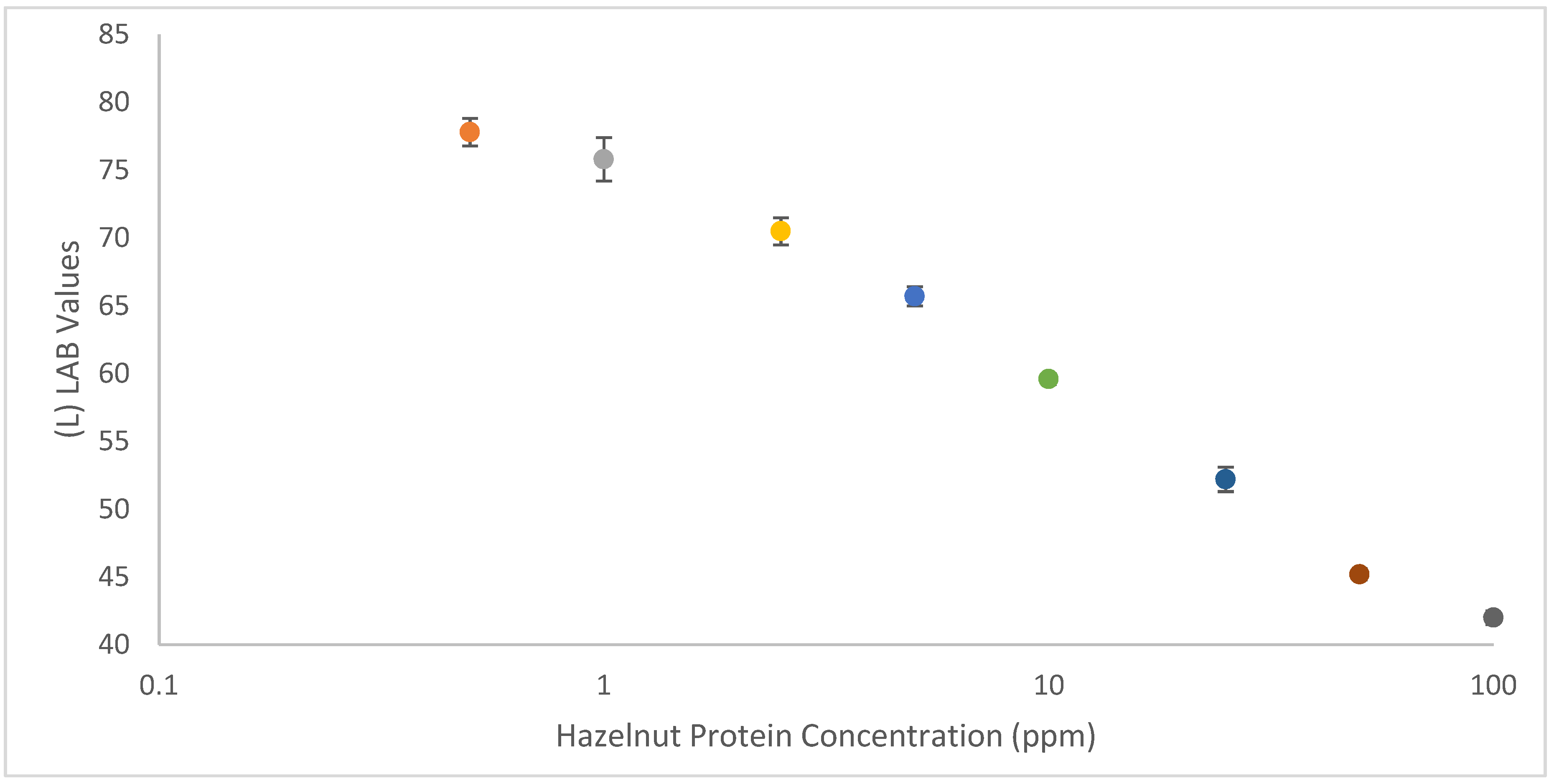
To obtain RGB/CMYK color values, each LFIA in a calibration range (100 ppm, 50 ppm, 25 ppm, 10 ppm, 5 ppm, 2.5 ppm, 1 ppm, 0.5 ppm, 0 ppm) was analysed, with ‘RGB Color Detector’ (version 1.035), by selecting a region of interest in the LFIA test line area using the crosshair function. To obtain fair color values, values were averaged from three distinct points on the test line of the strips (n = 3). Color values were also taken from the background (below the test line) to normalize the results. The ‘Nix Pro Color’ (version 1.28) sensor allows conversion between multiple different color spaces. Therefore, when plugging the RGB or CMYK values obtained in ‘RGB Color Detector’ into the ‘Nix Pro Color’ sensor, it is possible to select a conversion to LAB (or cieLAB) color space. Using the obtained LAB values, a calibration curve was plotted for LAB values vs hazelnut extract spiked into blank cookie extract using an ordinary spreadsheet program.
4. Discussion
Surface plasmon resonance was used to screen antibodies in their un-purified state based on their fast association, specificity and sensitivity towards hazelnut, for use in LFIA. This method saves significant time and resources compared with selecting mAbs by ELISA. In ELISA, it is preferred to use purified mAbs and the antibody purification process takes approximately one day for each antibody. Considering that in this study, 12 mAbs were ranked by SPR as an analysis tool, if these would have first needed purification, it would have taken over a week longer to get to the antibody assessment stage. As the method only requires small volumes of un-purified mAbs, it is possible to start assessing the mAb characteristics as early as the fusion stage. Additionally, as SPR is a label-free technique, even more time is saved by not having to perform additional labelling experiments, and more unequivocal information is obtained from SPR compared with ELISA.
The SPR results made it possible to select a very good and a less optimal antibody pair for application and comparison in a high-speed LFIA. The two prototype LFIAs displayed a significant difference in the timing of the appearance of the test line, with F-50-6B12’s test line appearing at least 20 s before the appearance of the test line on the S-50-5H9 strip. When considering moving towards consumer friendly food allergen detection, it is desirable to have LFIAs that give accurate, positive results, as quickly as possible, so that food can rapidly be assessed before its consumption. Quick allergen analysis can prevent unnecessary allergic reactions by allowing consumers to determine which portions of foods are safe to eat and which should be avoided. The proposed screening method could be extremely useful when trying to select antibodies of similar kinetics to use in a multiplex assay. In this way it would be possible to select capture/detector mAbs for a range of targets which have similar association rates towards their targets, so that when they are utilised in a multiplex assay, the T-lines appear within a similar temporal resolution. The optimal F-50-6B12 strips were able to detect the presence of hazelnut at trace levels in spiked buffer, spiked commodity and a real life hazelnut cookie, highlighting the LFIAs usefulness in real life. The F-50-6B12 LFIA is sensitive enough to protect even for the most sensitive hazelnut allergic individuals. Finally, a semi-quantitative smartphone readout was achieved by using simple and free color analysis apps to obtain device independent LAB values. This proves that even in the absence of additional light-control mechanisms, 3D-printed attachments and dedicated software apps, it is possible for anyone to obtain semi-quantitative LFIA results using their smartphones, provided that mAbs are labelled with carbon nanoparticles. Such apps could also be used to semi-quantify a multiplex assay. This study demonstrates a generically applicable proof-of-concept method for a novel association and sensitivity-based antibody selection procedure that can be applied to crude preparations for consequent application in LFIA with a visual or smartphone readout and an LOD in the low ppm range.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}